

1 Introduction
Most biological studies on post-transcriptional regulations have focused on one or only a few genes. This kind of approach is rapidly limited to scale up observed gene features to whole genome. Genome-wide analyses would necessitate taking in account regulations at both mRNA and protein levels. Indeed, a cell is a dynamic model where interactions between each level occur and lead to physiological effects. Integrative biology consists in linking different parts of the global cellular regulatory process to better embrace a biological event or response. Thus, the dogma of only gene expression determinating protein level is revisited by this approach. Protein concentrations in cells depend on the rates of degradation and dilution by growth of both mRNA and protein but also on the translational rate (Fig. 1). In this review, we describe mechanisms involved in mRNA and protein stability as well as in translation regulation in prokaryotes, and in particular, we emphasize their dependence on the growth phase and environmental growth conditions of the microorganism.
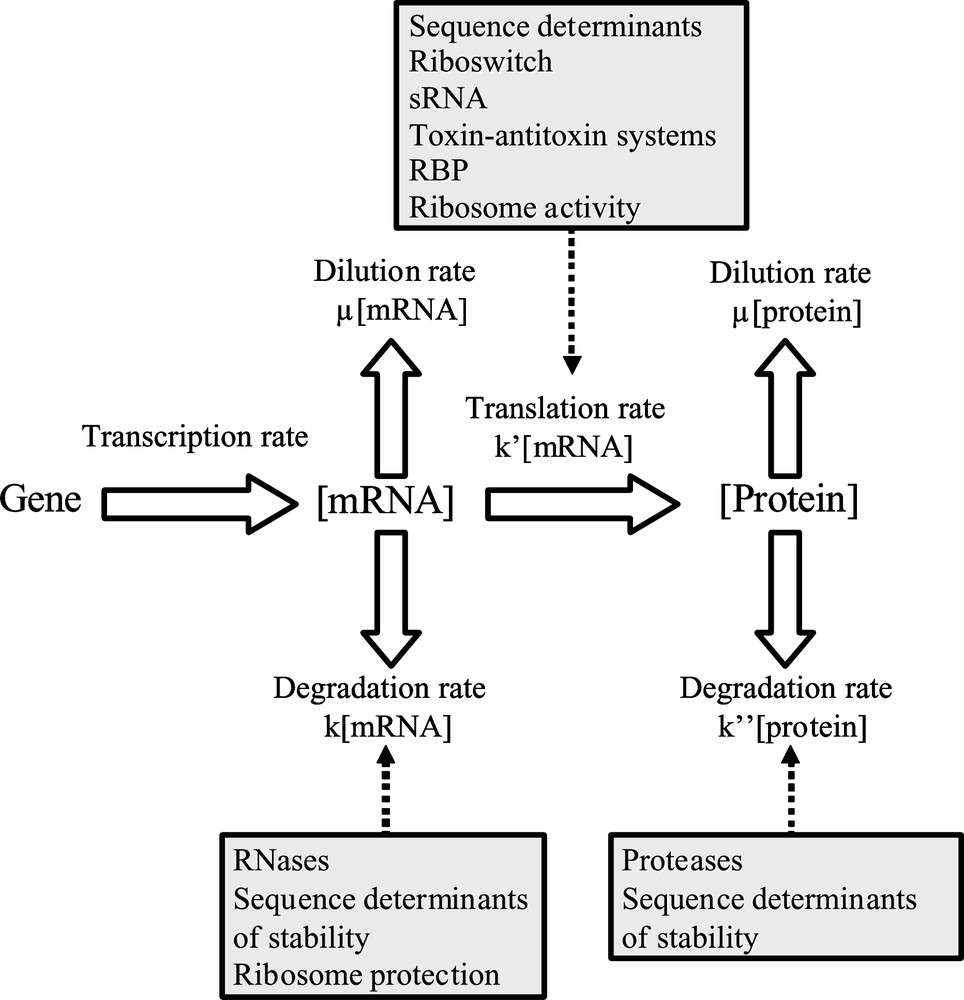
Schematic process of gene expression regulations. μ: growth rate; k: mRNA degradation constant; : translation efficiency; : protein degradation constant; sRNA: silencing RNA; RBP: RNA binding proteins; RNases: ribonucleases.
2 Mechanisms involved in mRNA/protein stability
2.1 mRNA stability
2.1.1 mRNA decay and RNases
RNA decay is responsible for variations in RNA concentrations and confers a low cost energetic means to adjust the mRNA concentration to cell needs. It is also involved in quality control of RNAs, in degrading faulty transcripts. Compared to eukaryotes, prokaryotic mRNA shows considerable instability with half-lives in the order of only a few minutes. This short stability allows prokaryote cells to respond rapidly to environmental changes. It is important to distinguish messenger RNAs, their steady-state concentration being strongly influenced by their half-life [1], from stable RNAs which are only degraded during stress or when a RNA molecule is defective [2]. It is now clear that mRNA decay involves a coordinated action between endoribonucleases (endoRNases) and exoribonucleases (exoRNases) (Table 1). Endonucleolytic cleavages create short RNA fragments that are subsequently degraded by exonucleases.
Summary of RNases found in E. coli and B. subtilis.
