

1 Abbreviations
2I61, Chain A
Depressant Anti-Insect Neurotoxin, Lqhit2 From Leiurus Quinquestriatus Hebraeus gi|122920541|pdb|2I61|A[122920541]
ANEPIIanti-neuroexcitation peptide II precursor [Mesobuthus martensii] gi|13506801|gb|AAK28341.1|AF242736_1[13506801]
DmNav1Sodium channel protein para gi|150421666|gb|P35500.3|P35500[150421666]
KvAPChain C, X-Ray Structure Of The Kvap Potassium Channel Voltage Sensor In Complex With An Fab gi|30749953|pdb|1ORS|C[30749953]
PDBProtein Data Bank
IDIdentity
BLASTBasic Local Alignment Search Tool
NCBINational Center for Biotechnology Information
MDmolecular dynamics
RMSDroot mean square derivation
2 Introduction
Scorpions, as the most ancient arthropods, have existed on Earth for more than 400 million years. They develop neurotoxins that affect various ion channels as defence weapons and to attack prey. Scorpion toxins affecting sodium channels (60 to 70 amino acid residues; four disulfide bridges) are divided into α and β classes according to their mechanism of action and binding properties [1]. Upon binding to receptor site-4, scorpion β-toxins shift the voltage dependence of channel activation in the hyperpolarizing direction [2]. The action may involve a voltage-sensor trapping mechanism as proposed by Cestèle et al. [3] and later modified to involve a two-step model [4]. The scorpion β-toxins are further divided into four groups according to their affinity for insect and mammalian sodium channels [1]:
- (1) anti-mammalian β-toxins;
- (2) β-toxins acting on both insect and mammalian sodium channels;
- (3) anti-insect excitatory toxins;
- (4) anti-insect depressant toxins.
Anti-insect depressant β-toxin anti-neuroexcitation peptide II (ANEPII) was screened out from the cDNA library of scorpion of Chinese Buthus martensii Karsch [5]. The gene of ANEPII was cloned to the expression plasmid and soluble expression of ANEPII was obtained in Escherichia coli (E. coli) [5]. The expressed scorpion peptide was separated and purified using a His-Bind resin liquid-chromatography column and a gel filtration column [5]. ANEPII was also integrated into the tobacco genomic DNA and expressed efficiently [6]. To date, no data have been reported about the functional positions of ANEPII.
Therefore, in the present study, we describe homology models for site-4 of the Drosophila melanogaster sodium channel (DmNav1) and ANEPII. These models were used as the starting points for nanosecond-duration molecular dynamics (MD) simulations. Finally, we attempted to predict the molecular interaction of DmNav1 with ANEPII, which would be useful for further recognition of the protein functional sites of ANEPII.
3 Materials and methods
3.1 Molecular modeling
The amino acid sequence of ANEPII was obtained from the databank in the National Center for Biotechnology Information. The protein BLAST program against Protein Data Bank available at NCBI was used to select a template structure for homology modeling of ANEPII. From the selected template, a three-dimensional model of ANEPII was obtained by homology modeling using the software package Discovery Studio 2.5.
The homology model of Drosophila melanogaster sodium channel site 4 (DmNav1) was also generated using Discovery Studio 2.5 with ClustalX alignment of the KvAP channel sequence and its crystal structure (PDB: 1ORS) as a template [7].
3.2 Dynamics and PROCHECK
The loop of the DmNav1 model was embedded in the SPC water while the other was embedded in a DPPC bilayer and used as the starting structure for nanosecond MD simulations. MD simulations were run using GROMACS-4.0 [8,9] (www.gromacs.org). The force-field was based on Gromos96 53a6 and the time step was 2fs. The LINCS algorithm was used to constrain bond lengths and normal pressure and temperature were used in the simulation. A constant pressure of 1 bar, independently in the z direction, was used with a coupling constant of τp = 2.0 ps. Water, lipid, and protein were coupled in a temperature bath at 323 K, using a coupling constant τt = 0.1 ps.
The ANEPII model was also embedded in the SPC water and combined with the OPLS-AA/L all-atom force field, employing the same parameters used previously for DmNav1, except for the temperature of the bath which was 300K.
