

1 Introduction
Most biologists are familiar with the interpretation of sequence alignments between different species. Substitutions, insertions and deletions that occurred since the last common ancestor are commonly noted and analyzed. Manipulating these evolutionary concepts is so habitual that it has probably become an unconscious process. However, capacities or rather deficiencies of the tools that generate alignments probably orient and bias our thought. Popular programs such as blast [1] or blat [2] compare nucleic or protein sequences and provide a measurement of local similarity through a symmetrical result often represented as pairwise alignments, but do not furnish a direct indication of duplications inside a single genome. However, nearly forty years ago in a landmark publication, Susumo Ohno proposed that gene duplications represent a major force in evolution. His basic premise is that by doubling the number of the genes, WGD (Whole Genome Duplications) would facilitate the emergence of new functions, and also promote radiations [3]. Progress in cytogenetic studies, followed by the recent explosion of the number of sequenced genomes has provided the opportunity to investigate the relics of such ancestral events. Several ancient polyploidization events have now been uncovered in Eukaryotes, and some of these are ancestral to many lineages. Even though the fraction of sequenced species remains marginal, repetitive findings tend to confirm a relatively high frequency of polyploidization during Eukaryote radiation. At present we have evidence for the existence of a panel of events from different ages and in different lineages. These findings have made Ohno's theory quite popular and duplicated genes from polyploidization have been called ohnologs by some authors [4–6]. In prokaryotes, however, despite a very large amount of genomic data available, the fraction of characterized genomes is probably lower, and no WGD event have been described to date. We can postulate many reasons for this, based on the major structural differences of DNA between prokaryotes and eukaryotes. Each polyploidization is characterized by an immediate amplification of the number of chromosomes. One of the possibilities is that the circularity and often single copy of DNA may be under constraint and could represent a major limitation.
Although the theme of gene duplications in evolution is usually attributed to Ohno, other authors had pointed it out earlier since the beginning of the 20th century (see [7] for a historical review). For example, in 1932 Haldane proposed the possible advantage for duplications to produce redundant copies that could lessen the risk due to deleterious mutations [8].
Several WGD have been characterized in at least three of the five supergroups of eukaryotes according to the cladistic by Keeling [9], Chromalveolates, Plantae and Unikonts (Figs. 1 and 2). Ever since such events have been described, and because each event is specific, new theories have been proposed to refine the original model of Ohno. These theories often concern the functional fate of duplicated genes (ohnologs). Beside their fundamental interest, these models have probably been motivated by a challenging conceptual problem. Suppose a cyclist in the “Tour de France” has two copies of all the components of his bicycle. To win, he can build one new bicycle; at least the same if is ahead in the race, or better, a more efficient one, but not two bicycles. He has the option of keeping a component in duplicate as a backup, or removing it, or innovating by using a component copy for a novel function, or a combination of these options.
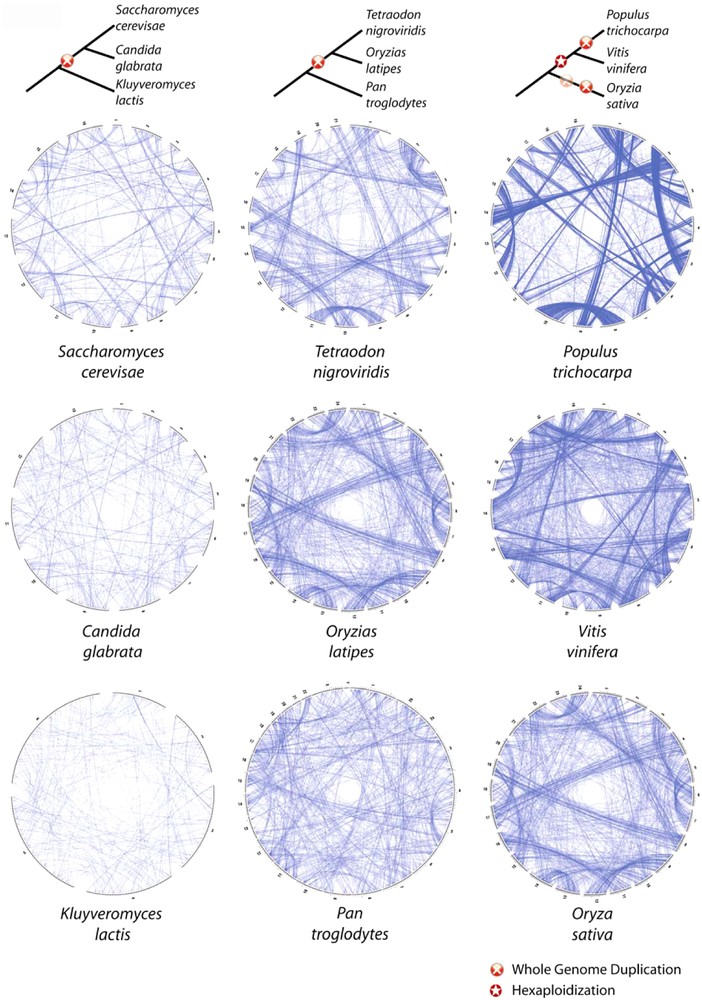
Comparative representation of chromosomal topology of paralogous genes in modern species. Trees indicate the relative date of polyploidization events in the lineages shown. Each circle represents chromosomes of one species, and each line connects two paralogous genes. For each species, a core set of paralogous genes was identified by an all-against-all comparison of the proteome of each species against itself using the Smith and Waterman algorithm [78]. Two genes, A and B were considered paralogs if B is the best match for gene A and if A is the best match of B (Best Reciprocal Hit). Paralogous genes which are found on the same chromosome (in most of the cases they arise from segmental duplications) were not drawn. Circular representations were produced using Circos (http://mkweb.bcgsc.ca/circos).