| Enzyme | E. coli members | B. subtilis members | Catalytic features | |
| Endonucleolytic enzymes | RNase E | Belongs to degradosome complex Initiation of most mRNA decay |
Essential enzyme | |
| RNase G | Homologue to RNase E Limited action in mRNA decay |
No essential enzyme | ||
| Endo and exonucleolytic enzymes | RNase J1 | Paralogue to RNase E Enzyme that couples 3′–5 endonucleolytic and 5′–3′ exonucleolytic activities |
Essential enzyme | |
| RNase J2 | Paralogue to RNase E 3′–5 endonucleolytic and exonucleolytic enzyme |
No essential enzyme | ||
| RNase M5 | 5S ribosomal RNA maturation | No essential enzyme | ||
| RNase Bsn | No substrate specificity | No essential enzyme | ||
| RNase H | Cleavage RNA–DNA hybrid molecules | Essential enzyme in B. subtilis in contrast to E. coli | ||
| RNase P | Cleavage of transfer RNA precursors for 5′ tRNA end maturation | Essential enzyme in E. coli and B. subtilis | ||
| RNase III | Acting on double-stranded structures | Essential enzyme in B. subtilis in contrast to E. coli | ||
| Exonucleolytic enzymes | RNase BN | 3′–5′ distributivea exonuclease | No essential enzyme | |
| RNase D | 3′–5′ distributivea exonuclease | No essential enzyme | ||
| RNase II | 3′–5′ processiveb exonuclease, predominant activity in E. coli | No essential enzyme | ||
| RNase T | 3′–5′ distributivea exonuclease | No essential enzyme | ||
| Oligoribonuclease | 3′–5′ distributivea exonuclease, specific for small oligoribonucleotides | Essential enzyme | ||
| YhamM | Acts on single stranded DNA and RNA | No essential enzyme | ||
| RNase R | 3′–5′ processiveb exonuclease Helps cleavage of secondary structures and is required for trans-translation |
No essential enzyme | ||
| PNPase | 3′–5′ processiveb phosphate dependent exonuclease | No essential enzyme | ||
| RNase PH | 3′–5′ distributivea phosphate dependent exonuclease | No essential enzyme |
a Distributive: enzyme that removes one nucleotide from one molecule, releases its product (a nucleotide shorter) and works on another molecule.
b Processive: enzyme that degrades entirely a molecule before working on a second molecule.
In Escherichia coli, initiation of RNA degradation starts with an endonucleolytic cleavage. Most of the mRNA turnover is mediated by a large protein complex called RNA degradosome, even if its assembly is not essential in E. coli [3]. Principal degradosome components are the following four enzymes: RNase E, polynucleotide phosphorylase (PNPase), RNA helicase B (RhlB) and enolase. RNase E is an endonuclease; its action is favored by the binding on its C-terminal half of the three other enzymes of the degradosome [1]. RNase E targets mRNA with an accessible single 5′ monophosphate extremity but is inhibited by triphosphate residues at the 5′ end [1]. RNase E scans mRNA in a 5′ to 3′ direction and then identifies and cuts its cleavage sites from 3′ to 5′ end [4]. Cleavage usually occurs in single-stranded A/U rich regions [1]. PNPase is a 3′–5′ exoribonuclease which degrades single-strand RNA to release 5′ phosphate nucleosides. RhlB helicase unwinds RNA duplex while the role of enolase in degradosome is not yet well defined [3]. This multi-protein complex was characterized in other proteobacteria but its composition differs between species [3]. RNase G also participates to a limited extend in mRNA decay in E. coli. This endoribonuclease shares 40% of homology with RNase E and presents the same cleavage site specificity [5].
In Bacillus subtilis, RNase E has no homologue. However, two RNase E paralogues, RNases J1 and J2 were identified [6]. They share functional homologies with RNase E in terms of cleavage mode and activity modulation by the nature of mRNA 5′ end [7]. RNase J1 is an essential enzyme in B. subtilis coupling endonucleolytic and exonucleolytic activities. Moreover, RNase J1 is highly conserved throughout bacterial and archaeal kingdoms [7] and an homologue is found in Lactococcus lactis [6].
Additional endoribonucleases, differing by their substrate specificity, were found in E. coli and B. subtilis such as RNase H which cleaves RNA–DNA hybrid molecules, RNase P that mainly plays a role on transfer RNA precursors and RNase III acting specifically on double-stranded structures [7]. RNase III was also identified in L. lactis [8]. Two RNases (RNases M5 and Bsn) from B. subtilis are also found in other low G+C Gram-positive bacteria but not in E. coli. RNase M5 is responsible of the 5S ribosomal RNA maturation while RNase Bsn does not seem to have any substrate specificity [9]. The importance of endonucleolytic cleavage on mRNA decay is well highlighted by two studies on citrate permease P (citP) mRNA stability, in both E. coli and L. lactis. It was shown that the lack of RNase III leads to strongly reduced citP mRNA decay in L. lactis [10]. In the same way, it was observed in E. coli RNase E or RNase III mutant strains a stabilization of citP mRNA (with a half-life more than 20 minutes instead of 3 minutes usually) [11].
In E. coli, six RNases (RNase BN, RNase D, RNase II, RNase PH, RNase T and PNPase) exhibit processive 3′ to 5′ exoribonuclease activity [12]. Oligoribonuclease, the only exonuclease essential for cell viability in E. coli, is required for complete degradation of mRNA into mononucleotides [13]. Particular exoribonucleases and hydrolases are not actually involved in the processive mRNA degradation but favor RNase E action, i.e. pyrophosphohydrolase removing pyrophosphate to 5′ end of triphosphorylated mRNAs [14] or RNase R helping the cleavage of secondary structures [2].
In addition to phosphorolytic PNPase, RNase PH and hydrolytic RNase R, B. subtilis contains another 3′–5′ exonuclease YhamM, capable to degrade single-stranded DNA and RNA [9]. Sequence homologues of YhamM were reported only in Gram-positive bacteria and in particular in L. lactis [15]. In the past, it was usually admitted that, contrary to eukaryotes, mRNA degradation in eubacteria only occurs in a 3′ to 5′ direction. Recently, RNase J1 of B. subtilis was shown to have in addition to its endonuclease function, a 5′–3′ processing exonuclease activity on ribosomal RNA and messenger RNA [16].
2.1.2 Sequence determinants of mRNA stability
Each mRNA coding sequence is flanked by untranslated regions (UTR) that contain regulating sequences of mRNA stability. Compared to eukaryotes, prokaryotes do not have a cap in the 5′ end of their messenger but an alternative protection via a triphosphorylated 5′ end was demonstrated in E. coli. Indeed, as we have previously mentioned, RNase E activity decreases when it meets a triphosphate residue at the 5′ end of an mRNA [1]. Presence of secondary structure close to 5′ end of an mRNA prevents also endonuclease cleavage. For example, 5′ terminal stem-loop could explain the unusual long half-life of E. coli ompA messenger [17] and influences B. subtilis aprE mRNA half-life [18]. An additional stabilizing element within the 5′ UTR is the ribosome binding site (RBS) sequence. Called also the Shine–Dalgarno sequence in prokaryotes, RBS is a purine-rich region localized around 10 bases upstream of the start codon. Initial anchoring of ribosomes onto mRNA depends on complementary pairing of the RBS sequence and a sequence close to the 3′ end of 16S rRNA in the 30S ribosomal subunit: higher is the complementary pairing, more efficient is the recruiting of ribosome. For example, higher mRNA stability of E. coli ompA [17] and B. subtilis aprE [18] was related to strong RBS/ribosome interaction. Sharp and Bechhofer [19] examined in E. coli the relationships between mRNA stability and the position and strength of a stabilizer-like element in 5′ UTR. According to them, strength and localization of RBS have a limited effect on mRNA stability compared to the presence of a secondary structure. In L. lactis mRNA stability was however related to the frequency of a RBS-like pattern in the 5′ UTR [20].
