

1 Introduction
The Usher syndrome (USH), an autosomal recessive genetic disease, is responsible for more than a half of the human cases of inherited deaf-blindness. This syndrome was first described by Albrecht von Graefe in 1858, but was named after Charles Usher, a Scottish ophthalmologist, who was the first to report the hereditary nature of the disorder (Usher, 1914) (see references [1,2]). Previous epidemiological studies have estimated the prevalence of USH as 1/25,000 [3], but more recent studies have proposed prevalence ranges from 1/6000 [4] to 1/10,000 [5] as there can be frequent misdiagnosis of the retinitis pigmentosa. USH is clinically and genetically heterogeneous. Studies concerning the properties of USH proteins, their interacting partners, and the phenotypes of USH deaf mutant mice have elucidated the molecular and cellular mechanisms underlying the USH hearing impairment and the roles played by the USH proteins in the cochlea (see references [6–8]). In contrast, little is known about the pathogenesis of vision impairment, as there is a lack of significant visual defect in most USH mutant mice (see references [2,6,7,9,10]). In this review, we focus on recent results that shed light on the pathogenesis of the USH retinal phenotype. We also discuss future therapeutic approaches aimed to prevent or delay the onset of retinitis pigmentosa in USH patients.
2 Clinical features and molecular diagnosis of Usher syndrome
Three types of USH (USH1, USH2, and USH3) have been distinguished clinically. These are defined according to the severity of the sensorineural hearing impairment, the presence or absence of vestibular defects, and the precocity of retinitis pigmentosa onset (Table 1) (see references [7,8,11–13]). USH1 is the most severe form. USH1 patients have severe to profound congenital bilateral hearing loss, which, if uncorrected, impedes speech acquisition. Vestibular dysfunction is also present from birth; USH1 children acquire the ability to take a sitting position and to walk later than usual. The USH1 retinopathy is classified as a rod-cone dystrophy, in which rod anomalies appear first and rapidly worsen, followed by a slow progressing cone dysfunction, and photoreceptor cell degeneration [9,13–17]. Night blindness may be detected during childhood, and this can be followed by a narrowing of the visual field (“tunnel vision”), which rapidly progresses to more severe blindness [2,18]. Retinal degeneration over the course of the disorder can be followed by the progressive reduction in electroretinogram (ERG) wave amplitudes and by fundus examination. Abnormal ERG responses may be recorded in patients prior to the symptoms of retinitis pigmentosa arising, and can help early disease diagnosis; the earliest age at which ERG abnormalities have been reported in USH1 children is 18 months [14–17,19]. USH2 patients display moderate to severe congenital hearing impairment (mostly affecting high sound frequencies). Speech is usually not affected, and vestibular function is normal. Symptoms of rod-cone dystrophy manifest later in USH2 patients than in USH1 patients, as the retinitis pigmentosa is usually diagnosed in USH2 patients between the ages of 10 and 40 [20,21]. USH3 is the least common type of USH but has a high prevalence in Finns and Ashkenazi Jews, likely as a result of founder effects (see reference [13]). In USH3 patients, hearing impairment starts before the age of 30, and is progressive. The progression speed is variable but, in most cases, patients ultimately become profoundly deaf. USH3 patients have well-developed speech. The vestibular defect is variable, and the onset of retinitis pigmentosa is postpubertal, usually starting from the age of 20.
Clinical characteristics of Usher syndrome (USH) subtypes. Three types of USH (USH1, USH2, and USH3) have been distinguished clinically. Indications regarding the evolution of speech acquisition by affected kids, their acquisition of the sitting position, and walking abilities should help to discriminate among the three USH subtypes.
| Hearing impairment | Vestibular dysfunction | Retinitis pigmentosa | |
| USH1 | Profound and congenital | Severe | Prepubertal onset |
| USH2 | Mild to severe and congenital | Absent | Postpubertal onset |
| USH3 | Mild and progressive | Variable | Postpubertal onset or variable |
Visual dysfunction is clinically diagnosed at an average age of 17 and 24 in USH1 and USH2 patients, respectively [14–17,19]. Such a late diagnosis has detrimental consequences for USH individuals. To date, about 14 different USH loci have been mapped on different chromosomes (see http://hereditaryhearingloss.org/), and 10 of the corresponding genes have been identified (Fig. 1). These genes encode six USH1 proteins – myosin VIIa (USH1B) [22], harmonin (USH1C) [23,24], cadherin-23 (USH1D) [25,26], protocadherin-15 (USH1F) [27,28], SANS (USH1G) [29], and CIB2 (USH1J) [30], three USH2 proteins – usherin (USH2A) [31], VLGR1 (USH2C) [32], and whirlin (USH2D) [33], and one USH3 protein, clarin-1 (USH3A) [34] (see Fig. 1). In addition, PDZD7 has been shown to act as a modifier of USH2 gene function [35]. Whether PDZD7 is also an USH2 causative gene remains to be established.