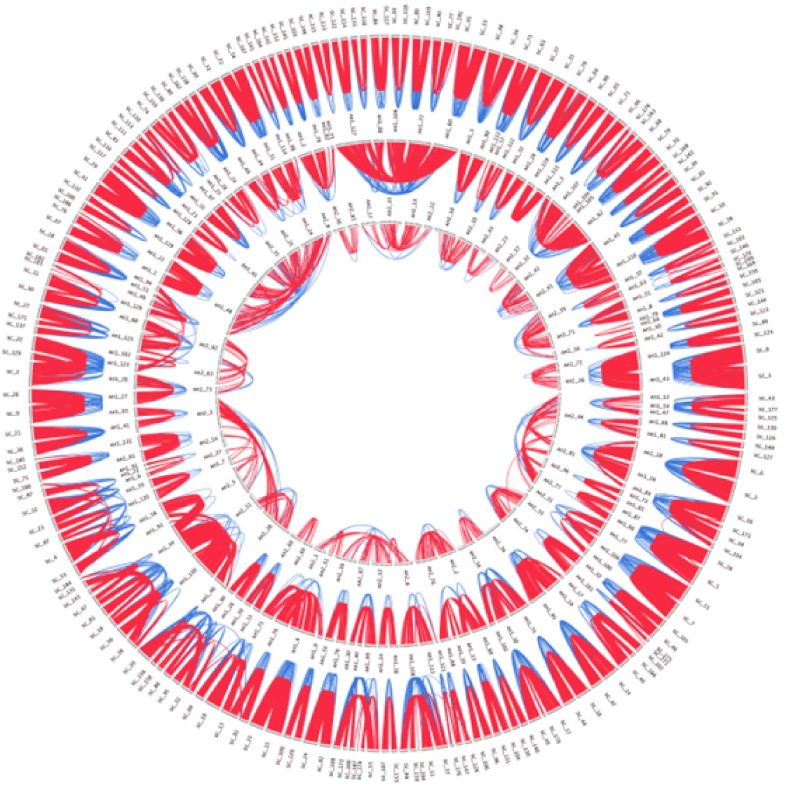
Representation of the successive duplications of the Paramecium genome. The exterior circle displays all chromosomes, and the two interior circles show the reconstructed sequences obtained by fusion of the paired sequences from each previous step. Paralogous genes connected by red lines are computed according to the BRH procedure as in Fig. 1. Blue lines link pairs of genes with a non-BRH match that were added on the basis of syntenic position. The position of an ancestral block is unrelated to the position of its constituents in the previous circle. See Ref. [46] for other details. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Readers can find previous specialized reviews in various aspects of polyploidizations [7,10–12]. Here we describe methodological and evolutionary insights about ancient polyploidization events that have come from recent programs of whole genome sequencing.
2 Revealing ancient events
Because ancient polyploidizations (PLZs) are de facto ancestral events, DNA from living species cannot provide “formal” proof of their existence in the past. Revealing an ancestral event always means providing a list of arguments that are consistent with one's hypothesis. Providing proof would require analysis of the genetic material of a fossil. This cannot be achieved today. Several methods have been used to demonstrate or confirm ancestral PLZ. Most of the time, ohnologs surviving from ancient PLZ represent a tiny fraction of the whole set of paralogs which is mainly composed of the results of numerous small local duplications. The rationales of the methods are different but their goals consist of differentiating relics of a large-scale event from this background noise.
Rejecting a hypothesis of PLZ is probably more difficult. Under the hypothesis of a PLZ, we expect that a small fraction of the genes can be maintained as duplicates (most would be lost), and the genome would return to a diploid state. We also expect intra- and inter-chromosome rearrangements. So, an apparent absence of any trace of PLZ in a genome cannot be completely sufficient for the rejection of the hypothesis of this event in the past. However, this problem is solvable indirectly via a related species where a PLZ could be demonstrated and dated previous to the radiation.
Lack of data is probably the main source of recurrent debates between experts. Ohno proposed that three rounds of WGD occurred at some phylogenetic positions in the evolution of vertebrates. The third round (called 3R) which is thought to be at the origin of the teleost fish lineage had been widely discussed before relatively recent efforts in genome sequencing of several vertebrate lineages. Indeed, this hypothesis was supported by several observations of gene families in which the number of members doubles at some major vertebrate radiations. The example the most frequently described is the family of Hox genes which is present in one copy in invertebrates, in four copies in mammals and in more than four in teleost fishes [13,14]. In teleosts, copies of genes may be lost or maintained depending on lineages. However, because these observations were based on partial data which may not be representative of a genome wide scale, the question remained controversial [15]. Thanks to the availability of both the nuclear DNA sequence of a teleost fish Tetraodon nigroviridis and of the human sequence which was used as an outgroup of the teleost lineage, convergent results in agreement with 3R were obtained at large scale and from two independent methods resulted in the resolution of this issue [16].
