

1 Inherited retinal degenerations that can benefit from gene therapy
Inherited retinal degenerations (IRD) display wide variation in their mode of inheritance, underlying genetic defects, age of onset, and phenotypic severity (https://sph.uth.edu/retnet/disease.htm). Despite growing knowledge on genetics, molecular mechanisms have not been delineated for all retinal diseases, and thus far treatment options are limited. Nevertheless, monogenic inheritance patterns exist and they can be autosomal dominant (i.e. certain forms of retinitis pigmentosa), recessive (LCA type II), X-linked (retinoschisis) or follow a mitochondrial inheritance pattern (i.e. Leber's hereditary optic neuropathy (LHON)). There are nine broad categories of IRD, with many subtypes under each category (Fig. 1).

(Color online) Percentage of broad categories of IRD and sub-categories of retinitis pigmentosa.
Adapted from [28].
Here, these inherited retinal diseases are discussed in view of the gene therapeutic approach that can be used to treat them. In view of the mechanisms of disease and genetics, there are a number of strategies where strong proof-of-concept laboratory studies have been obtained. We will place particular emphasis on these strategies.
2 Gene augmentation/replacement
Among monogenic diseases, those caused by recessive null mutations are the ones that are most amenable to gene therapy (Fig. 2). These mutations are mostly localized to the outer retina (photoreceptors and retinal pigment epithelium [RPE]).
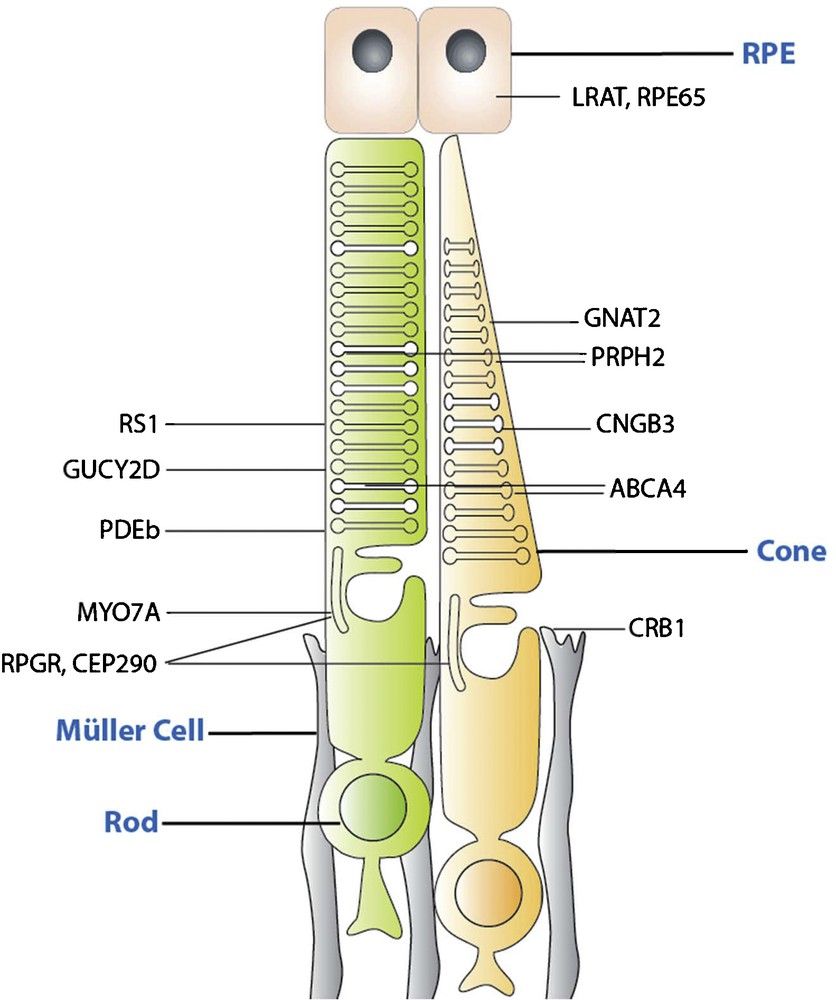
(Color online) Schematic drawing indicating the localization of proteins encoded by candidate genes for gene augmentation therapy. Gene defects in the corresponding genes lead to recessive phenotypes that are amenable to gene supplementation.