Finally, the obtained models were validated using PROCHECK [10].
3.3 Protein-protein docking
To generate the docking model for ANEPII and DmNav1, the ZDOCK [11] program in the Discovery Studio2.5 program package was used. ZDOCK is a rigid-body docking program that requires minimal information about the binding site. The program uses individual protein structures determined by experimental or computational methods as inputs and predicts the structure of a number of protein complexes. ZDOCK uses the Pairwise Shape Complementarity, desolvation and electrostatic energy methods to evaluate the proteins [12].
4 Results
4.1 Molecular modeling of ANEPII
Molecular modeling of ANEPII was carried out on the crystal structure of scorpion toxin from Leiurus quinquestriatus hebraeus (PDB ID: 2I61) chain A [13], which shares 81% sequence identity with ANEPII. The alignment between the ANEPII and the 2I61 is shown in Fig. 1(a). The three-dimensional structural model for the ANEPII was generated based on the 2I61 template using Discovery Studio 2.5 software.

(a) Sequence alignment result between ANEPII and the scorpion toxin from Leiurus Quinquestriatus Hebraeus (2I61); (b) Sequence alignment between the Drosophila melanogaster sodium channel (DmNav1) and the KvAP channel (PDB: 1ORS).
After MD simulation, analysis of 6 ns dynamics indicates that the ANEPII structure is stable and, after a rapid increase during the first 4 ns, the protein backbone RMSD over the last 2 ns of the ANEPII is ∼2.7 Å. Fig. 2(a) shows the evolution of the RMSD during the dynamic process. The potential energy of the system is shown in Fig. 2(b).
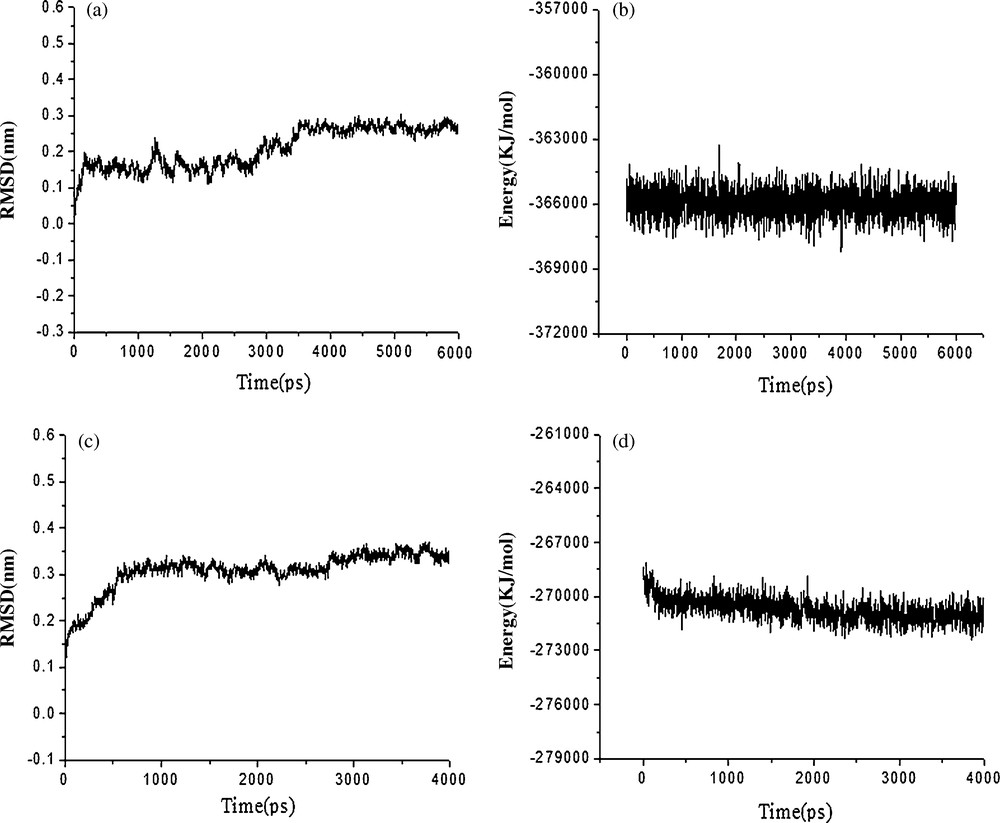
(a) RMSD with respect to the simulation time for a 6 ns molecular dynamics simulation of the ANEPII model; (b) The potential energy with respect to the simulation time for the 6 ns molecular dynamics simulation of the ANEPII model; (c) The RMSD with respect to the simulation time for the 4 ns molecular dynamics simulation of the DmNav1 model; (d) The potential energy with respect to the simulation time for the 4 ns molecular dynamics simulation of the DmNav1 model.