2.1 Using phylogenetic analysis
Phylogeny treats each gene family independently and reveals whether a putative duplication is anterior or posterior to a radiation. For each family of genes, the method requires only the sequences of 3 genes, 2 paralogous from one species and one from another species. Two paralogous genes that duplicated after the split between the two species should be branched closely. The paralogous distance is shorter than orthologous distance. In a situation where the duplication is older than the split, then the orthologous distance can be shorter than the paralogous distance (Fig. 3). The percentage of gene families respecting the first or the second topology provides an argument either in favor of, or against ancestral large scale duplication.
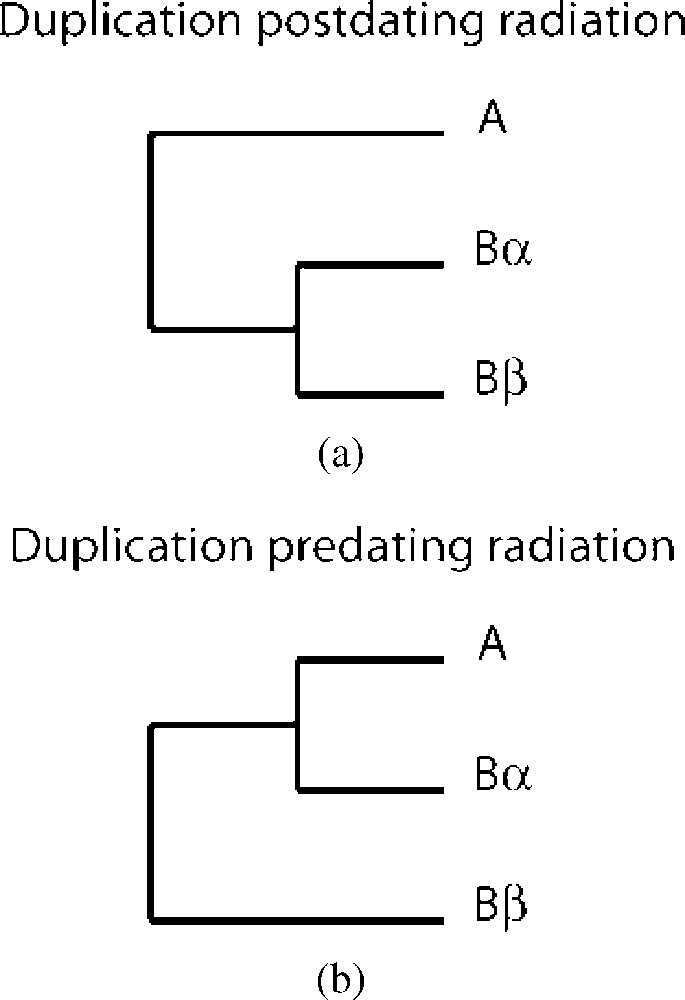
Examples of topology of phylogenetic trees to date a duplication event related to a radiation. Trees could be constructed using one gene which exists in two species (A and B) in different number of copies, one copy in A and two copies in B (Bα and Bβ). When duplication occurs after the split between A and B (tree a), a topology where Bα and Bβ are closer to each other than to A is expected. In this case, species A could considered as an outgroup. When duplication occurs before the split (tree b), if Aβ has been lost, Bα can be closer to A than to its paralogs Bβ.
This approach can be useful to test the hypothesis of a WGD with either a complete set of proteins [17] or a partial proteome of a species [15], and also when methods based on synteny are deficient (either due to a lack of data, or due to highly rearranged genomes). In a context where a WGD is admitted in a lineage, this method also permits dating the duplication of each pair of paralogous genes, either before or after the WGD [18]. After a WGD, a fraction of ohnologs possesses a similar rate of mutation in the two copies (symmetric evolution). However, in the other fraction, the evolution rate is significantly different between the two copies (asymmetric evolution). It has been demonstrated than phylogenetic trees reflect correctly the first type of situation, but tend to be misleading in the latter situation. An artifact named as “Long-Branch Attraction”, causes the dating of duplication too early, before the split with an outgroup species lacking the duplication [19]. Finally, this approach may be relevant for dating several PLZ in different lineages relative to each other (see below).
2.2 Using distributions of neutral substitution rates of genes
Since copies of genes resulting from a WGD have the same age, it is thus tempting to use this property. The rate of synonymous substitution between two paralogs can be used as a proxy to a relative date for their duplication. A significant fraction of paralogs with a similar rate of synonymous substitution would be an argument for a large-scale duplication. Because more than one substitution could occur at the same site, and therefore cannot be measured directly, different methods exist for providing estimations. The distribution of Ks (fraction of synonymous substitutions per synonymous site) between paralogs was used at large scale initially to evaluate the extent of duplication events in Arabidopsis [10,20–23]. Theoretically, the distribution of Ks follows a decreasing exponential curve, from low Ks values corresponding to recent duplications, to higher Ks values at the flat tail. The decay rate of exponential decrease depends on the rate of progressive losses of duplicated genes. The presence of a peak at low Ks values would be due to many and recent local duplications. Other peaks would correspond to bursts of gene duplications. In Arabidopsis, the shape of Ks distribution led to the conclusion that one recent event occurred and masked at least one earlier event. However, other authors concluded that the distribution is in agreement with 3 successive rounds of WGD along with the two earlier major radiations of the angiosperm lineage [24]. But some of these conclusions are in contradiction with more recent synteny data between various genomes of angiosperm [25]. This caveat from Ks analysis is due to some major limitations of this method. Old events are hardly distinguishable due to saturation of substitutions on synonymous sites. Mutation rates are possibly not constant over a long evolutionary time-scale and in different lineages. Inferring the level of polyploidy seems hazardous by this means. Also, sub-populations of ancient paralogous genes that underwent gene conversions would be characterized by low Ks values that would be interpreted as signals of recent duplication.
