

1 General presentation of inherited retinal dystrophies
Inherited retinal dystrophies (IRDs) belong to the vast group of neurodegenerations. They are Mendelian genetic conditions leading to the dysfunction and cell death of various retinal cell types, thus causing a progressive loss in vision. Cases where the functional defect is predominant are often considered as non-progressive congenital abnormalities, whereas those in which cell death predominates lead to irreversible vision loss (Fig. 1). In many of these diseases, there is progressive appearance of pigmentary deposits in the retina as a consequence of changes in the outermost layer of the retina called the retinal pigment epithelium (RPE), which is often secondary to the death of the retinal photoreceptors (PRs). Depending on the localization of the pigment deposits in the retina and of the accompanying retinal atrophy, one distinguishes the pigmentary retinopathies when the initial lesions are located in the peripheral regions of the retina, outside the central macula, and the macular dystrophies when the lesions are mainly located in the macula lutea. Thus, patients with pigmentary retinopathies typically undergo alterations in their peripheral visual field with moving difficulties, especially at low light levels, while they conserve until late in the course of the disease their reading capacities. In contrast, patients with macular dystrophies have early difficulties in reading and face recognition while they still move freely in their environment. When a severe degeneration is present at birth, usually both in retinal periphery and in the macula, the pigmentary retinopathy is called Leber congenital amaurosis. Pigmentary retinopathies are further subdivided in rod-cone dystrophies when the mutation has a direct pathogenic effect on the PRs rods, the cells which are sensitive to low light levels (night), and in cone-rod dystrophies when the PRs cones, which are sensitive to high light levels (daylight), are predominantly affected. Retinitis pigmentosa, the most frequent form of IRDs, corresponds to the rod-cone dystrophy. Less frequent are the cone dystrophies corresponding to a pathogenic effect only or mainly on cones.
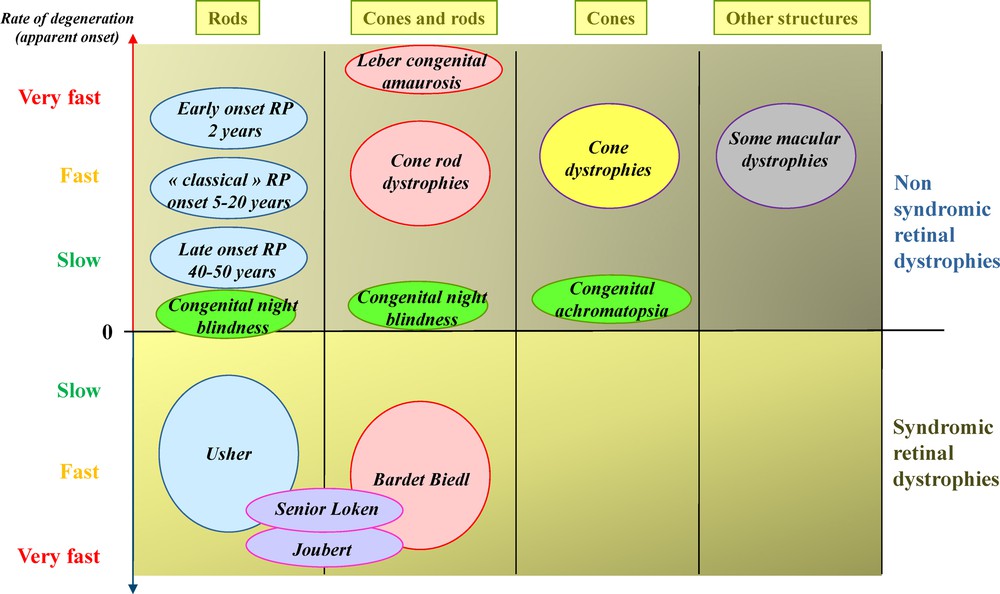
(Color online) Classification of inherited retinal dystrophies in function of the type of involved photoreceptors, rate of degeneration and syndromic/non-syndromic conditions.