Analysis of the Ramachandran plot of the average structure between 5 ns and 6 ns shows that 94.0% of the residues lie in the most favorable regions, with 6.0% in the additional allowed regions, which indicates that the model is suitable for structural studies. Fig. 3(a) shows the structure of ANEPII.
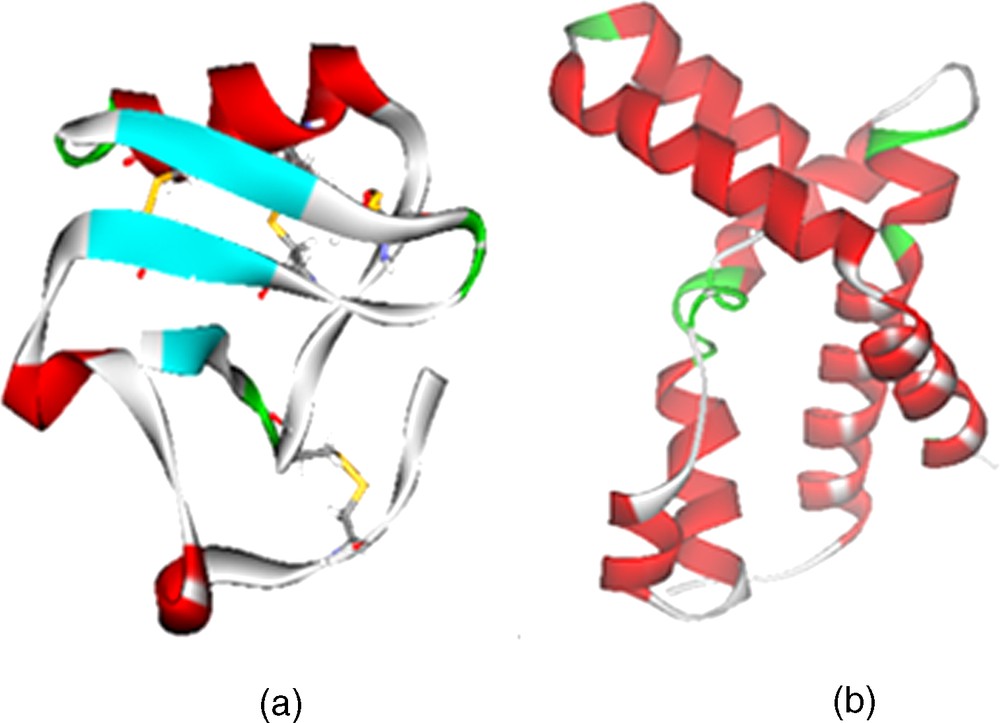
(a) Homology modeling of the ANEPII structure; (b) Homology modeling of the DmNav1 structure.
4.2 Molecular modeling of DmNav1
The model of DmNav1 is based on the crystal structure of KvAP that is a transmembrane ion channel and includes S1-S4 helices (with voltage sensor) cross the membrane. The sequence similarity of DmNav1 and KvAP is 44.4% (Fig. 1(b)). The three-dimensional structural model for the DmNav1 was generated by Discovery Studio 2.5 software.