2.3 Using synteny conservation with other species
Genomes descending from a common ancestor accumulate inter and intra-chromosomal rearrangements. Depending on both the time separating two species, and on the rate of genome shuffling, genomic regions conserving ancestral gene content are more or less short and numerous. By counting the number of events such as inversions and translocations that occurred since the last common ancestor, a genomic distance can be computed [26]. A WGD is comparable in the sense that these events occur in paralogous chromosomes instead of orthologous chromosomes. A linear conservation of gene order between two genomes is usually represented and can be noticed as clear lines in representations such as dot plot figures. However, a WGD exclusive to one of the two species may lead to fragmentation and blur lines because orthologous genes are projected on two distinct chromosomes (Fig. 4a). When duplicated genes are maintained, each pair can be connected to a single ortholog from an outgroup species, and we could obtain a significant number of relations of type 1-2, the partial signature of a WGD. But when loss of duplicates is massive after a WGD and is randomly distributed between each paralogous chromosome, we expect essentially relations of type 1-1 between paralogous genes.
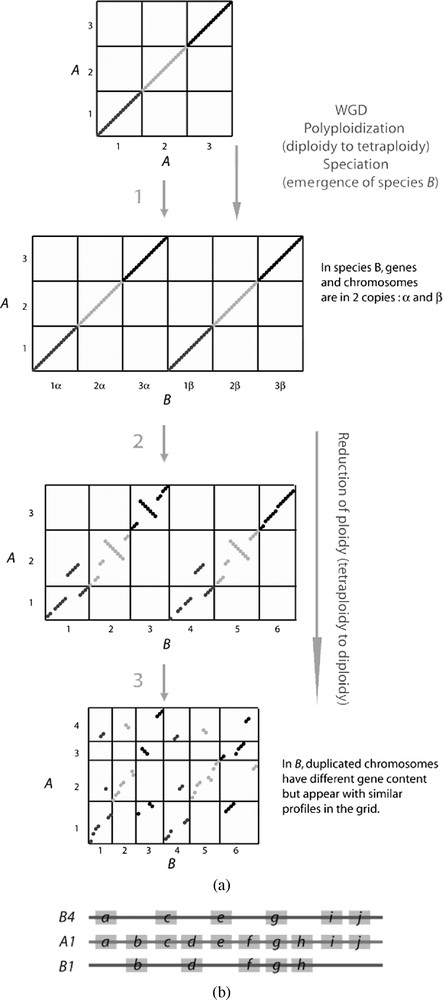
(a) Visualizing PLZ using macrosynteny grids. Four arbitrary evolutionary times are represented during which two distinct genomes diverge from a common ancestor through one WGD in one lineage. Columns and rows inside grids represent chromosomes and dots orthologous markers (genes, genomic regions…). The first grid corresponds to one genome A compared against itself leading to a clear diagonal crossing entirely the grid through every chromosome. In step 1, one WGD leads to a new lineage due to speciation (see text) and emergence of species B. At this step, by comparing A and B two diagonals would be represented if chromosomes are sorted. Genes and genomic regions in A and B, could be connected in relations of type 1:2. In steps 2 and 3, some duplicated genes in B are progressively pseudogenized or lost. More and more genes attain a relation of type 1:1 between A and B. Intra- and inter-chromosomal rearrangements contribute to obscure synteny. They affect 2 columns if they occur in A, but only one in B. Then, pairs of duplicated chromosomes in B, conserve similar profiles. For example, B3 and B6 correspond ancestrally to A3, but have orthologs in every chromosomes on A in the last grid, and these 2 columns (B3 and B6) must be visualized and compared entirely. So two duplicated chromosomes, or genomic regions in B, could have no gene maintained as a duplicate but could be seen as paralogous because they conserve similar profiles on A. (b) Representation of Double Conserved Synteny (DCS). Grey boxes indicate genes along one chromosomal region in a non-duplicated genome (A1), compared to two paralogous regions in a duplicated genome (B1 and B4). Because of many gene losses that occur after a WGD, a few genes remain in 2 copies in B. The ancestral order of the genes is conserved but B1 and B4 share very few genes.