In contrast with the blinding retinal degenerations described above, there are conditions due mainly to cell dysfunction in the retina. These are the congenital stationary night blindness in which the dysfunction takes place in the PRs rods or in the synapses of PRs with bipolar cells. In other conditions, the dysfunction occurs in cones leading to congenital achromatopsia and other forms of cone dysfunctions. Among IRDs, there are also vitreoretinopathies in which retinal glial cells and vessels are abnormal with as consequences retinal detachment, vitreous hemorrhages and PRs degeneration. When the choroid is affected, the conditions are named chorioretinopathies. There are subtypes of IRDs, relatively poorly defined, with difficulties in movement detection and adaptation to various light levels, which could be due to defects in specific subsets of retinal neurons like the amacrine cells. Finally, hereditary optic neuropathies, due to the degeneration of the retinal ganglion cells, are not classified among the IRDs because the major sign is the atrophy of the optic disc head visible in fundus whereas the retina has a normal aspect (Fig. 2).
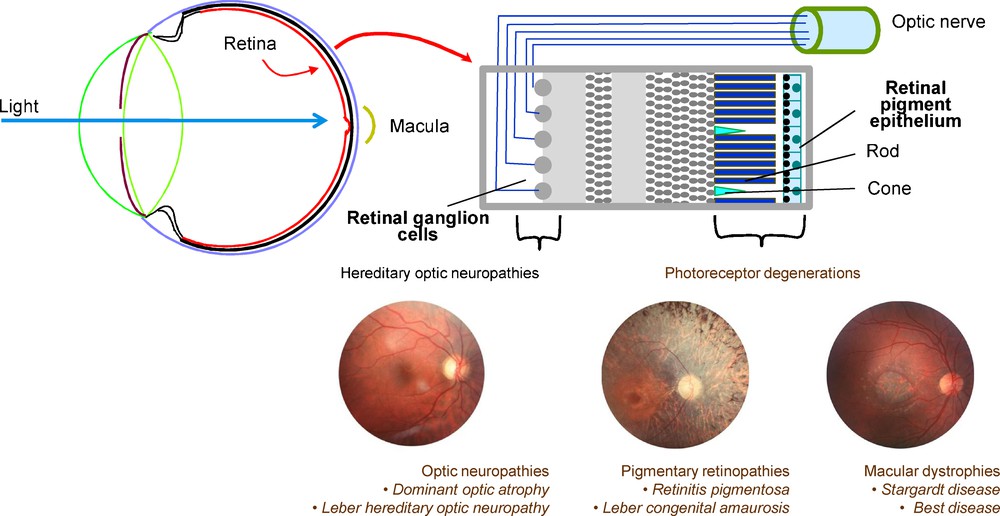
(Color online) Cellular structure of the retina and main groups of retinal dystrophies caused by the cell degeneration of the retina.
2 Overview of the genes involved in IRDs
In almost all groups of IRDs, the causative genes are many. Today, there are more 200 genes causing IRDs (https://sph.uth.edu/Retnet/sym-dis.htm). These genes code for proteins involved at many different levels in the retina (Fig. 3), being classified in function of the metabolism and of the cell type involved. The most important gene category is the one coding for proteins of the connecting cilium (ciliary proteins) and of the calical process of PRs (microvilli present at the apical end of the PRs inner segment), representing roughly 25% of all IRD genes. Many of them cause both non-syndromic pigmentary retinopathies and syndromic forms like the Usher (retinitis pigmentosa associated to deafness), Bardet Biedl, Joubert and Senior Loken syndromes. The genes coding for proteins of the visual transduction are not as many but they are responsible for non-syndromic forms in accordance with the specificity of the encoded proteins for PRs. The majority of the IRD genes codes for proteins acting directly in PRs. Yet, a few of them are expressed in the RPE, the supporting tissue necessary for proper functioning of the PRs. Another important gene category is the one coding for proteins involved in ubiquitous cell metabolisms, therefore not specific for the retina and thus frequently causing syndromic pigmentary retinopathies. However, it is of note that some genes coding for ubiquitously expressed splicing factors, are responsible for non-syndromic retinitis pigmentosa. In these cases, the restricted pathogenic effect to the retina might be explained in part by the intense synthesis of mRNA in PRs, considered as the highest mRNA synthesis in the body.