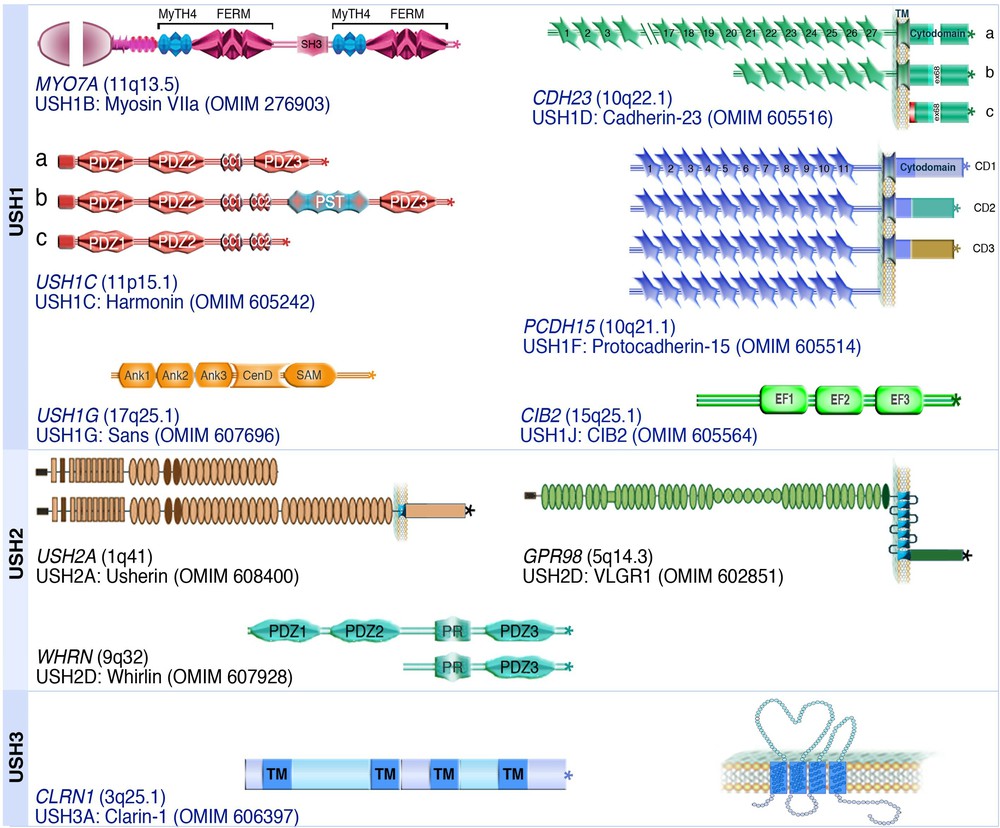
(Color online) Modular structure of the USH proteins. The USH proteins include a motor protein, myosin VIIa (USH1B), two PDZ-domain containing proteins, harmonin (USH1C) and whirlin (USH2D), a Ca2+ integrin-binding protein, CIB2 (USH1J), and five transmembrane proteins, cadherin-23 (USH1D), protocadherin-15 (USH1F), usherin (USH2A), VLGR1 (USH2C) and clarin-1 (USH3A). The OMIM website entries provide more information about USH proteins and associated diseases.
Private mutations are common in USH patients [36]; thanks to improving sequencing techniques, sequencing of the exons of all known USH genes is the best method for an early and reliable molecular diagnosis of the disease ([36–38]; see references [8,12,39]). Improvements in molecular diagnosis are still needed for all causative gene anomalies (such as large deletions, and modifications to introns and promoters) to be detected. The USH molecular diagnosis is critical for genetic counselling, educational orientation, the use of prosthesis (especially cochlear implants in USH1 patients to improve their speech acquisition), and the initiation of any new therapy under development. For example, patients may have learnt to use sign language (inefficient once they undergo visual loss), but could instead have benefited from early cochlear implantation (prosthetic electrode arrays that directly stimulate the primary auditory neurons) for oral language acquisition [40].