So when the number of duplicate losses is significantly higher than the number of chromosome rearrangements, relations of type 1-2 are computable not between genes, but rather between larger genomic segments. For example, two duplicated genomic segments descending from a single region with 6 genes [] before the duplication, and maintaining 3 genes each after the duplication, [] and [], can both be connected to a single segment [] that would exist in a related but non-duplicated genome (Fig. 4b). This kind of double conserved synteny (DCS) was initially described at a genome-wide scale to demonstrate an ancestral WGD in the yeast Saccharomyces cerevisiae by comparison with the non-duplicated Kluyveromyces waltii [27]. This was the first analysis using genome wide comparison between one species that undergone a WGD and an outgroup species. A posteriori it could seem surprising that this WGD was controversial, because 81% of the 5714 genes of S. cerevisiae are involved in DCS blocks. Only small genomic regions resulting from rearrangements and containing 3 genes on average do not show clear DCS patterns. But before the availability of any external non-duplicated genome sequences, only 457 gene pairs were characterized which could result from distinct local duplications. Similarly in vertebrates, to highlight the third round of whole genome duplication in the teleost fish lineage (3R), 6684 orthologous relations were computed between the genome sequence of Tetraodon nigroviridis and of Homo sapiens [16]. Analysis of the topology in the chromosomes of these relations revealed that 75% of orthologs are involved in DCS. Typically, along a single region of a human chromosome, series of genes are orthologs with Tetraodon genes located alternatively on two chromosomes. By comparison, 748 pairs of Tetraodon paralogous genes are maintained from the WGD (Fig. 1). So, at least in yeast and in teleost, the sequence of a non-duplicated genome provides 9–10 times more markers for revealing ancient WGDs.
Using synteny conservation to uncover ancestral WGD or PLZ is efficient using a non-duplicated genome as a reference. Conversely, comparing one genome known to be duplicated to another evolutionary related genome permits to test whether the event predates or not the split. An event predating the split would allow time for paralogous chromosomes to diverge sufficiently from each other before speciation that would lead to relations of type 1-1 after speciation. This rationale has been used to decipher the chronology of PLZ events in flowering plants. Large genome duplications and other polyploidizations seem to be more common, and better tolerated in plants than in animals and perhaps in protozoa [28,29]. Since the publication of the complete sequence of Arabidopsis thaliana, several studies led scientists to postulate that at least one WGD occurred in its evolution [22,30–32], and an old WGD would be common to many dicotyledons. Two other genome sequences of dicotyledonous plants are now available, Populus trichocarpa [33] and the grapevine Vitis vinifera [25]. The synteny analysis of the three possible pairwise comparisons makes it possible to define the number of polyploidization events that are unique to a lineage, or shared. The genome sequence of the grapevine revealed that this plant derives from one or more events that led to a hexaploid nuclear content. The current diploid state results from consecutive rearrangements that affected the original three components. Surprisingly, this event is dated earlier than the radiation between these three dicotyledonous species. Indeed single genomic regions of Arabidopsis and of the poplar are never syntenic with three other counterparts in grape but with only one. Conversely, an independent recent WGD in poplar [33] is clearly confirmed here by relations of type 1-2 with the grape genome entirely. The patterns of conservation of the grape genome with Arabidopsis are more fractioned but a clear correspondence of type 1-4 is established for many genomic regions. This result indicates that at least 2 WGDs occurred in the evolution of Arabidopsis after the formation of the paleo-hexaploid ancestor, and after the split with the grape.
Although the paleo-hexaploid ancestor was apparently common to many dicotyledons, it does not appear to be shared by rice Oryza sativa which is the only monocotyledon completely sequenced to date. In this case, constituents of grapevine triplets are orthologous to the same regions in rice.
Overall, these comparisons of plant genomes have pointed out that the type of polyploidization that may be at the origin of the dicotyledonous plant radiation is the formation of a hexaploid.
Because several aspects can be integrated at the same time, using synteny conservation to analyze WGDs and other polyploidizations is highly efficient. The orthologous relationships, thanks to the knowledge of their location on chromosomes, are used as markers of the dynamics of the genomes. But manipulating a large amount of data requires the availability of almost complete genome sequences with a significant level of anchorage on chromosomes.
3 Inferring ancestral genome organization predating WGD
A direct consequence of the analysis of WGD by synteny conservation between two species is the ability to infer more or less precisely an ancestral organization of the chromosomes. The principle commonly used is essentially based on parsimony. Genes which are topologically conserved between 2 species, i.e. a weak genomic distance, were also co-located on the sequence of the last common ancestor. Then, it is possible to infer the gene composition of ancestral linkage group, by clustering groups of genes which are conserved on identical chromosomes. Again, following a parsimony principle, with a third genome as an outgroup it is possible to cluster ancestral regions into large parts of ancestral chromosomes, as well as inferring some events that occurred in specific lineages such as translocations, chromosomal fusions or splits. The term paleogenomics has been proposed for this new discipline [34,35]. Notably, several efforts are concentrated on deciphering the sequences of ancestral mammals at different nodes that would help in understanding our recent evolution [36–40]. In that context, deciphering a pre-WGD situation is a special case in which only two species are needed. A non-duplicated genome must serve as outgroup, and is compared to the two components of the duplicated genome that are treated individually even if they are not independent. In the case of the WGD at the base of the teleost fish lineage, a protokaryotype of an ancestral vertebrate predating the WGD was inferred, firstly by comparing near complete sequences of Tetraodon with human as outgroup. Blocks of DCS (double conserved synteny) used to reveal the WGD were clustered into 12 types according to the chromosomes they connected between human and Tetraodon. Each of these 12 types of DCS, named A to L, would contain genes located ancestrally in the same linkage group [16]. The availability of a bird's sequence, another outgroup from teleost WGD, could refine this scenario. In fact, computing DCS between Tetraodon and the sequence of the chicken Gallus gallus [41] leads to the same 12 types of DCS (unpublished). However, some regions in Tetraodon that were too scrambled using the human outgroup, can be included in DCS. Due to the potentially high level of scrambling in gene order between ancestrally duplicated chromosomes, a statistical estimation of the accuracy of paralogous regions that are detected is important [11,17]. The accuracy of this kind of results depends greatly on the quality of the sequence assembly of living species, especially the fraction anchored on chromosomes. More than 90% of the chromosomes of the teleost fish medaka, Oryzias latipes, are now covered by its sequence assembly, contrasting with 61% for the Tetraodon. Blocks of DCS were also computed between Medaka and human with a similar strategy, and also by using the sequence of Tetraodon and the zebrafish [42]. These authors proposed a more precise scenario: rapidly after the WGD, the last common ancestor of these three fishes had 24 chromosomes that resulted from 8 major rearrangements. But the last common ancestor prior to WGD had probably 13 chromosomes A to M. Another group, by using partial data available but on more species, proposed instead a situation with 11 chromosomes [43]. Earlier studies based also on synteny analysis but lacking complete genome sequences suggested an ancestral karyotype of the vertebrate lineage with 12 or 13 chromosomes [44,45].