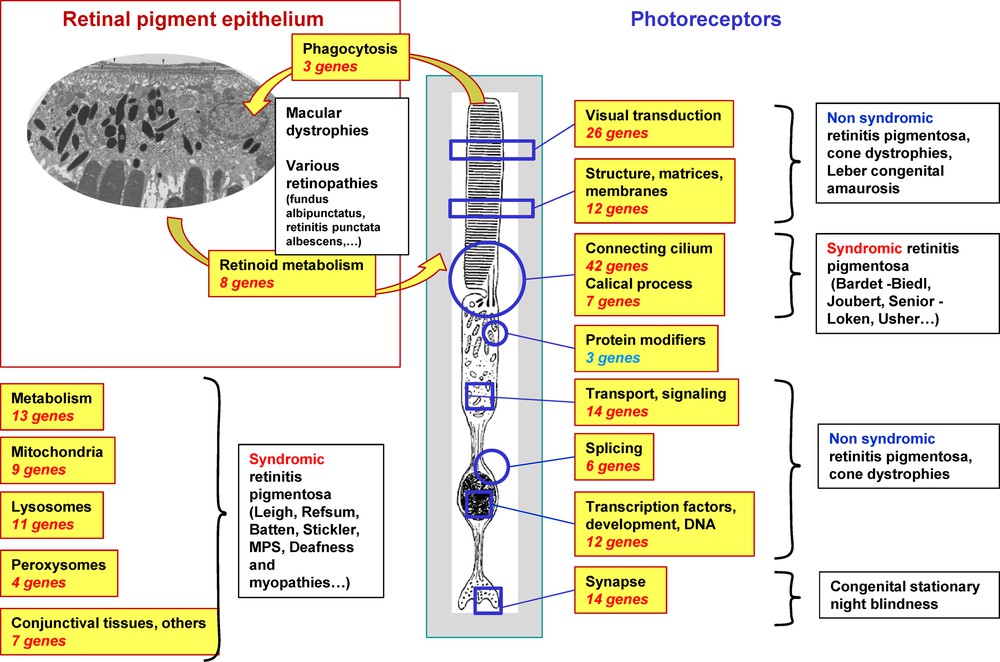
(Color online) Subgroups of genes involved in inherited retinal dystrophies.
Also interesting is the fact that a growing number of genes each cause more than one IRD form, and also both dominant and recessive forms. This is in part linked to the mutation type, with for example relatively mild pathogenic effects restricted to the macular area leading to macular dystrophies while more severe pathogenic effects will eventually cause more generalized PRs cell death, like the retinitis pigmentosa phenotype. The rate of cell death might also be dependent on the mutation type, so that very deleterious mutations will cause early onset degeneration diagnosed as Leber congenital amaurosis or juvenile retinitis pigmentosa.
3 Comprehensive screening of IRD patients in Montpellier
Over a 21-year period from 1990 to 2011, patients were recruited at our specialized outpatient clinic (Centre de référence – Maladies sensorielles génétiques du CHU de Montpellier). They underwent extensive clinical investigations and 107 genes were screened mostly through Sanger DNA sequencing [1]. There were 1957 IRD cases (1481 families) distributed in 70% of pigmentary retinopathy cases (56% non-syndromic, 14% syndromic), 20% maculopathies, 7% stationary conditions. Patients with retinitis pigmentosa (Fig. 4A) were the most frequent (47%) followed by Usher syndrome (10.8%). Among non-syndromic pigmentary retinopathy patients, 84% had rod-cone dystrophy, 8% cone-rod dystrophy and 5% Leber congenital amaurosis. Macular dystrophies were encountered in 398 cases (30% had Stargardt disease and 11% had Best disease). We could screen 609 families (not all families were screened because of lack of DNA or incomplete clinical records). Positive molecular results were obtained in 417/609 families (68.5%). The sequencing of the 5 most frequent genes (ABCA4, USH2A, MYO7A, RPGR and PRPH2) allowed for a positive molecular result in 48% of the 417 families with IRDs. Except for autosomal dominant and recessive retinitis pigmentosa in which less than half the families had positive molecular results, about 75% of the families with other forms of retinal conditions had a positive molecular diagnosis. Thus, although gene discovery considerably improved the molecular diagnosis in many subgroups of IRDs, retinitis pigmentosa, accounting for almost half of IRDs, remains only partly molecularly defined.