3 Molecular bases of the hearing and visual impairments in the Usher syndrome
3.1 USH proteins and the hearing impairment
To date, at least one mutant mouse model has been described for each genetic form of USH1 (with the exception of USH1J), USH2, or USH3. All currently available USH mutant mice faithfully mimic the human hearing impairment (see Table 2; [6,7]). Morphological, electrophysiological and molecular analyses of these animal models have identified the roles that the USH proteins play in the auditory sensory cells (hair cells). The hair bundle, the mechanoreceptive structure by which auditory hair cells respond to sound-evoked mechanical stimuli, is found to be disorganised in all USH mutant mice (see references [6,7]). A hair bundle consists of 50 to 300 F-actin-filled stereocilia; these are arranged in three rows that increase in height towards a genuine cilium (the kinocilium), and are held together by several types of fibrous links (Fig. 2A).
Phenotypes of USH mutant mice.
| USH protein USH gene Representative mouse models |
Type of mutation (genetic background) |
Phenotype | References | |
| Hearing loss | Visual defects | |||
| Myosin VIIa USH1B Myo7a 4626SB/4626SB |
p.Q720X (mixed 50% CBA/Ca; 50% BS, BALBc background) |
Congenital profound deafness & vestibular dysfunction | No retinal degeneration | [41,42] |
| Harmonin USH1C Ush1c –/– |
Exon 1 deletion (C57BL/6J background) |
Congenital profound deafness & vestibular dysfunction | No retinal degeneration | [43,44] |
| Harmonin USH1C Ush1c 216AA/216AA |
c.216G>A, introduces a cryptic splice site in exon 3 (mixed C57BL/6 and 129S6 background) |
Congenital profound deafness & vestibular dysfunction | 50% decrease in a- and b-wave amplitudes and retinal degeneration | [45] |
| Cadherin-23 USH1D Cdh23 v-2J/v-2J |
c.4104+1G>A (C57BL/6J background) |
Congenital profound deafness & vestibular dysfunction | No retinal degeneration (faster ERG responses) |
[46,47] |
| Protocadherin-15 USH1F Pcdh15 av-3J/av-3J |
p.E1373RfsX36 (C57BL/6J background) |
Congenital profound deafness & vestibular dysfunction | No retinal degeneration | [48–50] |
| Sans USH1G Ush1g js/js |
p.E228RfsX8 (C57BL/6J background) |
Congenital profound deafness & vestibular dysfunction | No retinal degeneration | [51] |
| Usherin USH2A Ush2a–/– |
Replacement of exon 5 (mixed 129/Sv and C57BL/6J background) |
Mild to severe congenital hearing impairment | Retinal degeneration | [52] |
| Vlgr1 USH2C Gpr98–/– |