The DmNav1 was energy-minimized to remove bad contacts derived from the homology modeling and to obtain a good starting structure to perform the molecular dynamics. After MD simulation, to assess the conformational drift of protein from its initial structure, we calculated RMSD of the backbone atoms of protein over 4 ns. In the DmNav1 simulation, there was an initial (over the first 3500 ps) steep increase in the RMSD to ∼3.5 Å, and then the value reached a plateau, with a final backbone RMSD of ∼3.5 Å after 3500 ps. Fig. 2(c) shows the evolution of the RMSD during the dynamic process and Fig. 2(d) shows the potential energy of the system.
The final refined model was evaluated by the Procheck program. As shown in the Ramachandran plot, 88.4% of the residues were in the core region and 11.6% in the allowed region. No residue was found in the disallowed region of the Ramachandran plot suggesting that no residue has a disallowed conformation. Fig. 3(b) shows the structure of DmNav1.
4.3 Scorpion neurotoxins docked with DmNav1
The modeled DmNav1 structure was checked against the known structure of scorpion depressant toxin LqhIT2. The interface residues of the DmNav1 receptor and the scorpion depressant toxin LqhIT2 involved in binding are shown in Table 1 and the complex structure is shown in Fig. 4(a). Previously, an exhaustive mutagenesis study of LqhIT2 assigned sites 11, 34, 36, 38, 53, 59 and 61 as key functional residues. This provides a good validation of the predicted structure of DmNav1.
Interface residues of the DmNav1 receptor and the scorpion depressant toxin LqhIT2 (The distance cutoff of the interface definition is 2.50 Å).
| Name | Interface residues involved in binding |
| DmNav1 | Met30, Asn31, Lys32, Met34, Glu84, Glu88, Gly89, Gln91 |
| LqhIT2 | Lys11, Tyr34, Trp36, Thr37, Trp38, Trp53, Thr59, Cys60, Gly61 |
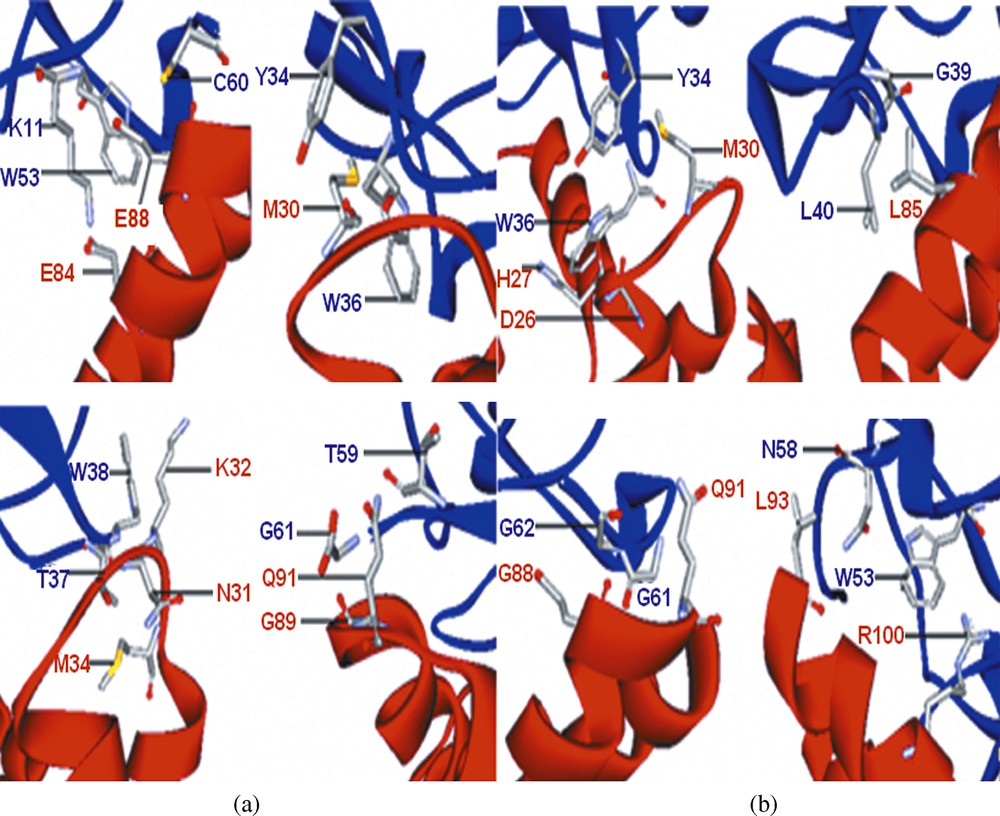
(a) Docked model of the LqhIT2 (blue) with the DmNav1(red) in which some key residues are highlighted; (b) Docked model of the ANEPII (blue) with the DmNav1 (red) in which some key residues are highlighted.