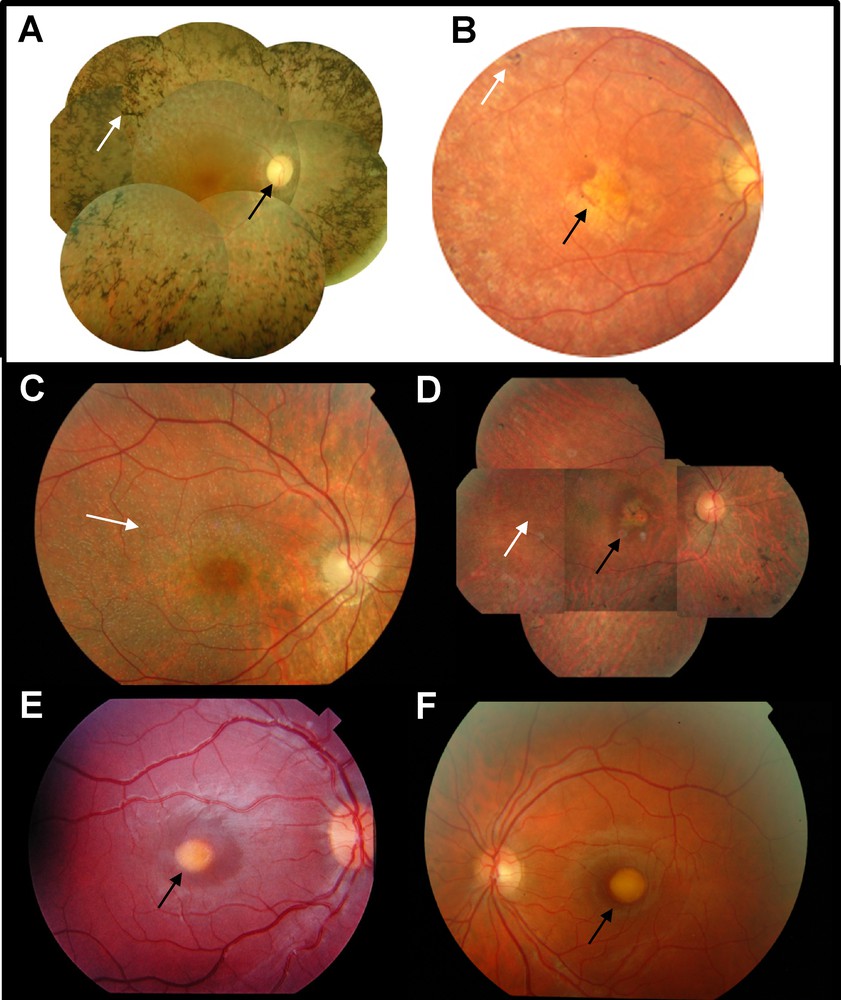
(Color online) Fundus photographs of patients with inherited retinal dystrophies. A. Typical retinitis pigmentosa fundus, showing many bone-spicule-shaped pigment deposits in the periphery of the retina (white arrow). The optic disc is pale (black arrow) and the retinal vessels are highly attenuated. B. Fundus of a patient with Leber congenital amaurosis carrying RPE65 mutations. Pigment deposits are rare (white arrow) and the macular area is atrophic (black arrow). C. Fundus of a patient with fundus albipunctatus and RDH5 mutations showing typical white dot deposits (white arrow). D. Fundus of a patient with retinitis punctata albescens and RLBP1 mutation. White dot deposits are visible (white arrow) and there are some pigment deposits with a macular atrophy (black arrow). E. fundus of a child with Best macular dystrophy due to a BEST1 mutation and showing a typical “egg yolk” yellow deposit in the macula (black arrow). F. Fundus of an adult with vitelliform macular dystrophy due to the recurrent p.Leu238Arg mutation in IMPG1 showing a macular vitelliform deposit (black arrow).