The genome sequence of the ciliate Paramecium tetraurelia provides strong evidence for a highly conserved WGD. A majority of the genes, around 68% are present in 2 copies. Moreover, the location of the genes is so preserved that it is possible to infer almost exactly their ancestral order without the need of another genome. A two-step procedure has been developed to find traces of ancestral WGDs and to infer the ancestral order of genes. Recursively, this method has been applied three times revealing at least three WGDs, which occurred successively, but at separate time during the evolution of Paramecium [46] (Fig. 2). In terms of protein conservation the age of the third WGD can be estimated at an ancient time point in the evolution of the ciliate clade. This is a unique situation so far, in which it has been possible to access a very ancient genomic organization without an external non-duplicated genome.
4 Consequences of WGD
4.1 Structural modifications due to WGD
4.1.1 Speciation
PLZ can lead to speciation at two distinct stages:
- – Fixation of the polyploid organism;
- – Emergence of numerous species.
Starting from a diploid species, a WGD creates a tetraploid genome. A cross between diploid and tetraploid would create triploid having a high probability of sterility (odd number of chromosomes leading to problems during segregation). Thus, tetraploid species are reproductively isolated. Coyne and Orr say that “The discovery of polyploidy speciation represented the first major triumph in the genetics of speciation” and underlines the note of Haldane that speciation by polyploidy represents “the most important correction which must be made to Darwin's theory of the origin of species” [47].
By doubling the chromosomes and thus the genes, PLZ can raise some constraints that affect the structure of the genome. Notably, chromosomal exchanges between paralogous arms can be facilitated due to high similarity. Thus, ancestral structure would be modified when local rearrangements or genes losses occurred in the meantime inside only one paralogous arm. Overall, rearrangements and gene losses tend to decrease similarity and colinearity between paralogous chromosomes. As time goes by, reproductive isolation may become firmly established, in favor of the emergence of new species. But the principal factor of speciation is probably the consequence of reciprocal gene loss (RGL) which occurs when one copy of an essential gene is lost independently in two sister groups that descend from the same WGD. In this model, the two sisters lose the same copy in half the cases, and lose the reciprocal copy in the other half. Thus, a double null homozygote, lethal, would be produced in 1/16 of F2 hybrids, and naturally the reduction of hybrid fertility is proportional to the number of gene silencing [23,48]. This passive mechanism would contribute to speciation in agreement with the Bateson–Dobzhansky–Muller model.
Analyses of RGL have been performed by comparing different species descended from a WGD in yeast and in teleost fishes. Two different studies that compared zebrafish with Tetraodon or with medaka respectively showed evidence of RGL after the two speciations. The rate of ancestral genes that underwent RGL would be about 8% [44,49]. In yeast, a similar rate has been measured between S. cerevisiae and S. castelli (∼6%) and between C. glabrata and S. castelli (∼7%), but a lower rate was observed between C. glabrata and S. cerevisiae (∼4%). Some of essential genes of S. cerevisiae correspond to half of RGL situations with S. castelli, enabling estimation of the reduction of viability for hypothetical hybrid spores to be [50].
The level of chromosomal rearrangements may also play a role in speciation. In yeast, some species of the Saccharomyces genus can be crossed but produce sterile hybrids. But in some cases at least, sterility seems to be due to differences in chromosome organization rather than in gene content. After modifying the order of genes of one yeast species in order to obtain colinearity with another species, hybrid spores are viable [51]. However macrosyntenic rearrangements do not seem to be a prerequisite for speciation in yeast [52].
During mitosis, the mismatch repair system prevents recombination between dispersed repeated sequences and therefore contributes to a reduction in the risk of lethal rearrangements and deletions. But it has been shown in yeast that the same mechanism acts as a post-zygotic barrier [53]. Crosses between different strains of Saccharomyces cerevisiae, or of Saccharomyces paradoxus which are supposed to be species which diverged a long-time ago are partially sterile. However, disruption of the mismatch repair system reduces reproductive isolation. These probable roles of this system in speciation, but also in gene conversion during meiosis could act as well after WGD to complement RGL.