Stem-loops in 3′ UTR are also present in most prokaryotic mRNA and protect them against 3′ to 5′ exonucleases. Like in eukaryotes at their 5′ end, prokaryotic mRNAs are polyadenylated at their 3′ end (polyadenylation is not restricted to mRNA but it could take place on any accessible 3′ end of RNA). Surprisingly, in contrast to eukaryotes, adding 3′ end poly(A) tail destabilizes bacterial mRNA [21]. This signal is useful for RNases such as PNPase (which belongs to degradosome) and RNase R that need some unpaired nucleotides to attack 3′ loop [2]. Two types of poly (A) tails exist in E. coli [21]. First, a 10–40 nucleotide-long tail with exclusively A residues is added by a poly(A) polymerase I (PAPI) and found after Rho-independent transcription terminator or attached to degraded mRNA. During exponential growth, it was observed than more of 90% of E. coli open reading frames were polyadenylated by PAPI [21]. An alternative longer (about 100 nucleotide-long) and heteropolymeric tail can also be synthesized by PNPase. PNPase, primarily acting as the phosphorolytic nuclease of E. coli degradosome, is indeed bifunctional. Since the phosphorolysis reaction is close to equilibrium, PNPase can catalyse both mRNA degradation and 3′ end polyadenylation [22]. In B. subtilis, shorter homogeneous poly(A) tails (5 nucleotides average length) and heteropolymeric tails (composed to close 90% adenosine) up to 113 nucleotides length were observed. However, contrary to E. coli, it appears that PNPase is not responsible for heteropolymeric tails since they were observed in pnpA mutant strains [23].
In addition, in E. coli, it was established by Mohanty and Kushner [21] a correlation between poly(A) levels and mRNA decay rates: increasing PAPI level leads to increased poly(A) level and consequently to reduced mRNA stability. It was also found that polyadenylation of transcripts in E. coli is a sensing signal to adjust specifically RNase E and PNPase levels. RNase E and poly(A) tails do not interact in vivo but polyadenylation at the cell level is involved in autoregulation of RNase E protein level via transcript stability [24]. Indeed Mohanty and Kushner showed that half-lives of PNPase and RNase E (but not of RNases II and III) transcripts depended on poly(A) mRNA levels [21].
2.1.3 mRNA stability and mRNA translation
Ribosome binding on mRNA influences mRNA stability. In E. coli, ribosome association on mRNA does not stabilize downstream sequence but only protects mRNA in the vicinity of ribosome binding against RNases [25]. Close spaced ribosomes can also mask RNase E cleavage sites. However, in this case, the translation must not be interrupted since ribosome stalling leads to activation of trans-translation and thus provokes the decay of reading mRNA (for further details on trans-translation, see Section 3.6 on ribosome activity). Carrier and Keasling [26] established a mechanistic model to understand mRNA decay. They determined that for a successful cleavage by RNase E, the space between ribosomes should be more than 24 nucleotides. Examples of ribosome binding in the 5′ UTR enhancing stability of downstream mRNA sequence were reported in studies on ermA, ermC and phage82 mRNAs [9]. In these cases, ribosome binding prevents action of 5′–3′ exonucleases and 5′ end endonucleases.
Some variances emerge from ribosome influence on mRNA half-life in E. coli and B. subtilis: while translation seems to be required to inhibit cleavage by RNase E in E. coli, static ribosome can inhibit cleavage by the B. subtilis equivalent [9]. Sharp and Bechhofer [27] were in agreement on B. subtilis but were less conclusive on E. coli, for them ribosome binding and translation initiation rather than translation itself increased mRNA stability in E. coli.
2.1.4 Genome-wide determinations of mRNA half-lives
Few studies on mRNA stability at a genome-wide level were reported in prokaryotes by using microarrays. First a transcriptomic study of mRNA half-lives in E. coli, was performed by Bernstein et al. [28]. A wide range of mRNA stabilities was reported but 80% of mRNA half-lives were low and comprised between 3 and 8 minutes. The mRNA abundance was not a predictor to its stability. Moreover, the authors did not succeed to link mRNA stability to sequence features (such as open reading frame (ORF) length, predicted secondary structure in UTRs or density of RNase E cleavage sites) but they determined a correlation between mRNA stability and functional categories. A second study by Selinger et al. [29] used microarrays to identify global RNA degradation patterns in mid-log phase. mRNA half-life was also dependent on functional categories and an average mRNA half-life of about 7 minutes was observed. Study of the degradosome action at a genomic scale highlighted differences in mRNA decay in E. coli: some mRNA degradations were mediated by degradosome contrary to others that did not [30].
In B. subtilis, Hambraeus et al. [31] examined mRNA decay rates in early stationary-phase cultures. About 80% of mRNAs exhibited a half-life lower than 7 minutes. Surprisingly, no correlation existed between mRNA stability and neither gene encoded function nor sequence features in 5′ region.
In L. lactis, most of mRNA half-lives ranged in exponential phase between 1.5 and 10 minutes (Fig. 2) [20]. Contrary to previous studies, mRNA half-lives were correlated negatively with ORF length. Genes with the highest expression were globally the less stable and a relationship between mRNA stability and gene function (house keeping genes were stable and those involved in stress adaptation unstable) was demonstrated. Nevertheless, values of mRNA half-lives were significantly lower than the generation time (in the range of hour), indicating that degradation of mRNA was the main process of mRNA disappearance and dilution by growth could thus be neglected in such conditions (Fig. 1).