Replacement of exons 2–4 with a neomycine cassette (C57BL/6J background) |
Mild to severe congenital hearing impairment + audiogenic seizure susceptibility | No retinal degeneration | [53,54] |
| Vlgr1 USH2C Gpr98 del7™/del7™ |
Replacement of exon 82 (mixed C57BL/6J and 129X1/Sv background) |
Mild to severe congenital hearing impairment | No retinal degeneration (mildly abnormal ERG responses) |
[55] |
| Whirlin USH2D Whrn L–/L– (long isoform) |
Partial replacement of exon 1 (C57BL/6J background) |
Mild to severe congenital hearing impairment | Retinal degeneration | [56] |
| Whirlin USH2D Whrn–/– |
592-bp deletion (p.H433fsX58mutation) (C57BL/6Jbackground) |
Mild to severe congenital hearing impairment | No retinal degeneration | [57] |
| Clarin-1 USH3A Ush3a –/– |
Disruption and deletion of promoter and exon 1 (C57BL/6Jbackground) |
Mild and progressive hearing impairment, & variable vestibular defect | No retinal degeneration | [58] |
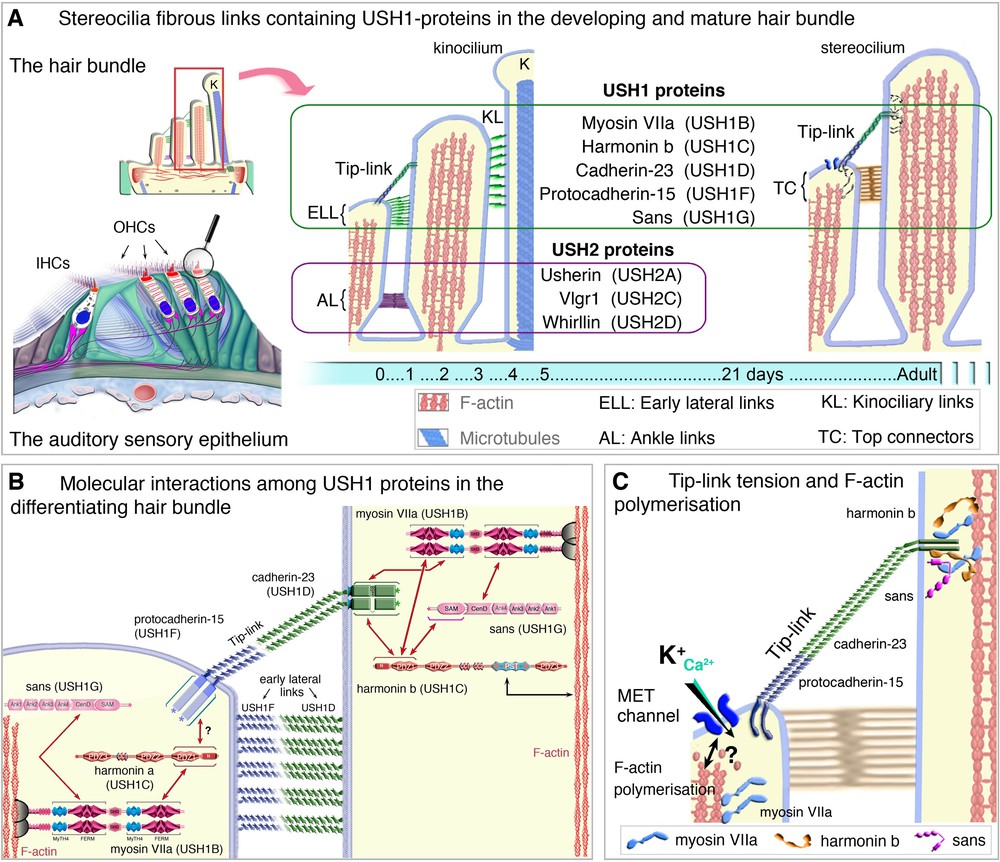
(Color online) USH proteins in the hair bundle of mammalian auditory hair cells. A. During development, early transient lateral links, baso-lateral ankle links and tip-links connect the growing stereocilia. At mature stages, the top connectors (in outer hair cells) and the tip-links hold the hair bundle stereocilia together (A). B. USH1 proteins form the early lateral links (ELL) between stereocilia, the stereocilia-kinociliary (KL) links and the tip-link. USH2 proteins form the transient ankle links (C, D) interactions among the USH1 proteins at the stereocilia tip (C), and the link between the mechano-electrical transduction (MET) machinery, USH1 proteins and F-actin polymerisation (D).
Adapted from Caberlotto et al. [59] and El-Amraoui et al. [60].
Numerous direct interactions between USH proteins [7,29,59,61–69] have been detected in vitro [7,8,29,53,59,61–65,69]. In the hair bundles, USH1 proteins are progressively restricted to the tips of the stereocilia in the differentiating and mature hair cells [43,59,70–72]. When an USH1 gene is inactivated in a mouse model, at least one other USH1 protein is absent or mislocated in the hair bundle, indicating that the USH1 proteins interact also in vivo [43,59,61,62,73]. USH2 proteins are transiently located in the baso-lateral region of the stereocilia during hair bundle development, contributing to the formation of the ankle links [53,74] (see Fig. 2A). The spatio-temporal distribution of USH3a in hair cells is still unknown [75,76].
