

1 Introduction
Comparative genomics tells us much about the evolution of regulatory networks. The deciphering of the history of genome architectures based on gene synteny and sequence homology allows the identification of orthology links. Orthology defines the relationship between genes in different species that originate from a single gene in the last common ancestor of these species. Such gene pairs are most likely to share the same function and hence provide good insights into the conservation of regulatory functions among related organisms [1–5]. The comparison of gene noncoding sequences to identify conserved cisregulatory elements in different genomes (giving rise to the “orthomotif” concept) provides valuable information on the evolution of regulatory networks [6–8]. However, it is clear that this information alone is not sufficient to explain the phenotypic diversity that is observed among closely related species. This is the “kapla concept” that illustrates the fact that, different species with quite similar genomic toolboxes can eventually have very specific physiological features and lifestyles. In other words, subtle genomic changes in cisregulatory elements and in the amino acid composition of transregulatory proteins can result in large rewiring of regulatory networks and gene expression patterns. This observation, together with the rapid increase of available genome sequences from the whole tree of life and the development of costless and efficient multispecies transcriptomic platforms, led to the emergence of a new discipline called comparative functional genomics [9]. Among eucaryotes, the Hemiascomycota yeast phylum is a valuable model to study the evolution of gene expression regulation [10]. Thanks to international and national initiatives, from which Génolevures has been a successful example and a great source of inspiration, the genomes of about 20 different yeast species have been sequenced, spanning 300 million years of evolution [2,11]. These species exhibit significant genomic changes and diverse lifestyles. Yet they have enough genomic similarity to allow the definition of orthology links with a reasonably high level of confidence. Yeast species have compact genomes, compatible with the easy design of specific microarrays. They can grow easily in laboratory conditions and most of them are genetically tractable.
2 Multispecies comparison of gene expression profiles
The standard approach in yeast comparative functional genomics consists in performing transcriptome analyses in closely related yeast species grown in similar environmental conditions. The resulting gene expression profiles are then compared with various information, including orthology links, functional annotations from the gene ontology (GO) and the conservation of known cisregulatory elements in orthologous promoters. To do so, several methodologies have been published, including the direct comparison of gene expression profiles from orthologous genes [12–15], the quantification of the coexpression conservation of clusters of genes [16,17] or multispecies fuzzy clustering using orthology links as a constraint to optimize GO enrichment in the final clusters [18]. To our knowledge, the most impressive work of this kind has been performed by the laboratory of Aviv Regev and her collaborators (Dawn Anne Thompson, personal communication). They conducted genome-wide gene expression analyses of the diauxic shift [19] in 15 yeast species (13 Hemiascomycetes and 2 Schizosaccharomyces). Gene expression profiles were compared based on orthology links, GO annotation and the conservation of orthomotifs. These comparisons allowed them to decipher the evolution of particular functional modules and to infer the ancestral regulatory networks for each phylogenetic node.
All these analyses successfully provided comprehensive views of the evolution of the transcriptional networks connected for instance with ribosomal protein expression [12,14], basal carbon metabolism and energy generation [14], and responses to environmental stresses [20] or antifungal drugs [18]. More generally, they have unraveled part of the plasticity of regulatory networks among yeast species.
However, comparative transcriptomic approaches have several limitations. First, orthology links are actually fuzzy, sometimes difficult to establish. The first ranked orthologue of a gene may not be its real functional homologue. Furthermore, species specific genes (i.e. genes without any clear orthologue) are generally put aside in most of these studies. Second, for most yeast species, GO annotations are mostly proposed based on the annotations of the Saccharomyces cerevisiae homologous genes, instead of using direct experimental evidence. This creates an obvious bias in multispecies functional analyses. With the same rationale, most of the cisregulatory elements used in these analyses had been characterized in S. cerevisiae. Therefore, the absence of these sequences in one species may not reflect changes in regulatory interactions but rather the divergence of the sequence that is recognized by the same, orthologous, regulator in the different species. Third, the conservation of gene expression profiles does not necessarily implies that the underlying regulatory networks are conserved, even when similar cisregulatory elements are present in the orthologous promoters. These pitfalls in comparative transcriptomics were nicely pointed out by the team of N. Barkai, which showed that there is actually no global correlation between promoter sequence evolution and the divergence of gene expression [15].