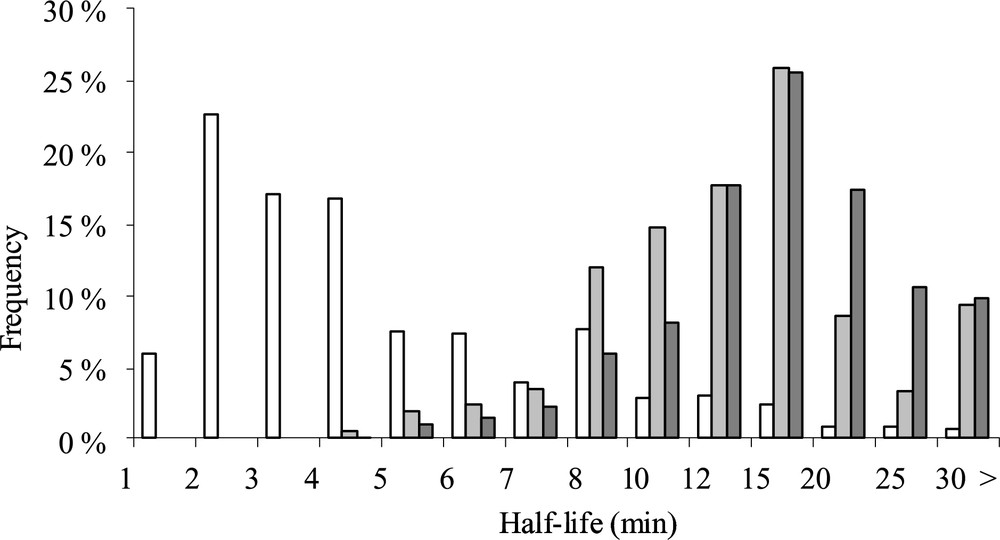
Half-life frequency distribution in exponential (open column), deceleration (light gray), and starvation (dark gray) phases during carbon starvation adaptation of L. lactis.
(Figure is from Ref. [20].)
2.2 Protein stability
2.2.1 Protein degradation and proteases
Protein degradation is an energy-demanding process requiring ATPase activity in both eukaryotes and prokaryotes. In eukaryotes, protein breakdown is mainly an ubiquitin-dependent pathway and involves the proteolytic complex 26S proteasome. Even if eukaryotic and prokaryotic proteases share similar barrel-shaped structure, proteasome assembly is not observed in bacteria [32]. Only mechanisms related to proteolysis in bacteria will be described hereafter.
The main proteases are part of the Clp (caseinolytic protease) family. This family is widespread in bacteria and found in E. coli and B. subtilis [33] but also in L. lactis [34]. In E. coli, proteolytic complexes of two Clp proteases are formed, e.g. ClpAP, ClpXP and ClpYQ, one Clp protease is an ATPase (ClpA, ClpX or ClpY) while the second is involved in proteolysis (ClpP or ClpQ). The subunits of proteolytic complexes are associated in rings forming barrel-shaped complex. The upper ring is the ATPase that belongs to ATPases Associated with diverse cellular Activity (AAA) family. In the proteolytic complex, ATPase denatures and translocates substrates for degradation [35]. When Clp ATPase is not docked to a proteolytic complex, it displays a chaperone activity [36]. The substrate specificity of the degradation complex is linked to the ATPase compartment and can be increased by specific adaptor binding on ATPase rings. Access to the proteolytic active site buried in the lower ring is restricted to unfolded proteins due to a narrow entrance. When the proteolytic core is ClpQ (or Heat shock locus V, HslV) a double ring of six identical subunits is formed and the associated ATPase is ClpY (or Heat shock locus U, HslU) [35]. In proteolytic complex containing ClpP, the lower ring is composed of fourteen subunits stacked in two heptamer rings that could interact with either ATPase ClpA or ClpX [32]. The proteolytic complex ClpXP is involved in degradation of most of all SsrA-tagged proteins resulting from trans-translation (for further details on trans-translation, see Section 3.6 on ribosome activity). Adaptator protein SspB increases ClpXP affinity for C-terminal SsrA-tagged proteins [37]. ClpXP recognizes five classes of peptide motifs, two peptide sequences are localized in C-terminal position and the others in the N-terminal part [38]. A specific ClpXP adaptor, RssB, is required for degradation of the starvation factor RpoS [39]. The second proteolytic complex ClpAP degrades less efficiently SsrA-tagged proteins and is mainly involved in degradation of proteins bearing N-degrons (N-end rule substrates), with the help of adaptor protein ClpS (for further details on N-degrons, see Section 2.2.2 on N-end rule). More precisely, depending on cell needs, binding of ClpS to ClpA N-terminus occurs to regulate ClpA autodegradation and to modify ClpA substrate specificity [40].
In addition to the Clp protease family, degradation of protein can involve the cytoplasmic serine protease Lon. Lon is a ring-shaped hexamer containing like Clp protease an ATPase domain and an internal proteolytic chamber [35]. Lon was observed in E. coli, B. subtilis and L. lactis [41]. In E. coli, this protease participates not so much in the degradation of misfolded proteins (its activity is 30 times lower than ClpXP on SsrA-tagged proteins [37]). Lon implication in regulations of biological processes was reported in E. coli [42]. FtsH (or High-frequency lysogenization, HflB) carries ATPase and proteolytic sites in the same polypeptide chain as Lon. FtsH is a Zn metalloprotease which differs from other proteases in E. coli by its localization in the inner membrane. FtsH was also discovered in B. subtilis [35] and in L. lactis [43]. FtsH degrades both soluble and membrane-associated proteins [35]. Two membrane proteins, HflK and HflC form a stable heterodimer HflKC that can associate FtsH to form the FtsH holoenzyme. This association might change FtsH specificity from membrane protein to soluble substrates but little is known about FtsH function in vivo [44].