The USH1 proteins have critical roles in the hair bundle both during development and at mature stages (Fig. 2A–C). Cadherin-23 and protocadherin-15 form heteromeric structures required for the formation of transient fibrous apical links interconnecting the stereocilia and the adjoining kinocilium [43,61,63,71,72] (see Fig. 2B). In mature hair cells, cadherin-23 and protocadherin-15 form the tip-link [77]. The other three USH1 proteins, harmonin, sans and myosin VIIa, anchor these links to the actin filaments of the stereocilia [59,61,62,78–80] (Fig. 2B,C). USH1 proteins also form the core of the mechano-electrical transduction machinery [59,77–80] (see Fig. 2B,C). A role of USH proteins at the hair cell synapse has also been proposed [76], but this has currently only been shown for harmonin [81].
3.2 USH proteins and vision impairment
The age at onset of retinitis pigmentosa varies between USH patients, but they all ultimately undergo a vision loss. However, in the corresponding mouse models, the visual phenotype differs greatly according to the USH clinical subtype (see Table 2). Retinal cell degeneration and vision loss have been unambiguously reported only in USH2 mutant mice lacking usherin [52] or the whirlin long isoform [56]. The three USH2 proteins have been detected in a spatially restricted inner segment membrane region that surrounds the photoreceptor connecting cilium [52,56,82,83]. This region is known as the periciliary ridge complex region [84], and is where vesicles dock with the plasma membrane during translocation to the outer segment. It has been suggested that USH2 proteins contribute to the trafficking of cargos moving from the inner segment to the outer segment of photoreceptor cells [82]. However, phototransduction proteins are known to transit from the inner to the outer segment of photoreceptor cells, and no obvious defect in their localisation has so far been observed in any of the USH2 mutant mice. It has also been suggested that the two transmembrane proteins usherin and Vlgr1 form fibrous links between the connecting cilium and the surrounding plasma membrane in the periciliary ridge complex region; these links are absent in the photoreceptor cells of Vlgr1 deficient mice [82].
With the exception of harmonin mutant mice (Ush1c/c.216G>A knock-in) [45] (see Table 2 and below), none of the other USH1 [41,46,48,49,85] or USH3a [58] mutant mouse models exhibit retinal degeneration (Table 2). USH3a transcripts have been detected in the inner nuclear layer of the retina [58], but it is still unknown whether the USH3a protein, of unknown function, is present in bipolar cells, amacrine cells, horizontal cells or glial Muller cells. The USH1proteins have been detected in different types of retinal cells, including photoreceptor cells [17,82,86–90]. Thorough analysis has shown that the only significant morphological difference between USH1 mutant and wild-type mice is an aberrant positioning of the melanosomes and phagosomes in the retinal pigment epithelium (RPE) cells of myosin VIIa-deficient mice [91–93]. RPE cells from myosin VIIa-deficient mice have been found to display lower amounts of RPE65, a key metalloenzyme in the retinoid visual cycle that is necessary for maintaining visual function [94]. Functional myosin VIIa may be necessary for the light-dependent translocation of RPE65 to the cell centres, thereby preventing their degradation [94]. Myosin VIIa is the only USH1 protein present in RPE cells [86,90]. The impact of an RPE cell defect on the progression of the retinal dystrophy in USH1B patients remains to be understood. Some insight into the role of USH1 proteins in photoreceptor cells has been provided by detailed analyses of mutant mice and USH1 patients. Optical imaging of the retina and analyses of the visual function in USH1B individuals have shown that photoreceptor cells are the primary target cells [19]. A role of all USH1 proteins in the photoreceptor synaptic region has been proposed [82,87], but no significant retinal synaptic defect has been observed in USH1 mutant mice [9].
High opsin concentration in the connecting cilia of photoreceptor cells is associated with an absence of functional myosin VIIa [95,96]. This suggests a defect in myosin VIIa-mediated trafficking of this protein between the inner and outer segments. Spectrin βV, the mammalian β-heavy spectrin [97,98], has been identified as a myosin VIIa and rhodopsin binding partner in photoreceptor cells. This spectrin associates also with two other USH1 proteins (sans and harmonin) and co-distributes with myosin VIIa from the Golgi apparatus to the base of the outer segment, matching the trafficking route of opsin and other phototransduction proteins [99]. Formation of a spectrin βV-rhodopsin complex has been detected in the differentiating photoreceptors as soon as their outer segment emerges. This suggests that a failure of the spectrin βV-mediated coupling between myosin VIIa and opsin molecules accounts for the opsin transport delay in myosin VIIa-deficient mice [99].