4 Discovery and prevalence of genes of the visual cycle
The visual cycle is the metabolic process through which the visual pigment (rhodopsin in rods and cone-opsins in cones) is regenerated after light activation, in order to maintain continuous vision (Fig. 5). Light isomerizes 11-cis retinal, the unique chromophore of all vertebrate opsins, into all-trans-retinal, changing the conformation of the visual pigment which becomes activated. Subsequent inactivation of the visual pigment through phosphorylation and steric hindrance of the transducin binding site yields apo-opsin and all-trans-retinal. All-trans-retinal must then be isomerized back to 11-cis retinal through several steps occurring in the photoreceptors, adjacent RPE and Muller cells. Kinetics of the visual cycle is controlled by several key enzymes and binding proteins. Defects in these proteins can slow down or interrupt the visual cycle causing a lack of chromophore, which as a consequence leads to various retinal diseases including relatively moderate functional visual impairment or very severe blindness.
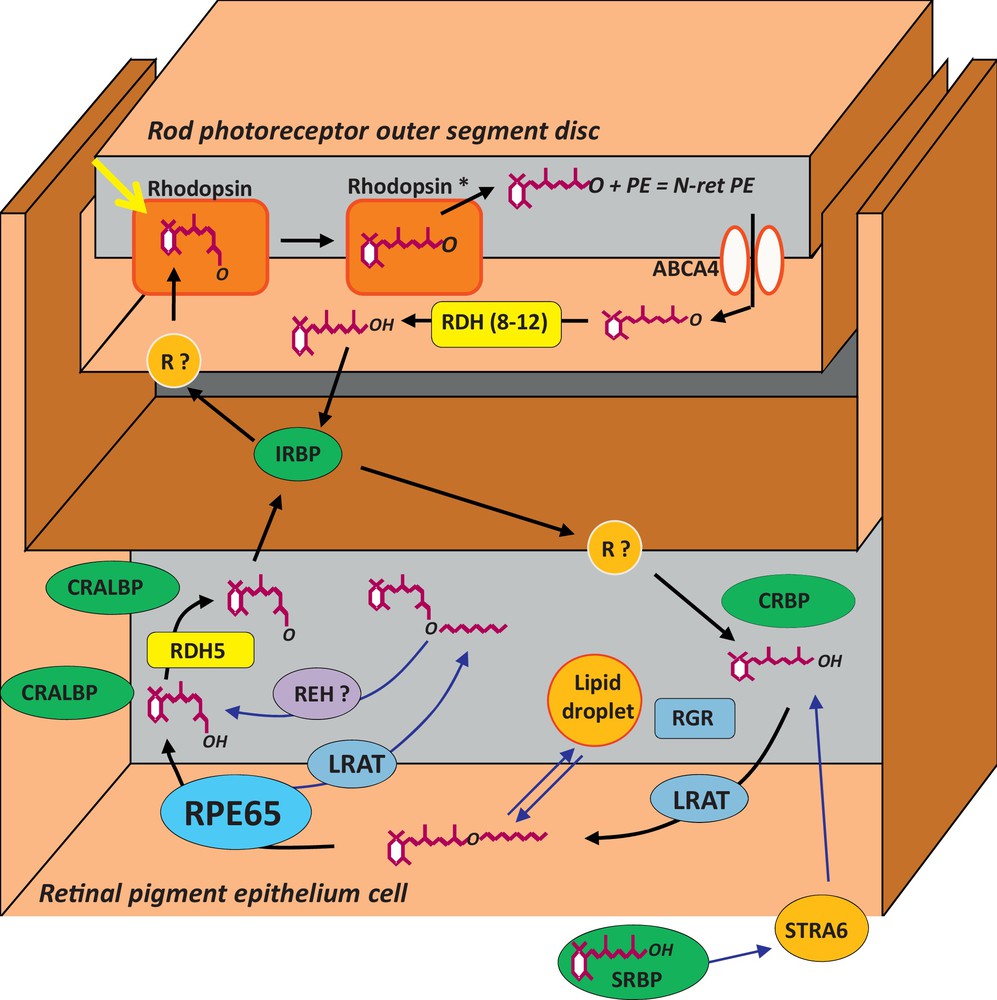
(Color online) The visual cycle. R? means an unknown retinoid receptor. Refer to the text for details on the name of proteins.