2.2.2 N-end rule and protein stability
N-end rule pathway determines protein stability with the nature of N-terminal residues classified as stabilizing or destabilizing residues. Observed in prokaryotes and eukaryotes, classification of residues depends on species. In E. coli, hydrophobic (leucine, phenylalanine, tryptophan and tyrosine) amino acids are primary destabilizing residues; basic amino acids (arginine and lysine) are secondary destabilizing residues while other residues are stabilizing amino acids [45]. Secondary residues could be conjugated to primary residues by leucyl/phenylalanyl-tRNA transferase [46]. In fact, ClpAPS is mainly the protease which degrades N-end rule substrates [47]. The N-motif rule could explain the observed variability between protein half-lives.
2.2.3 Large-scale determinations of protein half-lives
No large-scale study of protein half-lives in prokaryotes has been published until now. In yeast, two studies present contradictory conclusions on measured protein half-lives. In the report of Pratt et al. [48], 50 protein turnover rates were determined by mass spectrometry using stable labeled amino acids in chemostat culture. Very heterogeneous degradation rates were measured and an average protein half-life was established around 31 hours. In a proteome-wide study, Belle et al. [49] employed a direct measure of protein half-lives during exponential phase, using TAP-tagged proteins, and the average calculated protein turnover was much more lower around 45 minutes. It seems that the range of protein half-life is similar in prokaryotes, since modeling of protein half-lives in L. lactis established a median protein turnover from 23 to 224 minutes depending on growth conditions. Unlike mRNA half-lives, protein half-lives are in the same range or even higher than the generation time, indicating that dilution by growth cannot be neglected at all (Fig. 1). This point highlights the importance to integrate the dynamic dimension in protein regulation studies.
3 Translation regulations
Translational regulations depend on intrinsic mRNA control elements (sequence and structure features), and involve binding of different types of effectors, proteins (RNA binding proteins and ribosomes), RNAs and metabolites.
3.1 Sequence determinants
Contributions of sequence features in the efficiency of translation initiation, elongation and termination were investigated in several bacteria. The strength of RBS/ribosome interaction has been used to estimate the efficiency of translation initiation in E. coli, B. subtilis [50] and D. vulgaris [51]. It is interesting to note that in E. coli, 30S ribosomal subunit attachment is mediated by a ribosomal protein S1. No homologue of S1 protein was found in B. subtilis, suggesting an increasing importance of RBS strength in this bacterium [52]. A translational enhancer, called downstream box because of its localization after start codon is supposed to interact with the 16S subunit for improving translation in E. coli. However, Rocha et al. [52] failed to find the same importance of this box in B. subtilis. The influence of the start codon nature and context on translation initiation has also been investigated. In D. vulgaris, Nie et al. [51] determined that the most frequent start codon was ATG and that ATG was also the most efficient in this strain. According to Rocha et al. [52] translation initiation is the limiting rate most of time. So RBS and start codon should play major role in translation efficiency. In addition, negative effect on translation initiation of secondary structure that sequestered RBS was reported in L. lactis [53].
Concerning translation elongation, analysis of amino acid composition and gene expression levels in E. coli and B. subtilis proteomes shows an increasing usage of less energetically costly amino acids in abundant proteins [54]. A similar conclusion was reported in D. vulgaris [51]. In the three species, codon usage is biased towards “major” codons that are generally recognized by abundant tRNAs in order to enhance translational elongation rates [51,54]. Thus, elongation could become the rate-limiting step when rare codons are used or amino acid availability is limited [52]. Using a modeling approach, Mitarai et al. [55] worked on the assumption that translation rate depends on codon, and therefore that ribosome on a fast codon could collide with the preceding ribosome translating a slow codon. So, regarding codon bias, the authors concluded that translation time wasted because of ribosome–ribosome collisions could be reduced by generating mRNA with highly selected codon usage.
In B. subtilis, Rocha et al. [52] reported different recognition efficiencies of release factors as a function of stop codon identity. In the same way, Nie et al. [51] showed in D. vulgaris the presence of an optimal stop signal composed of base C following the preferential stop codon, TAG and concluded that the importance of the stop codon context in affecting the translation efficiency should not be neglected.
3.2 Riboswitch
Riboswitches are non-coding RNA secondary structures that regulate gene expression at both transcriptional and translational levels. They respond to changes in concentrations of small molecule ligands, for instance nuclear bases, amino acids or sugars. Most of the time riboswitches are located in the 5′ UTR of gene encoding protein related to the metabolism of their ligands [56]. The binding of a specific metabolite allows an allosteric rearrangement of the riboswitch structure that leads to structuring of disordered regions. A riboswitch is composed of an aptamer region where ligand binds a platform region containing the RBS sequence. Inhibition of mRNA translation initiation occurs when RBS is sequestered in a stem-loop structure [57]. Riboswitches are widespread in bacteria, they mainly affect transcription in Gram-positive bacteria whereas in Gram-negative bacteria they predominantly act on translation repression [56]. For example, Winkler et al. [58] showed in E. coli that translation of mRNA encoding enzymes involved in thiamine synthesis is regulated by a riboswitch with an aptamer highly selective for its target ligand, thiamine.
3.3 sRNA
Non-coding RNAs (ncRNA) or small RNAs (sRNA) are modulators of gene expression and are grouped in three classes. Lower than 500 nucleotide-length, both first and second classes regroup sRNAs which act at the translational level. After pairing with the target mRNA, they induce mRNA structural rearrangements generating new cleavage sites leading to mRNA degradation. The first class regroups cis-sRNAs [59] that present a perfect complementarity to their target mRNA sequence (in RBS or coding region). They either inhibit translation or destabilize mRNA. Cis-sRNAs usually do not require cofactors and are localized on plasmid or on bacterial chromosome. The second class includes trans-sRNAs which are encoded by distinct loci from their target transcript. Trans-sRNAs bind to non-coding sequences and so exert indirect regulation on their target mRNA. Most of them require proteins such as Hfq. The third and last class of sRNA differs from others by acting at the transcriptional level. At first, sRNA were all considered as repressors but nowadays it is known that some sRNAs promote translation [60] such as DsrA (a cold shock protein) in E. coli which disrupts stem-loop structure by binding to its target, rpoS mRNA.