As aforementioned, the anomalies detected in USH1 mutant mice do not result in abnormal visual phenotype. The discrepancy between the mouse and human retinal phenotypes has made it particularly difficult to understand the mechanism of the vision loss in USH1. Explanations for this phenotypic discrepancy include the shorter lifespan of the mouse, interspecies differences in light exposure, functional redundancy of USH1 proteins in the mouse retina, and intrinsic differences in the photoreceptor cell architecture [1,6]. The origin of the hearing impairment in USH1 patients has been unravelled by exploring a holistic molecular mechanism involving the five defective USH1 proteins (myosin VIIa, harmonin, cadherin-23, protocadherin-15 and SANS) as all USH1 genetic forms have the same auditory phenotype [7,8]. Likewise, a clue to the pathogenesis of the USH1 retinal dystrophy came from postulating that USH1 proteins probably act together in a similar complex in photoreceptor cells, and by considering that the presence and absence of retinal degeneration in USH1 patients and mouse models, respectively, may be informative as to the role of the USH1 proteins complex [90].
3.3 Major structural differences in the architecture of rodent and primate photoreceptor cells
Comparative studies to determine the exact subcellular distributions of five USH1 proteins (myosin VIIa, harmonin, cadherin-23, protocadherin-15 and SANS) have recently been carried out in mouse, monkey, and human retinas [90]. Myosin VIIa, SANS and, to a lesser extent, protocadherin-15 have been detected in the murine photoreceptor inner segment, but immunostaining experiments did not detect harmonin or cadherin-23. However, similar explorations in monkey and human retinas revealed a strong staining for the five USH1 proteins at the junction between the inner and outer segments of photoreceptor cells [90]. This interspecies discrepancy in protein distribution is specific to USH1 proteins as all three USH2 proteins were detected in the periciliary membrane complex region of photoreceptor cells in all species analysed (including xenopus, mouse, monkey, and man) [90]. Structural analyses of mouse and monkey photoreceptor cells showed marked differences in the intermediate region between the outer and inner segments. Microvilli-like structures that emerge in the apical region of the inner segment and form a collar around the base of the outer disks were observed in the monkey, but were absent or vestigial in the mouse (see Fig. 3A, B). These calyceal processes were first described 50 years ago [100,101], but their role is still unknown. They are extremely labile and are preserved under particular fixation conditions of the retina [90]. This may explain their inconsistent identification pattern, and the absence of USH1 proteins detection in these structures in photoreceptors thus far [70,89,90,102]. Large F-actin bundles extend downwards, from the F-actin core of the calyceal processes, underneath the plasma membrane and along the apical half of the inner segment (see Fig. 3B–D). These F-actin-labelled roots, absent in mouse and rat photoreceptors, can be regarded as stable landmarks of the labile calyceal processes (Fig. 3A, B). Calyceal processes and their F-actin roots are thought to form a continuous cytoskeletal girdle, which may strengthen the interface between the outer and inner segments [90].
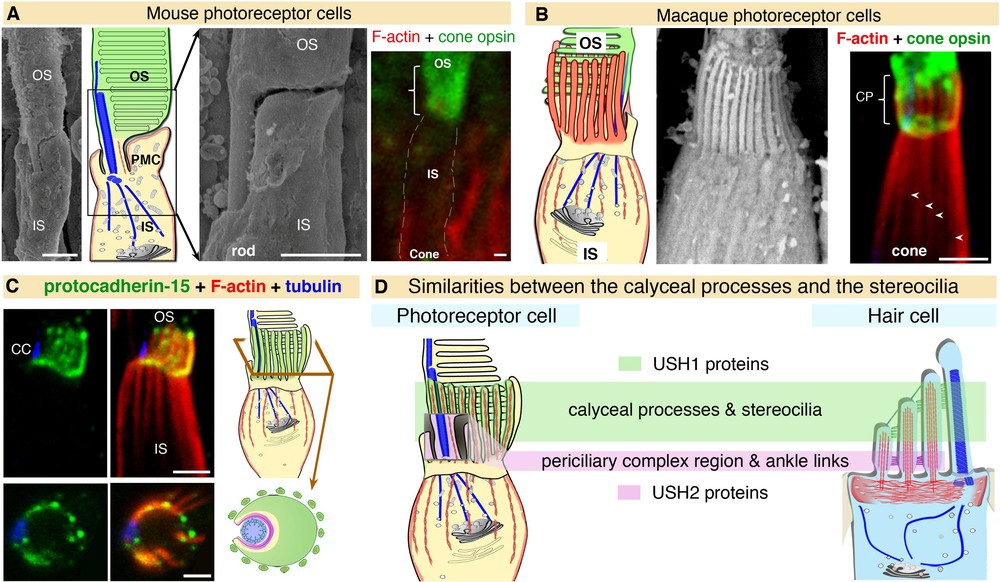
(Color online) A,B. Detailed analysis of the retina by scanning electron microscopy reveals structural differences between mouse and macaque at the interface between the inner segment (IS) and the outer segment (OS), the calyceal processes (CP) being absent in the mouse (A) but well-developed in the macaque (B). Calyceal processes are long microvilli that emerge from the apical region of the inner segment and ensheathe the basal third of the opsin-labelled outer segment. The F-actin-labelled (red) roots (arrowheads) can be used as stable landmarks for the presence of the calyceal processes, as they are less sensitive to tissue fixation conditions (A, B). C. USH proteins in the photoreceptor cells of primate retinas. All the USH1 proteins (illustrated here for protocadherin-15) are present at the junction between the inner and outer segments in macaque photoreceptor cells. D. The distribution of USH1 proteins in structures rich in F-actin, both in hair cells and photoreceptor cells, highlights unsuspected similarity between the two sensory cell types.