The purpose of docking studies was to identify the functional positions of anti-neuroexcitation peptide II (ANEPII). ZDOCK of ANEPII was carried out on the active site of DmNav1. We found that ANEPII binds with the predicted structure of DmNav1 (Fig. 4(b)) and the interface residues are shown in Table 2. The residues, Tyr34, Trp36, Gly39, Leu40, Trp53, Asn58, Gly61, and Gly62, of ANEPII act as functional residues.
Interface residues of the DmNav1 receptor and the ANEPII (The distance cutoff of the interface definition is 2.50 Å).
| Name | Interface residues involved in binding |
| DmNav1 | Asp26, His27, Met30, Leu85, Glu88, Gln91, Leu93, Arg100 |
| ANEPII | Tyr34, Trp36, Gly39, Leu40, Trp53, Asn58, Gly61, Gly62 |
5 Discussion
Voltage-gated sodium channels are the signature channels of excitable cells. They are the molecular target for several groups of neurotoxins, which bind to different receptor sites and affect voltage-dependent activation, conductance, and inactivation [14,15]. Voltage-gated sodium channels consist of a pore forming α-subunit and auxiliary β-subunits. Their principal α subunit is composed of four homologous domains (I-IV), each containing six probable transmembrane α-helices(S1–S6), and a re-entrant pore loop(P) between S5 and S6 [2]. The S4 segment of each domain, which contains 4–8 positively charged residues at three-residue intervals, functions as a voltage-sensor. DmNav1 has only been generated with the MODELLER software and was not simulated by molecular dynamics [16] but, in our study, this model in a DPPC bilayer was simulated by nanosecond-duration molecular dynamics to examine the rationality of the model.
The neurotoxic polypeptide ANEPII derived from β-scorpion peptide, which exhibits flaccid paralysis properties in insect models, acted on the voltage-gated sodium channel site-4. We initially identified the functional residues of ANEPII using bioinformatic techniques. From the results obtained in this work, the residues, Tyr34, Trp36, Gly39, Leu40, Trp53, Asn58, Gly61, Gly62, of ANEPII docked with DmNav1 and acted as functional residues. Most of them are hydrophobic residues.
Some studies have shown that β-scorpion toxins, which enhance sodium channel activation by shifting its voltage dependence to more negative membrane potentials, bind to a receptor site near the extracellular end of the transmembrane segment [3]. Toxin binding alone has no effect on activation but, when the channel is activated by depolarization, the bound toxin enhances activation by negatively shifting the voltage dependence. This effect is thought to be mediated by trapping the activated S4 voltage sensor in its outward, activated position by binding to the toxin sitting in its receptor site on the extracellular surface of the channel protein. So, we believe that the functional residues of ANEPII bind to the resting state of DmNav1 and, subsequently, the bound toxin traps the voltage sensor in its outward. Also, the homology model of DmNav1 developed above used crystal structure of KvAP (1ORS) as template for resting conformation, KvAP is properly a resting conformation of potassium channel structure with the highest known resolution (1.90 Å).
We hope that our model will be useful for designing new mutants of ANEPII and work in this field is in progress.
Acknowledgments
This work was supported by the grants from Liaoning Provincial Natural Science Foundation of China (No. 20082060), Scientific Research Fund of Liaoning Provincial Education (2009A689), Personnel Abroad's Excellence Science Technology Fund of P.R.China Ministry of Personnel (LXZZ2007003) and Shenyang Pharmaceutical University Research Initiation Funds for the Returned Personnel.
Vous devez vous connecter pour continuer.
S'authentifier