The hypothesis that major evolutionary lineages emerge from PLZ events is becoming increasingly accepted. In plants, ∼235 000 angiosperm species could be descended from one or several successive common PLZ [25,28], and cereals would share an ancient PLZ [54]. The diversification of the ∼12 000 species of homosporous pteridophytes with high chromosome numbers may be related to an ancient PLZ [48,55,56]. Among protozoa, at least two of the three WGDs of Paramecium can be placed at the base of a radiation [46]. Similar co-occurrences have been discussed in yeast [27,57–59]. Among vertebrates, the Euteleostei group derives from the 3R WGD in vertebrates and represents in terms of number of species (24 000) and of variety of morphological adaptations, the largest phylum [60]. Also, in parallel, several lines of evidence indicate that the 3R WGD is not present in non-euteleostei fishes [61,62]. The hypothesis of two rounds of WGD at the base of the vertebrates is more and more supported by genomic data [63]. The final statement about this question came from the genome sequence of Amphioxus, Branchiostoma floridae. A pattern of genome-wide quadruple conserved synteny with vertebrates has been shown [64] thus confirming the intuition of Ohno “It is our contention that the ancestors or reptiles, birds, and mammals have experienced at least one tetraploid evolution either at the stage of fish or at the stage of amphibians” [3]. An overview of the relics of major events in yeast, plant and vertebrate evolution is displayed in Fig. 1 with comparative examples from duplicated genomes and outgroups in each lineage.
4.1.2 Gene loss and pseudogenization
As we discussed previously, lack of data represents a major limitation in finding the trace of an ancestral WGD. The most well-known studied WGDs, in vertebrates and in plants are old, and these organisms have lost a large majority of their duplicated genes. From a technical point of view, synteny breakage due to gene loss complicates genome-wide comparative analysis between species having different PLZ.
Reduction of ploidy of a duplicated genome (tetraploidy to diploidy for example) is one consequence of gene loss. Potential structural and functional biases in the ways in which genes are lost are open questions. Among the duplicated chromosomes from the most recent WGD in Paramecium (Fig. 2), the size of the genomic regions that are maintained as single copies, corresponds to the lost sibling. The pattern is compatible with a mechanism which acts at the gene level or at least on a small scale. The range of decay state observed indicates that pseudogenization is probably a progressive process rather an abrupt phenomena immediately after WGD. However, comparative analyses of yeast genomes suggest a rapid phase of gene loss immediately after WGD [50]. One might expect a random distribution of gene deletions between duplicated chromosomes as is the case in the sequences of various teleosts and in Paramecium [16,44,46]. However, different species of yeast tend to lose the same duplicate (orthologs rather than paralogs) independently. This leads to the conclusion that different pressures affect the two paralogs [65]. Nevertheless a topological bias exists in the Arabidopsis sequence where Thomas et al. showed clusters of genes preferentially retained in two copies [66]. Genes that are lost or maintained in duplicate shape the emerging species functionally. Moreover, duplicates are more or less preferentially lost over the short term depending on some functional biases (see below). Massive gene loss is the more visible long term effect and seems to be the most predictable fate that has been noticed in every lineage concerned by PLZ.
4.1.3 Chromosomal rearrangements
Whole genome comparisons between species provide clues about differences in the frequencies of rearrangements in lineages and even in some chromosomes. In the same manner, analysis of the chromosomal topology of ohnologs highlights type of rearrangements that occurred post-WGD. This kind of event seems to spare some chromosomes more or less. In the poplar, chromosomes PtVIII and PtX have remained stable since the recent WGD, with no large inter-chromosomal translocation, whereas PtI is a combination of 4 ancestral linkage groups [33] (Fig. 1). Surprisingly, such differences in structural evolution of chromosomes do not diminish with time but persist. After 400 million years of evolution since the WGD, paralogs of Tetraodon chromosome Tn14 are almost exclusively located on Tn10, which is, however, connected essentially to Tn1, Tn7, Tn14 and Tn21 (Fig. 1 and Ref. [16]). In Paramecium, the level of conservation at the proteic level between paralogs of the recent WGD is comparable to that observed between humans and mice. But whereas hundreds of large blocks of synteny separate the two mammal orthologous sequences [67], in Paramecium the rate of rearrangements is so low that it would indicate a more general constraint that affects the chromosome structure [68]. Depending on the phylum, we observe different patterns of conservation of the ancestral structure of the chromosomes during the evolutionary transition from polyploïdy to diploïdy, probably as a result of forces of different intensities (compaction, transposon activities, population size, generation time, etc.). It has been suggested that the rate of rearrangements would be accelerated after WGD but this hypothesis cannot be rejected or accepted at the moment due to the small amount of data available [69].