Adapted from Sahly et al. [90].
This structural difference between species coincides with a differential distribution of USH1 proteins in the region. In primates, the five USH1 proteins co-localise at membrane-membrane connection sites between the outer segment of the photoreceptor cells and the calyceal processes (in both rods and cones), and the inner segment (in rods only) (see Fig. 3C). The conserved distribution of USH1 proteins in the calyceal processes of frogs and humans, which are evolutionarily distant by about 300 million years, suggest that they have a pivotal role in this microvillar crown. The USH1 protein complex is thought to form an adhesion belt, which connects the outer segment basal region to the surrounding structures [90]. By cupping the base of the outer segment, in which the nascent outer disks fold [103–105] (see Fig. 3C), the USH1 protein-mediated adhesion complex would contribute to the daily renewal of photoreceptor outer segments. The absence of typical calyceal processes and low or absent expression of most USH1 proteins in mouse photoreceptor cells may explain why defects in USH1 proteins do not impair vision in this species. A defect in the USH1 protein network-mediated membrane-membrane coupling between the photoreceptor outer segment and the surrounding subcellular compartments is probably the cause of the USH1 retinal dystrophy in humans [90].
Auditory and visual sensory cells have many common structural features. The synapses of both cell types share structural and functional features. The presence of ribbons, to which synaptic vesicles are attached, and which are specialised for substantial and sustained neurotransmitter release [106,107], is one example. Both cell types harbor microvilli or microvillus-cilium structures, which are interconnected by the USH1 protein network (Fig. 3D) [90]. Both the photoreceptor outer segment and the kinocilium of inner ear sensory cells are classified as modified sensory cilia (Fig. 3D) [90]. This raises the possibility of a mechanosensory role of the USH1-associated network also in the calyceal processes, reminiscent of that operating in the stereocilia of auditory sensory cells.
Future studies should address how differences in the organisation of the outer segment of rods and cones (see [108,109]) impact on their relationship with the calyceal processes. Rods have pinched off apposed disks surrounded by the outer segment external plasma membrane, but the outer disk lamellae of cones are formed by simple evaginations of the plasma membrane which are directly exposed to the extracellular environment and thus to the calyceal processes. Future work should consider the function of the calyceal processes and whether they may, for example, provide a rigid structure to guide the folding of the outer disks. Studies could explore whether these processes have a physiological role and are involved in diurnal vision.
4 Multiple therapeutic approaches for the treatment of USH visual impairment
USH patients can greatly benefit from early cochlear implantation, which improves their hearing and communication abilities [40,110]. This is despite some limitations in device ability to understand speech in a noisy environment. In contrast, there is currently no way to prevent retinitis pigmentosa or restore vision. Unlike the hearing impairment, which is congenital in almost all USH patients, photoreceptor cell degeneration generally begins around or after puberty, which is within a suitable timeframe for therapeutic gene therapy. The development of an appropriate USH1 animal model will however be needed to enable specific therapies and related preclinical tests to be carried out [90].