In 1993, we described RPE65, a new abundant and specific 65-kDa protein of the RPE [2,3]. Several years later, we discovered that mutations in RPE65 were responsible for 5 to 10% of the cases of LCA and a few cases of arRP [4–6] (Fig. 4B). Later on, several groups in USA demonstrated that RPE65 was the key isomerohydrolase restoring the 11-cis retinoid levels after light stimulation of the retina [7–9]. This shed light on the role of the RPE and of the visual cycle genes in IRDs and we subsequently looked for involvement of other visual cycle genes in IRDs. We searched for RPE65 protein partners but none of these were found to be involved in IRDs until now [10]. As others, we screened the gene just upstream RPE65 in the metabolic pathway of the visual cycle, coding for the LRAT which esterifies all-trans-retinol to all-trans retinyl esters and we discovered a homozygous deletion [11,12] in a young boy with a LCA phenotype similar to that of RPE65. Then, we screened several dehydrogenases (RDH5, RDH8, RDH10) which are abundant in the RPE but only mutations in RDH5, previously known to lead to the fundus albipunctatus, were found (Fig. 4C) (unpublished results). Finally, we also screened the retinoid binding proteins, including RLBP1 (coding the 11-cis retinoid binding protein CRALBP), RBP3 (coding the interphotoreceptor matrix binding protein IRBP), RBP1 (coding the cellular retinol binding protein) and the opsin-related proteins RGR and RRH. We found several families with a large 3′ deletion of RLBP1 originating from Morocco in the very characteristic form of IRD named retinitis punctata albescens [13] (Fig. 4D) as well as other mutations in European families [14]. No mutations were found in the other genes [15,16].
5 Prevalence of the genes in the autosomal forms of retinitis pigmentosa
Retinitis pigmentosa (RP) is the most frequent form of IRDs, therefore we focused on this particular disease. In 2009, we began a study to evaluate the prevalence of known genes in the autosomal dominant retinitis pigmentosa (adRP) and to search for new genes in this inheritance form. Of the 23 genes currently described, we screened only the 10 most frequent genes, either by sequencing all exons, or by sequencing only the hot spots. A new locus for autosomal dominant cone-rod dystrophy was found [17]. We sequenced 283 probands of adRP families and found that 50% of the families had a mutation. We then submitted to exome sequencing 12 families, which were negative for the first screening. We found that 8/12 of these “negative” families had in fact mutations which were either missed by Sanger sequencing or present in gene parts unscreened in the primary screening. By extrapolation of the results of the 12 families, we expect that about 20% of the adRP families do not have mutations in the known genes. We are currently analyzing the exome sequence of several members of the last four families.
At about the same time, we also screened consanguineous families for autosomal recessive RP (arRP). We first found a new mutation in the MERTK gene [18] and found that RP1 mutations are present in arRP families [19]. We genotyped 31 families with microsatellite markers for the two frequent genes USH2A and EYS, and found that one third were homozygous for one of the two genes. We then selected 15 families, which were not homozygous for the markers and we subjected the patients to 250 K SNP genotyping to search for homozygous regions [20]. In 10/15 families, we found homozygous regions in which known arRP genes were described and we indeed found mutations in RLBP1 (2 families), RP1 (2 families), IMPG2, CNGB1, PDE6A, PDE6B, C2ORF71 and NR2E3 [21]. The 5 negative families have been subjected to exome sequencing. For two of them, a large homozygous region contains a candidate gene, which is under investigation.