3.4 Toxin–antitoxin system
Toxin level in cell can be under translational regulation via toxin–antitoxin systems. Toxin–antitoxin system consists in a stable toxin and an unstable antitoxin (a cis-acting sRNA or protein) encoded by the same operon. When both are expressed, antitoxin binds to its corresponding toxin and neutralizes it [61]. For example, Gerdes and Wagner [62] studied the hok/sok toxin–antitoxin system in E. coli. Translation of the “host killing” (hok) gene encoding a cell membrane toxin is indirectly blocked by binding of the “suppression killing” (sok) cis-sRNA antitoxin sRNA to a third gene, “modulation of killing” (mok) gene, that encompasses hok RBS sequence. When the global translation machinery is defective, cis-sRNA is quickly degraded whereas toxin with a higher stability exerts harmful effects on cell. Same process is observed when antitoxin is a protein: in this case it is rapidly degraded by proteases such as ClpXP or Lon [61]. Pandey and Gerdes [63] searched in more than one hundred bacterial genomes the presence of all known toxin–antitoxin loci. They noticed that host-associated organisms did not have such a locus in contrast to most of free-living prokaryotes. Interestingly, it appeared that L. lactis was devoid of it, maybe because of a limited exposure to severe stress due to its rich medium requirement for growth.
3.5 RNA binding proteins (RBP)
In E. coli, when ribosomal proteins are in excess compared to ribosomal RNA, some regulatory ribosomal proteins function as translational repressors [60,64]. Regulatory ribosomal proteins non-assembled in ribosome bind to their own mRNA. This binding causes transcript attenuation or rapid mRNA degradation. Since many ribosomal proteins are organized in operon in E. coli, repression of the regulatory ribosomal protein translation leads to translational inhibition of all downstream ribosomal protein genes. This feedback control is called retro-regulation.
Another well-known example of a translational regulatory function of a RNA binding protein is the one of the hexameric Host Factor I (HFI or Hfq). Hfq regulates its synthesis by binding to two sites in the 5′ UTR of its own mRNA and inhibits formation of translation initiation complex. In addition, Hfq is involved as a cofactor in post-transcriptional regulation by helping at least 20 of the 46 trans-acting sRNAs known in E. coli [65]. Hfq interacts with sRNA and mRNA at an A/U-rich single-stranded region preferentially followed by a RNA helix in order to stabilize sRNA and to facilitate recognition of its target mRNA [66]. For instance, Hfq is involved in ompA mRNA destabilization, ompA being the major outer membrane protein in E. coli. In this regulation, Hfq and sRNA micA bind to their target ompA mRNA and block translation initiation [67]. The resulting absence of ribosome binding on ompA mRNA leads to RNase E action and transcript degradation.
3.6 Ribosome activity
When translating ribosome stalls for a long time on mRNA because of aberrant mRNA or presence of rare codons, trans-translation takes place. Trans-translation is a protein quality control mechanism during translation allowing ribosome recycling and degradation of both mRNA and native peptides which could be deleterious for the cell [68]. The centerpiece of this mechanism is the small stable RNA A (ssrA) or transfer-messenger RNA (tmRNA). The tmRNA is composed of an mRNA-like domain, with a short peptide reading frame, linked by a pseudoknot I to a tRNA-like domain with an acceptor stem [69]. This acceptor stem is recognized and charged by alanyl-tRNA synthetase. Two other protein factors are required for tmRNA activity: small protein B (smpB) and elongation factor Tu (EF-Tu). The first has a critical role in the first trans-translation step: smpB binds to tmRNA and to each ribosomal subunit, and so allows tmRNA entrance through ribosome. The second, EF-Tu, binds to amino acid acceptor arm to protect ester linkage of aminoacylated molecule. Stalled ribosome has a free A site that allows tmRNA binding. tmRNA replaces mRNA which will be rapidly degraded by RNase H. Alanine on tmRNA is then transferred to nascent peptide sequence. Hence, translation continues (without translation initiation factor binding) with tmRNA own reading frame sequence encoding a degradation motif. When translation ends, tagged protein is degraded by protease complex and ribosomes are released [69]. Trans-translation is a eubacterial wide-spread mechanism not found in archaebacteria and eukaryotes. It is present in E. coli, B. subtilis and also L. lactis [70]. Composition and length of tmRNA reading frame are function of bacterial species, from 8 to 35 residues, the first amino acid being always alanine.
In addition to trans-translation, ribosomal pausing on a rare codon can lead to reading frame shift of one nucleotide (+1 or −1 frame shift) to obtain a new codon with a corresponding tRNA more abundant in E. coli. Alternatively, ribosome could skip a part of mRNA sequence and so can reduce peptide sequence [71].
Ribosome synthesis is another regulatory level of protein synthesis. A first mechanism called retro-regulation, to adjust ribosomal protein synthesis to rRNA synthesis in E. coli was already described in Section 3.5 on RNA binding proteins. In addition, the synthesis of ribosomal proteins can be controlled via a feedback loop in which guanosine tetraphosphate (ppGpp) acts as a feedback signal [64]. For instance, in E. coli, when amino acid pools are too low, ppGpp is synthesized via the ppGpp synthetase and accumulates. Higher ppGpp levels decrease the strength of rrnP1 promoter (promoter of rRNA operons), turning off ribosomal RNA synthesis and thus reducing de novo ribosome synthesis.
3.7 Large-scale determinations of translation efficiency
Most of the translation regulations described above are studied at the local level on particular mRNA. They are examples of specific translational control of particular mRNA models. It is thus difficult to project how substantial is the role of these translational regulations at the global level. Are these regulations widely or not extended in the whole genome and are they active in vivo? A first estimation in B. subtilis reveals that only 4% of its genes are regulated at least in part by translation, by RNA control elements residing within mRNAs and involving bound effectors (proteins, RNAs and metabolites) [72].