Several strategies to cure retinitis pigmentosa are being developed, and some target most retinal dystrophies, regardless of their genetic cause (see references [8,111–113]). Efforts to develop and improve artificial retinal implants, both those in contact with the photoreceptor cell layer (microchips) and those implanted in the inner retina in contact of ganglion cells (electrode arrays), are underway [114,115]. Several patients with isolated retinitis pigmentosa, USH2, USH3 or choroideremia have had implanted capsules of Human NTC-201 Cells (see http://clinicaltrials.gov/ct2/show/NCT00447980). These encapsulated cells provide subretinal delivery of a ciliary neurotrophic factor (CNTF), and aim to prevent the degeneration of photoreceptor cells. Similarly, the transplantation of forebrain-derived progenitor cells to the subretinal space in an Ush2a knock-out mouse has been shown to sustain visual function and protect photoreceptor cells from degeneration [116]. An optogenetic approach has also been shown to restore light sensitivity in mouse models of retinitis pigmentosa and reactivate light-insensitive photoreceptors in human retinal explants [117,118]. The success of gene therapy in some human retinopathies [113,119–121] raises hope for the development of more specific approaches to treat the retinal defects of USH. Oxford Biomedica are leading a clinical trial to evaluate the safety of subretinally-delivered lentiviral vectors containing the gene encoding myosin VIIa (http://clinicaltrials.gov/show/NCT01505062). This was initiated after a demonstration that melanosome apical localisation in RPE cells and opsin transport in photoreceptor cells could be restored in myosin VIIa-defective mice following a subretinal injection of an equine lentivirus carrying the full-length myosin VIIa cDNA [122]. However, lentiviral transfection of photoreceptor cells is not very efficient, and since photoreceptors are the primary target cells of the USH1 retinopathy, alternative strategies are likely to be needed. New vectors targeting visual and auditory sensory cells, and allowing the successful transfer of cDNAs of different sizes is needed, especially since some of the USH proteins are very large. The targeting of photoreceptor cells using adeno-associated viruses (AAV)(either oversized viruses, or a combination of two AAVs containing parts of the USHc DNA, for reconstitution of the full protein in vivo), has been recently undertaken for myosin VIIa and also other USH proteins [122–126]. Some potential therapeutic approaches have recently been developed in some USH mouse models. Synthetic aminoglycoside compounds were used to induce stop codon read-through of the USH1C p.R31X nonsense mutation [127]. Two compounds with good biocompatibility, NB54 and PTC124, have been successfully used to restore the production of functional full-length harmonin proteins in vitro and in vivo [128]. In a further study, the c.216G>A knock-in mouse model that displays combined deafness and retinal degeneration was used [45]. Antisense oligonucleotides (ASO) have also been shown to correct defective pre-mRNA splicing of USH1C transcripts harbouring the c.216G>A mutation [129]. Treating newborn mice with a single systemic dose of ASO was shown to partially correct the splicing defect, increase protein expression, improve stereocilia organisation in the cochlea, and rescue cochlear hair cells, vestibular function and low-frequency hearing. It is not known whether the retinal degeneration observed in these mice was also corrected [129].
5 Concluding remarks
The parallel between calyceal processes and the mechanosensitive stereocilia (see Fig. 3D) suggests that mechanical inputs may apply also to photoreceptor cells. The issue of the USH proteins being part of functional units acting as sensors and conveyors of mechanical forces in sensory systems, and how, individually and collectively, they fulfil their expected functions warrants further consideration. As regards the USH1 retinopathy, the absence of the calyceal processes in mouse photoreceptor cells calls for appropriate animal model(s) of the human retinal disease. This is very timely given that diverse therapeutic approaches are currently developed to treat the visual defect of Usher syndrome.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We thank J.-P. Hardelin for his critical comments on the manuscript. This work was supported by European Union Seventh Framework Programme, under grant agreement HEALTH-F2-2010-242013 (TREATRUSH), LHW-Stiftung, Fondation Raymonde & Guy Strittmatter, Fighting Blindness, FAUN Stiftung (Suchert Foundation), Conny Maeva Charitable Foundation, Fondation Orange, ERC grant 294570-hair bundle, the French State program “Investissements d’Avenir” managed by the Agence Nationale de la Recherche [ANR-10-LABX-65], “The Foundation Fighting Blindness Paris Center Grant”, and the Fondation Voir et Entendre.