3 Multispecies experimental investigations of gene network structures
An alternative approach in comparative functional genomics uses all the available functional genomics tools (mainly transcriptome analyses of knock-out strains and genome-wide chromatine immunoprecipitation (ChIP-chip)), to directly investigate the structure of regulatory networks in each yeast species, instead of inferring it from the S. cerevisiae knowledge. Fig. 1 presents an example of such strategy applied to the characterization of the AP-1 regulatory module (involved in the genomic response to the antifungal drug benomyl) in three yeast species (S. cerevisiae, Candida glabrata, Candida albicans) (Goudot et al., in press). Such approach will benefit from the building of collections of knocked-out or tagged strains for each regulator gene of each yeast species, on the same model than what was done for S. cerevisiae at the end of the 1990s [21].
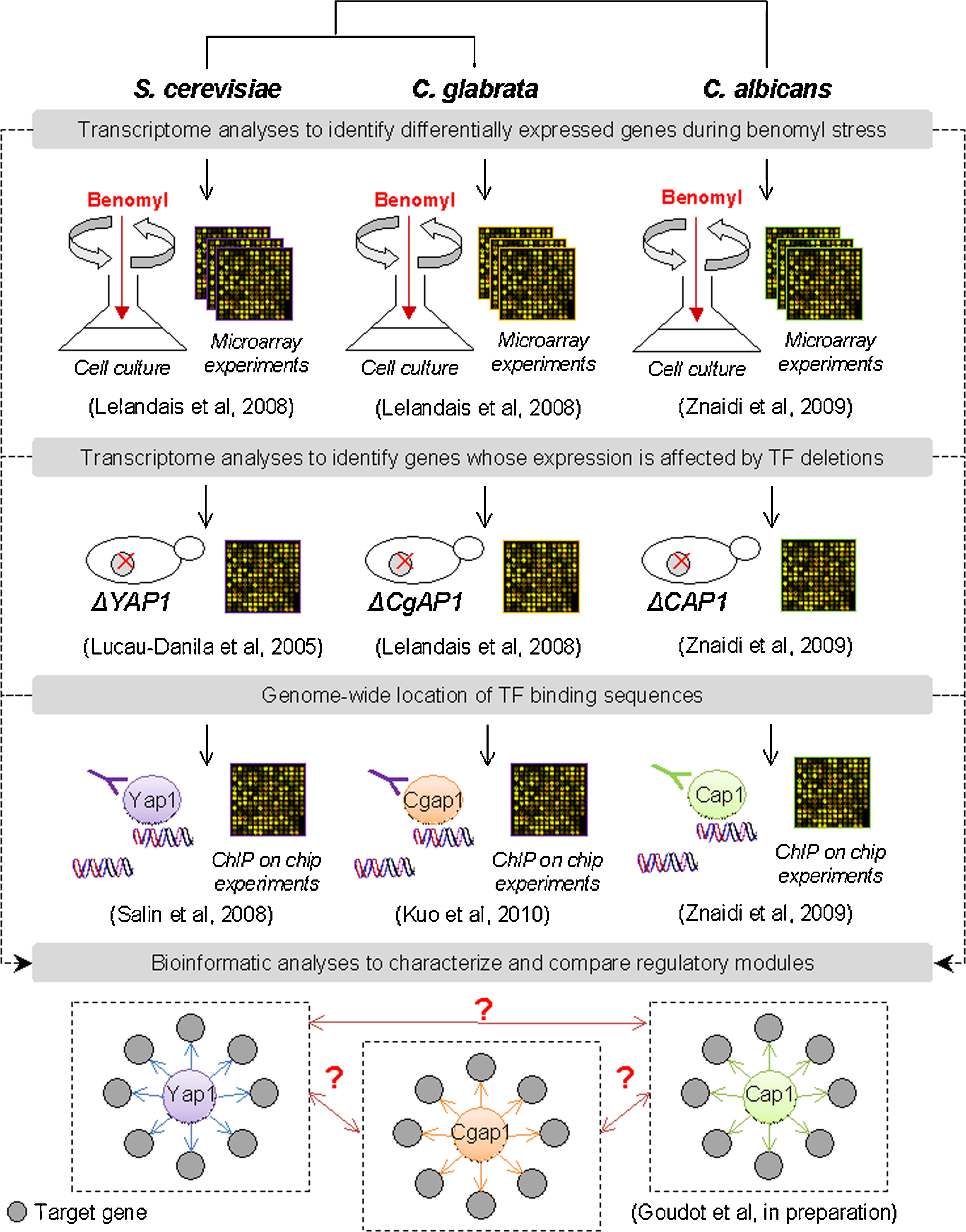
General strategy to characterize a transcriptional regulatory module. Characterizing a transcriptional regulatory module consists in identifying all the genes whose transcription is modulated by a particular transcription factor. Applied to the case study of the AP-1 regulon in yeasts during benomyl induced-stress, the general strategy shown here proceeds in several successive steps, using datasets which focus on different levels of transcriptional regulation: (1) identification of differentially expressed genes during benomyl stress, (2) identification of genes whose benomyl induction was affected by the deletion of the gene encoding the AP-1 transcription factor, and (3) genome-wide location of the AP-1 transcription factor, as determined by ChIP-chip analyses. The characterization of the AP-1 regulon in three yeast species (Saccharomyces cerevisiae, Candida glabrata and C. albicans) was performed independently using datasets collected from the literature [17,22,30,73,74]. Finally, the inferred transcriptional regulatory modules were compared using bioinformatics analyses of GO terms, cis-regulatory motifs, etc.