Translation regulations can be evidenced at the genome scale by the comparison of the mRNA and protein levels. Early studies of correlation between mRNA and protein expression levels on a genome-wide scale were first performed in the yeast Saccharomyces cerevisiae. The lack of correlation observed was explained by experimental errors, differences in in vivo protein half-lives and translational regulations [73–76]. Recently, large-scale analyses of mRNA expression and protein abundance data showed that the correlation between mRNA and protein abundance is also weak in prokaryotes. In E. coli, large-scale absolute protein expression measurements showed that only 47% of protein abundance could be explained by mRNA concentration [77]. In D. vulgaris, Nie et al. [78] used a multiple regression approach in order to determine correlation between mRNA and protein abundance. mRNA data came from whole-genome microarray analysis and protein concentrations were obtained by a LC/MS approach. According to them, mRNA abundance can explain only 20–28% of the total variation of protein abundance, suggesting that mRNA–protein correlation cannot be determined by mRNA abundance alone. Among various factors affecting the mRNA–protein correlation, analytical variations in mRNA and protein abundance contributed to 34–44% of the total variation of mRNA–protein correlation, mRNA sequence features to 15–26%, and protein and mRNA stabilities to 5% and 2%, respectively [51,78]. In L. lactis, mRNA and protein abundances were weakly correlated independently of cell physiology state (Fig. 3). Spearman coefficients of 0.42, 0.27 and 0.25 were found during exponential, deceleration and stationary phases, respectively.
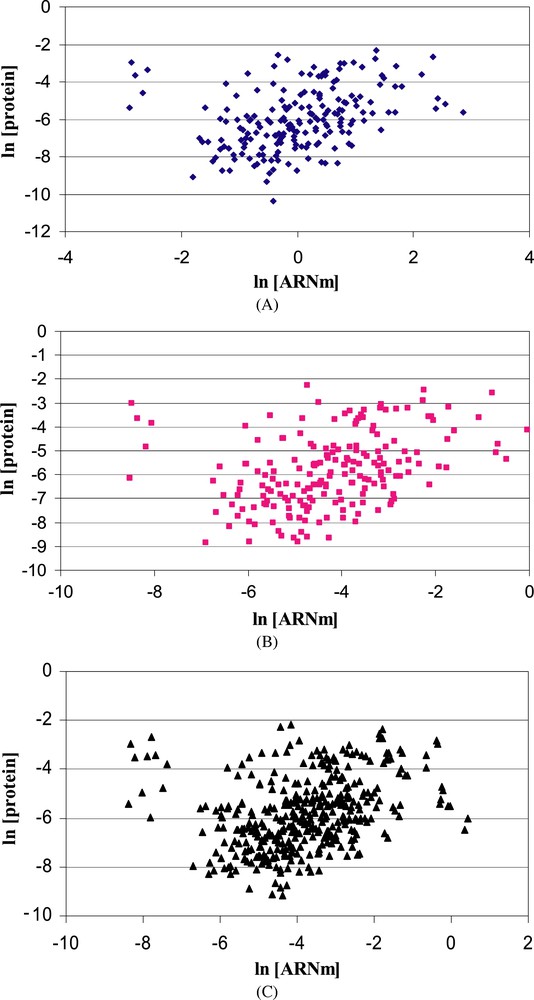
Correlation of mRNA and protein abundances in exponential (A), deceleration (B) and stationary (C) phases during batch culture of L. lactis. Both deceleration and stationary phases were obtained during isoleucine deprivation.
These weak correlations between mRNA and protein levels tend to show the importance of translation regulations in prokaryotes. In order to get a comprehensive overall view of translation efficiency and regulation, simultaneous analysis of the translational status of thousands mRNAs must be performed. Polysome profile analysis at a genomic level reflects in vivo translatability of each mRNA, by giving assess to each mRNA ribosome occupancy and ribosome density. Ribosome occupancy is defined as the fraction of a given mRNA with at least one bound ribosome while ribosomal density is the number of ribosomes on active mRNAs divided by the transcript length. Large scale polysomal analysis was developed in yeast by combining polysome (ribosome/mRNA complex) fractionation and transcriptomic techniques [79,80]. Up to now, the only genome-wide polysomal analysis in prokaryotes was performed in archaea [81]. Lange et al. [81] reported that 20% of Halobacterium salinarum transcripts and 12% of Haloferax volcanii ones were translated with non-average efficiencies. Mechanistic model developed for E. coli by Zouridis and Hatzimanikatis [82] highlights polysome influence during translation. First, they pointed out that increased polysome size led to increased translation rate and suggested that polysomes self-organize to achieve maximum translation rates and second, they noticed that translation limiting step (initiation, elongation or termination) depends on estimated polysome size.
In the future, clustering of mRNA in subsets according to their translation efficiency will help to quantify the in vivo involvement of the different translation regulation mechanisms, for instance via in silico search of consensus sequences to riboswitch or sRNA in specific mRNA subsets. In addition, development of ribonomics approach [83] in prokaryotes could identify mRNA subpopulations associated with specific mRNA–RBP complexes. Comparison of those mRNA subpopulations with mRNA subsets generated via polysomal studies will provide in vivo implication level of RBP-based mechanisms in translation regulation.
4 Growth phase and stress effects on mRNA/protein stability and translation efficiency
4.1 mRNA stability and mRNA translation initiation
As a function of growth phase or in response to changes in environmental growth conditions, turnover of a specific mRNA or a group of mRNAs can be modified. mRNA stability of some genes depends on growth phase. According to Kuzj et al. [84], three mRNA classes could be defined as a function of their stability during cell entry in stationary phase. A first class includes transcripts, such as cat mRNA, with increased stability in the stationary phase. A second class regroups mRNAs such as ompA mRNA which have a shorter half-life in stationary phase than in exponential phase. Finally, a third class contains mRNA with no change in stability during growth cycle.
tmRNA has an important role for growth during stress in both E. coli and B. subtilis. During amino acid starvation, faulty mRNAs are recognized and eliminated by trans-translation in E. coli [85]. In the same way, Muto et al. [86] studied ssrA depletion under several stress (high temperature, high concentration of ethanol or cadmium chloride) and concluded that trans-translation mediated by tmRNA is essential for growth under stress. A third study realized in B. subtilis completes these observations by showing that trans-translation is necessary for growth at low temperature [87].