4.2 Functional consequences at the gene level
One of the most fascinating theoretical consequence of PLZ is the potential for functional innovation inherent in ohnologs [3]. It seems evident that this fate could only be achieved if both paralogs are maintained (no gene lost). But maintaining genes in several copies may have functional consequences which affect the metabolism globally. Historically, Susumo Ohno assumed that one copy could maintain the ancestral function while its sibling could accumulate mutations until eventually being selected with new function. This scenario for novelty acquisition is known as neofunctionalization. Conversely, under the subfunctionalization model, distinct functions from the pre-duplicated gene are distributed, with more or less overlap, between the two sister genes. The most common situation, nonfunctionalization concerns copies that firstly become pseudogenes and are then lost. A formal description of this model, Duplication-Degeneration-Complementation (DDC model) was presented by Force and co-authors [70,71]. One surprise from the various descriptions of PLZ is the paucity of functions created by neofunctionalization that have been demonstrated to date. However, beyond the rare cases in which neofunctionalization has been functionally tested, some insight about the real importance of this type of gene evolution is provided by computational simulations. Indeed, two paralogs of proteins that evolved to neofunctionalization may retain a trace of an asymmetrical evolution. The signal which indicates an asymmetrical rate of mutations is measurable, using the length of branches in a phylogenetic tree, at least if an outgroup is available. The fraction of paralogous proteins that have experienced asymmetrical evolution in Paramecium, plants, and yeast is significant and tends to increase with time [46,72,73]. This tendency can also be observed in teleost fishes despite the low number of unambiguous ohnologs [18,74].
In the model of subfunctionalization, separation of ancestral functions between two sister genes could be either in space, in time or in functions. Numerous cases are characterized by analysis of the level of gene expression at different stages or in different tissues of an organism. In the teleost zebrafish, for example, the engrailed gene is present in two copies, eng1a and eng1b. In vertebrate species outside the 3R lineage, such as mouse and chicken, eng1 is a single gene and is expressed both in the hindbrain and in spinal neurons whereas in zebrafish, the expression of each copy is specific to one area [71].
However, functional innovation would not be the main cause for retention of ohnologs. Rather, most ohnologs would be retained as a side effect of their function. Several functional biases have been observed among the ohnologs maintained. In particular, some types of functions such as signaling molecules and transcription factors are preferentially retained for a long time after a WGD in plants, yeast and paramecium, but are not enriched among local duplications [24,46,65,73,75]. Several models exist to predict or to explain such biases in function of genes maintained as duplicates after a WGD and analysis of the genome sequence of Paramecium confirmed two wide effects that had been predicted (the models are nicely reviewed in [12]). First, interactions, pathways, networks or complexes formed by proteins would create constraints on their stochiometry which is noticeable at the gene level. Indeed such genes are preferentially co-retained at first, and co-lost a long time after the WGD. Disrupting the equilibrium of stochiometry would be then counter-selected, leading to a rule of the “all or none” type. This effect previously proposed as a “dosage imbalance effect” has been also observed in yeast [76,77] and would explain the difference in functional biases of the genes that are retained in duplicate after PLZ or after small-scale duplications. Second, highly expressed genes are also preferentially retained in duplicate, both short and long periods after the WGD. We can postulate that a high expression level of some genes may be selected due to a gain of fitness. But in certain cases, the expression level may reach the upper limit of the transcription machinery efficiency. Then, having another functional copy would make it possible to raise this limit. Some authors have also noticed a fraction of ohnologs with a very low evolutionary rate, possibly resulting from gene conversions. Here again, those genes are functionally biased.
5 Conclusion
In considering the evolution of species, certain types of events are correlated with the emergence of large radiations. Endosymbiosis, at the base of several major lineage, is one of these. Their impact is genetic, they provide a pool of potential functions but may also be structural when they provide a supplementary compartmentalization.
Other events lead to increased genetic variability such as sexuality and meiosis. In a diploid species, each gene, or at least most of them are present in 2 allelic forms. The possession of a double genome per individual allows a genetic mixing of the population. The repertoire of alleles that exists in a population at a given time represents opportunities for adaptations and for the emergence of new functions by mutations.
Whole Genome Duplication leads also to doubling each gene of an individual, but over a long time and under constraints such as gene dosage, and other types, certain copies are lost or maintained. Some of the retained copies tend to a specialization or to a new function via sub- or neo-functionalization. Gradually, the genome returns to diploïdy from a transitional tetrapoïdy status. All of these steps are relatively long, and mainly due to differential gene losses, emergence of a new species is facilitated. Each hybridization between sub-populations having different losses of duplicates, is a genetic combination that may lead to speciation and may then be potentially innovative.
Whereas diploïdy and sexuality allows a genetic mixing between each generation of a species and between every individual, WGD makes the stoichiometry of the genes more complex and bootstraps the genetic pool favouring emergence of new species. A genetic variability could then be exploited between individuals but also between emerging lineages which are still inter-fertile.
Half a century of fundamental hypotheses about the impact of polyploidizations on evolution has begun to be confronted with observations. Darwin's theory of natural selection has never been seriously rejected but has been refined to take into account new findings, such as the neutral evolution theory for example. Similarly, current authors support the vision of Susumo Ohno about facilitation of emergence of new functions and of species by duplications, but refine it with a supplementary hypothesis about short-term regulation of the forces that lead to these long-term results. It is not unexpected that these questions can be addressed within a genomic framework. Recent findings depend on comparisons between complete DNA sequences of chromosomes from model species. In the near future, we have the technical possibility to be confronted with a range of complete sequences from genomes with different separation times since PLZ that would furnish clues about the regulation of gene repertoire shaping over time. We must not forget that the human species is a result of ancient PLZ too. Future scientific plans must continue in the path of obtaining excellent genetic maps and excellent quality sequences. No major finding about nature could emerge from data which is too partial.