Many groups already used multispecies ChIP-chip and/or knock-out analyses to accurately study the evolution of regulatory modules associated to one or few transcription factors [13,17,22–28]. Several interesting conclusions arose from these different studies. First, the binding patterns and motif preferences of most transcription factors evolve much faster than initially hypothesized. This was demonstrated in a study of Ste12 genome-wide location in three closely related Saccharomyces species, which pointed out significant differences at relatively low evolutionary distances [23]. This was also nicely exemplified by a study of Mcm1 binding patterns in three distant yeast species [25]. While the canonical, S. cerevisiae-like, DNA binding motif of Mcm1 was still recognized in each species, it is present in different target promoters, making the core of ancestral Mcm1 bound genes remarkably small. Moreover, new regulatory interactions were characterized, suggesting that additional DNA motifs are recognized by Mcm1 in different species, due to species-specific combinations with other transcription factors [25].
Second, there are few correlations between the evolution of gene expression and the evolution of gene sequence [29]. Indeed, several cases of highly conserved gene expression patterns resulting from highly variable regulatory interactions have been described [15,17,22,24,26–28]. This can be explained by the important level of functional redundancy in the regulatory networks and the high plasticity of DNA/protein and protein/protein interactions. Thus, several combinations of transcription factors can be used to compensate cis- and trans-mutations and to obtain just the same gene expression pattern. This is for instance the case of Gal4, which transcriptionally controls the galactose pathway genes in S. cerevisiae, but which has completely different roles in C. albicans, where the galactose-inducible genes are controlled by Cph1 [28]. A less drastic situation is presented by the Yap1 transcription factor, which is the main regulator of the oxidative stress response in S. cerevisiae. While the global response to oxidative stress is conserved from S. cerevisiae to C. albicans, the preferred DNA binding site of the C. glabrata orthologue of Yap1 (called Cgap1) is slightly different from the S. cerevisiae canonical Yap response element [17]. These mutations in cis have been partially compensated in trans by mutations in the DNA binding motif of Cgap1, thus maintaining a significant role of this factor in oxidative stress response [17,22]. However, among the 90 genes which promoters bound Cgap1 under stress, only 17 have orthologues that bind Yap1 in S. cerevisiae [22]. Moreover, the inactivation of the Cgap1 decreased the induction of a smaller number of stress responsive genes than that of Yap1 [17]. In conclusion, although Yap1 and Cgap1 conserved a significant overlap in their regulatory activities, their DNA binding patterns and their functional roles diverged.
Third, the evolution of specific gene regulatory modules may not follow the phylogeny established from global comparative genomics. For instance, the functioning of the Yap1 regulon mentioned above is more similar to the functioning of the Cap1 regulon of C. albicans than to the Cgap1 regulon, although C. glabrata is phylogenetically closer to S. cerevisiae (Goudot et al., in press) [17,22,30]. This is not the only particularity of C. glabrata which also has a unique pattern of regulation for ribosomal protein encoding genes [12].