6 Discovery of a new gene causing vitelliform macular dystrophies
Macular dystrophies are inherited retinal dystrophies in which various forms of deposits, pigmentary changes and atrophic lesions are observed in the macula lutea, the cone-rich region of the human central retina. Mutations in several genes, expressed in either the photoreceptor rods or cones (ABCA4 [MIM 601691], ELOVL4 [MIM 605512], PRPH2 [MIM 179605], PROM1 [MIM 604365]) or in the retinal pigment epithelium (BEST1 [MIM 607854], CDH3 [MIM 114021], TIMP3 [MIM 188826]), the photoreceptor supporting tissue, are associated with inherited macular dystrophies.
Vitelliform macular dystrophies (VMD) form a subset of macular dystrophies characterized by yellow, round deposits usually at the centre of the macula and containing lipofuscin, a chemically heterogeneous pigment visualized by autofluorescence imaging of the fundus. Best macular dystrophy (BMD [MIM 153700]) is a juvenile form of VMD inherited as an autosomal dominant trait leading to visual acuity loss and featuring in many cases a characteristic decrease in the Arden ratio obtained from the electrooculogram (Fig. 5E). Many BMD families harbor a heterozygous mutation in BEST1 [22], which is specifically expressed in the retinal pigment epithelium. In addition to BMD, there are cases of adult-onset vitelliform macular dystrophy (AVMD [MIM 608161]), frequently simplex and featuring a small vitelliform disc on the fovea, moderate loss in visual acuity and normal Arden ratio. In AVMD families with autosomal dominant inheritance, mutations in BEST1 or in PRPH2 are sometimes found. However, most cases of AVMD and some BMD cases do not harbor mutations in either BEST1 or PRPH2 [23]. The involvement of both genes was also excluded from a form of autosomal dominant atypical VMD described in one family (VMD1 [MIM 153840]) [24]. Therefore a subset of VMD individuals must harbor mutations in other genes.
Starting from a French family with autosomal dominant vitelliform dystrophy (Fig. 4F), microsatellite analysis revealed a 95.2 Mb haplotype on chromosome 6p12.1-q24.3. By exome sequencing of several patients of the family, we found a c.713T>G leading to p.Leu238Arg in the IMPG1 gene [25]. The same mutation was also found in another French family and in a Spanish family with the same phenotype. We then sequenced the 17 exons and flanking intronic regions of IMPG1 in 144 probands with VMD and various forms of macular dystrophy. We found two additional autosomal recessive vitelliform dystrophy families with either homozygous (family from Italy) or compound heterozygous (family from France) mutations. Interestingly, affected patients often have multifocal spots of lipofuscin accumulation in the retina. IMPG1 (Interphotoreceptor Matrix ProteoGlycan 1) encodes SPACR, a 150-kDa Sialo Protein Associated with photoreceptor Cones and Rods protein, a component of the interphotoreceptor matrix located in the subretinal space in which photoreceptors and the RPE are in close apposition. The role of insoluble protein SPACR is to form a scaffold around outer segments of rods and cones, probably to exert a constraint helping in the maintenance of the elongated shape of these structures. Future studies are necessary to investigate how the mutations in IMPG1 lead to accumulation of lipofuscin in the retina.
The finding of IMPG1/SPARC deficient cases introduces, besides the photoreceptors and the RPE, a third actor in the causes of VMD, namely the interphotoreceptor matrix that joins together the photoreceptors and the RPE. In this respect, it is noteworthy that most IMPG1-related VMD cases had a normal or moderately decreased EOG Arden ratio, which distinguishes them from the BEST1-related VMD cases due to a dysfunction of the RPE, thus indicating different mechanisms of lipofuscin accumulation. It will be of interest to further explore other cases of VMD, unlinked to IMPG1, to test whether other components of the interphotoreceptor matrix are involved in the VMDs.
Disclosure of interest
The author declares that he has no conflicts of interest concerning this article.