Only two genome-wide analyses dealing with the response of mRNA stability to growth conditions in microorganisms are reported in the literature. The first one, the only study in prokaryotes, was performed in L. lactis during carbon starvation [20], the second concerns yeast mRNA stabilomes compared between two stress conditions that differ in cell response time [88]. During genome-wide adaptation of L. lactis to carbon starvation, an important increase of median half-lives in the deceleration and starvation phases (Fig. 2) was observed indicating that mRNAs were globally stabilized in response to carbon starvation [20]. This study highlights the importance of the mRNA stability control in gene expression response to adverse growth conditions.
mRNA translation initiation is a second point of modulation for cell adaptation to environmental changes and during growth phases. At low temperature, mRNA tends to form secondary structures which disturb mRNA translation. To prevent formation of secondary structure in mRNA until ribosome binding and translation initiation, RNA chaperones are required. Among nine CSPs (Cold Shock Proteins) found in E. coli, three of them are RNA chaperones induced at low temperature. Moreover, these CSPs seem to interact with two cold shock-induced RNA helicases CshA and CshB in B. subtilis [89]. During adaptation to temperature, other proteins that belong to cold shock response, CsdA and RbfA, help ribosomal function. CsdA unwinds double stranded RNA favoring ribosome function [90]. RbfA is involved in ribosomal 16S maturation preventing polysome dissociation [91]. In addition, sRNAs (such as DsrA, OxyS, RhyB) often provide environmental signals for translation regulation under stress growth or suboptimal conditions [92] and cells could use sRNA for a rapid and transient activation of some genes in response to stress [93]. For example, DsrA sRNA is base pairing at low temperature to rpoS mRNA for enhancing translation of this gene which codes for a stationary phase sigma factor [92].
Analogically to riboswitch, mRNA translation could be also modulated by secondary structure (that sequesters RBS) sensitive to temperature. For instance, thermosensitive structure regulates translation of the major and best characterized cold shock protein CspA [60].
4.2 Protein stability
Under stress conditions, ClpXP participates in protein quality control and SspB helps adjustment to stress response: in absence of SspB, induction of extracytoplasmic stress response is reduced and delayed [94]. In parallel, synthesis of chaperone proteins is induced. In E. coli, three major chaperone systems are involved in folding nascent peptides: trigger factor linked to ribosome, DnaK assisting translation, and GroEL which is involved in protein folding after ribosome release.
During high temperature stress, another protease family is required, called High temperature requirement A (HtrA) in E. coli. HtrA homologues are found in other Gram-negative as well as Gram-positive bacteria such as L. lactis [95]. Three proteases are involved, called DegP, DegS and DegQ. These heat shock-induced proteases have neither ATP binding domain nor regulatory component. DegP function switches according to temperature: at low temperature DegP acts as a chaperone whereas at high temperature DegP degrades (unfolded) regulatory proteins involved in signaling pathway control. DegQ is poorly studied but its activity is vital and its substrate specificity seems to be the same than DegP. DegS differs from the two other proteins since DegS is a membrane-anchored protease and plays an important role in stress response [96]. stress response is induced when cells are exposed to extreme temperature causing most of protein synthesis arrest.
4.3 Translation efficiency
Entry to stationary phase corresponds to formation of 100S particles in E. coli. 100S particle results of dimerization of 70S ribosomal complexes followed by Ribosome Modulation Factor (RMF) binding. 100S particle has no translational activity [97]. Wada [98] noticed that translation inhibition was parallel to 100S formation in vitro and that RMF expression was inversely proportional to growth rate. 100S association is reversible: cell transfer in stationary phase to fresh media causes 100S dissociation, rapid RMF degradation within one minute and translation activation. Other proteins could bind in addition to 100S particles: YfiA, Hibernation Promoting Factor (HBF or YhbH) and Stationary-phase-induced ribosome-associated-protein (SRA or s22) [97]. YfiA and HBF share 40% of sequence homology, YfiA binds to either 70S ribosomal complex or 100S particles in stationary phase whereas HBF exclusively binds to 100S particles. Surprisingly, RMF binds to ribosomal dimers independently of these proteins [99]. Study of HBF function in vitro suggests that HBF must promote and stabilize 100S particles [100]. SRA binds exclusively to 30S ribosomal subunits during stationary phase. SRA protein synthesis is regulated by several global regulators of which ppGpp but its function remains unknown [101]. RMF was studied in cells under several stress. In nutritional stress, Izutsu et al. [102] found that ppGpp but not induces RMF synthesis. RMF expression is also found during osmotic stress (cells in log phase) [103], heat stress (cells in stationary phase) [104] and under acidic conditions in exponential phase [105].
A global study of translation efficiency as a function of growth conditions was reported in L. lactis [106,107]. For all genes encoding enzymes of glycolysis and lactate and mixed-acid fermentative pathways, rate modeling shows an average of threefold increase in translational efficiency in cells grown on glucose compared to cells grown on galactose. In addition, under acid stress conditions, translational regulation has a major influence (compared to transcriptional regulation) in the change of glycolytic enzyme concentrations. At low pH, the calculated translational efficiency increases confirming that the translation apparatus of L. lactis is optimized under acidic growth conditions. More recently, a genome-wide comparison of translational regulation between growing and stationary phase archaea cells was reported [81]. In H. salinarum, translation of 1% of all the genes is specifically repressed in either of the two growth phases. Specifically in exponential phase, almost 20% of all transcripts analyzed are translated above-average efficiency and include genes for many ribosomal proteins, RNA polymerase subunits, enzymes and chemotaxis proteins. This high number of genes with coordinate differential translational regulation indicates that a common regulatory mechanism may exist.
5 Conclusion
Gene expression regulation involves a multilevel process including two major steps, transcription of genes into mRNAs and their translation into proteins (Fig. 1). In the past, translation regulation was often neglected in bacteria and until recently, most of the studies dedicated to translation regulations at the genomic scale were related to eukaryotes. However, recent integrative analyses of transcriptomic and proteomic data in prokaryotes have revealed also a modest correlation between mRNA expression and protein abundance, raising the importance of translational regulations also in prokaryotes. This review underlined the considerable progress made recently in our understanding of prokaryotic post-transcriptional regulations. Genome-wide analyses regarding mRNA turnover, protein stability and translation efficiency are now or will be soon available in bacteria. To gain the most from these genomic-scale studies, next challenge will be to integrate all those data related to different aspects of translational regulation to the complex process of gene expression regulation. This integrative approach will enable a deep understanding of the global cellular adaptation process.
Acknowledgement
This work was supported by the INRA Integrative Biology Program (agroBI).