Epigenetic changes may partially explain some of the observations listed above. A correlation between promoter structure, chromatine organization and the expression divergence of genes had been established based on sequence analyses and gene expression profiles [31,32]. Two recent studies have analyzed experimentally the nucleosome positioning and its impact on gene expression in two closely related Saccharomyces species [33] and in 12 different hemiascomycetes species [34], respectively. They both concluded that divergences in the relative positions of nucleosome free regions and cisregulatory motifs significantly participate to the evolution of gene regulation, putting forward the importance of taking into account the chromatine organization level in such studies. For instance, the DNA motifs of Ste12 and Ume6, which control mating and meiosis, respectively, are found almost perfectly conserved in the promoters of the same genes from S. cerevisiae to C. glabrata. However, while these cisregulatory signals are located at the nucleosome free regions in most Saccharomyces species, they are occluded by nucleosomes in C. glabrata [34]. This may explain why C. glabrata has never been observed to mate.
Still, most of these works have been conducted on a transcriptional and DNA-centered point of view. An interesting challenge is to take into account the posttranscriptional aspects of gene expression regulation, by studying the RNA regulon concept [35] in an evolutionary perspective. While several studies have considered the RNA binding proteins in their analyses of regulatory networks in S. cerevisiae [36–38], few experimentally addressed these RNA regulons on a multispecies scale in Hemiascomycetes [39]. In this frame, the recently unraveled and largely unexplored world of noncoding RNAs in yeasts is particularly exciting [40–47]. The laboratory of Aviv Regev recently published a study of the expression of 6 antisense transcripts in 6 different yeast species [48], which opens the way to systematic analyses of the evolution of antisense transcription-driven regulatory processes.
4 Microevolution of regulatory networks
Comparative functional genomics raises the question of the mechanisms and forces that drive the parallel evolution of genome sequence, epigenetic features and regulatory interactions. Paradoxically, multispecies analyses are not so efficient to unravel these mechanisms, because the evolutionary distances considered are too large to actually elucidate the detailed dynamics of the observed events. On this guise, studies that consider smaller evolutionary time scales may be more informative. Several ways can be investigated. For instance, interspecies mating is efficient between closely related yeasts and the creation of such hybrids proved to be a valuable tool to study the cis and trans basis of expression divergence between species [49,50]. The analyses of intraspecies variations in genomic sequences [51,52], in gene expression [53] or in transcription factor DNA binding patterns [54] provide information on genotypic and phenotypic polymorphism of regulatory networks on microevolutionary scales. Furthermore, gene expression or the genome-wide location of a particular transcription factor can be considered as quantitative traits, which allows powerful genome-wide studies in order to detect trans and cis acting loci (eQTL) and to dissect the complex polygenic events involved in gene regulation changes [54–58]. Laboratory-driven evolution is an even more dynamic way to reveal the genomic events and the molecular mechanisms that can cause the adaptation of the regulatory networks to environmental or genetic constraints [59–63]. With the same rationale, synthetic biology and the artificial perturbation of cellular pathways underlined the fantastic plasticity of these networks [64]. The next step is to investigate at the single cell level the genomic changes arising in monoclonal populations at each generation and their impact on gene expression and phenotypic variability. Finally, the in silico evolution of artificial regulatory networks [65] provides comprehensive scenarios to estimate, for instance, the topological and functional impact of gene duplication [66,67].
5 Conclusions
These last five years, functional comparative genomic studies have drawn a highly complex picture of the evolution of regulatory networks in yeasts (Fig. 2). The less intuitive finding is certainly that apparent phenotypic uniformity, possibly reflecting traits under high selective constraints, often hides a large variety in the functioning of the corresponding regulatory networks. This obviously emerged from the balance between the genetic divergence due to structural variations, sequence mutations and horizontal gene transfers [68,69], which permanently reshape the regulatory pathways, and other evolutionary forces (selection and drift), which tend to reduce this polymorphism. Compensatory mutations between cis- and transregulatory actors is one way to maintain core functional modules, while allowing some plasticity [17,22,24]. However, this sole mechanism can hardly explain dramatic rewiring of large and essential functional modules, like the reprogramming of the ribosomal protein encoding genes between fermentative and nonfermentative yeast species [14]. Gene duplication is a major cause of neo- and subfunctionalization, therefore allowing the corewiring of large transcriptional modules [1,12,70,71]. Indeed, functional redundancy, either caused by gene duplication and families of paralogous regulatory factors or by the polygenic nature of most regulatory pathways, tends to promote a certain “regulatory instability”, by allowing the divergence of regulator properties and transcription factor substitutions, while maintaining essential gene coregulations [25,27,49,72].
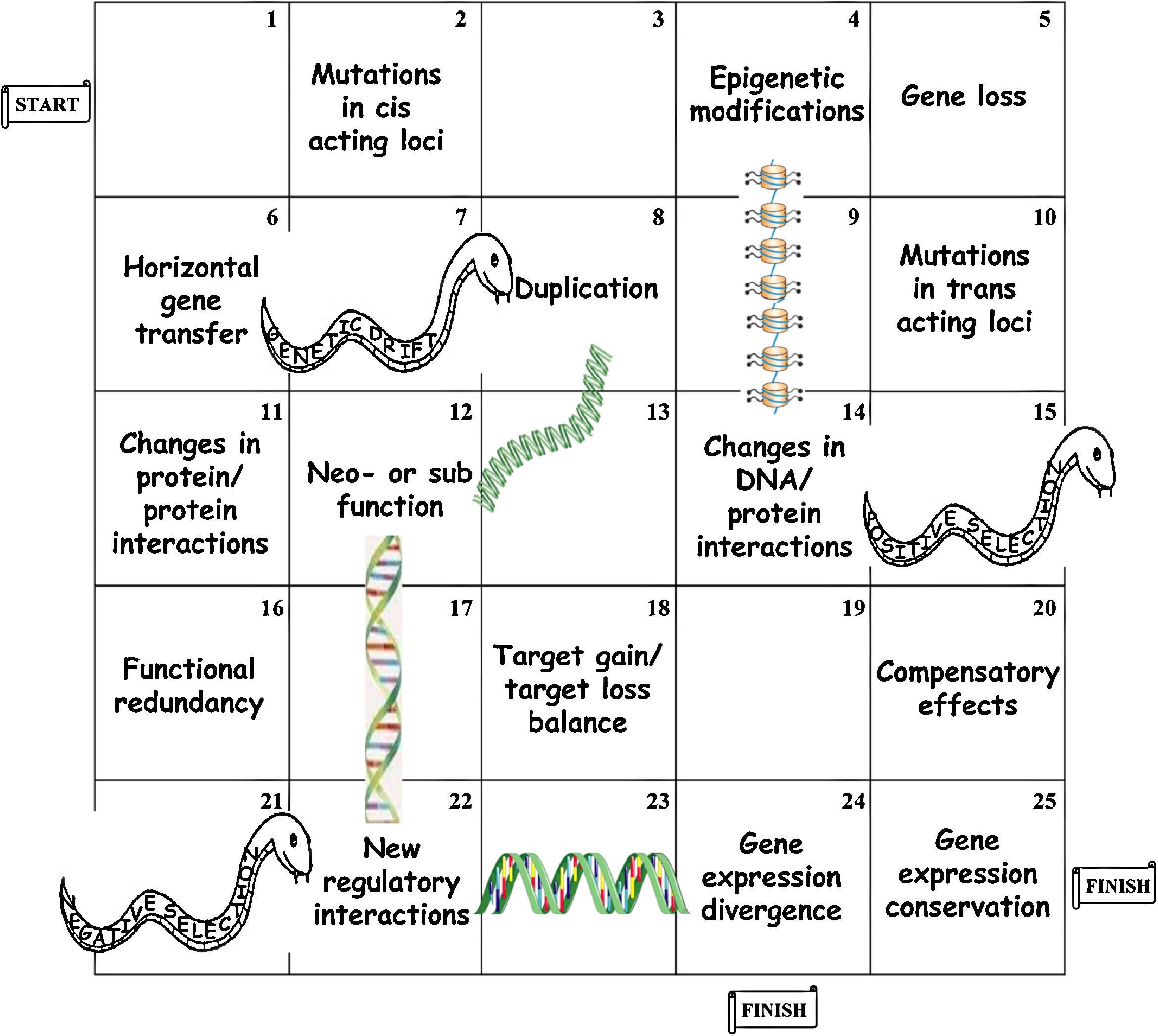
The “snakes and ladders” game of regulatory network evolution. SNP-like and structural variations in the genomic sequence, together with horizontal gene transfer and epigenetic changes, permanently challenge the functioning of regulatory networks by altering DNA-protein and protein/protein interaction patterns. Compensatory effects and functional redundancy buffer these changes at the level of gene expression, while allowing sub- and neofunctionalization and therefore divergence of the regulatory interactions. This polymorphism in transcriptional and posttranscriptional pathways eventually provides the conditions for significant rewiring of gene expression.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We are grateful to Dawn Anne Thompson for stimulating discussion and for allowing us to mention her work prior to publication.