Cell type targeting, desired gene expression level and cell-to-cell variation of gene expression are important considerations in identifying disease targets, which can benefit from gene therapy. In most cases, recessive and X-linked mutations cause an absence of protein, or production of functionally null protein, and consequently, the expression of wild-type protein is likely to significantly ameliorate the disease phenotype. LCA 2, choroideremia, Stargardt's disease and retinoschisis are examples of monogenic recessive diseases, where there has been multiple proof-of-concept preclinical or clinical data showing promise [1]. Recessive genotypes where gene replacement would be appropriate include, but are not limited to, RS1 (XLRS: X-linked retinoschisis), CNGA3 and CNGB3 (achromatopsia), GUCY2D (LCA1), and RPE65 (LCA2), MYO7A (Usher 1B) and ABCA4 (Stargardt's), REP-1 (Choroideremia), and RPGR (X-linked Retinits pigmentosa, RP). Of these, LCA2, Usher 1B, Stargardt's disease and choroideremia are already in clinical trials, with a clinical trial on achromatopsia starting soon [2]. As previously mentioned, LCA2 was the first one of these monogenic recessive diseases to come to clinical application [3–5].
2.1 Lessons learned from clinical trials
Several groups have set-up a phase 1 clinical trial for the treatment of LCA2 with four active clinical trials that are ongoing currently (clinical trial numbers: NCT00749957, NCT01496040, NCT00516477, NCT01208389). The data from these clinical trials collectively point to the safety of gene transfer with sustained improvement in retinal and/or visual function in all of the patients having undergone treatment. It is important to note that the greatest improvement was noted in children, all of who gained ambulatory vision (ClinicalTrials.gov, number NCT05164770). Follow-up has surpassed 5 years for the first three groups and to date no adverse events (inflammation, complications…) have been reported in this period. Patients received a single subretinal injection of AAV bearing functional copies of the RPE65 gene where one eye was treated months or years before the second eye [6]. Sequential bilateral injection was shown to cause minimal inflammation and improved visual function in three LCA2 patients. Thus, subretinal re-administration of AAV2 is safe and effective, even in the setting of pre-existing immunity to the vector, a parameter that has been used to exclude patients from gene therapy trials. One unexpected outcome of the LCA2 trial was about the implications of the surgical administration route. It has been shown that RPE65-LCA gene therapy is sufficiently safe and substantially efficacious to the extrafoveal retina. However, there is no benefit but some risk in treating the fovea [7]. These findings might be specific to retinas with LCA2 or they might have to do with the particular nature of the attachment between the foveal photoreceptors and their underlying retinal pigment epithelium (RPE). The apical RPE membrane in the primate wraps around the photoreceptor outer segments with an intricate structure (unlike the rodent retina where apical RPE lacks inter-digitation with outer segments) [8]. In humans, the interaction between foveal photoreceptor outer segments is very strong, which might explain why the reattachment process after detachment might be more complicated in this specialized region. No evidence of age-dependent effects was found in this respect. In other instances, patients who have received a peri-foveal administration developed a pseudo-fovea. Lastly, it has been shown that despite the gene therapy disease progression continues, implying that gene replacement alone might not be sufficient to stop the progression of the disease [9]. This is a particularly important finding as the same result occurs in the canine model when treatment is at the disease stage equivalent to humans, establishing the need for combinatorial therapy to improve vision in the short-term but also slow retinal degeneration in the long-term. Among the possibilities for combinatorial therapy, there is delivery of agents to prevent the loss of retinal cells, such as neuroprotective, prosurvival, or anti-apoptotic factors or antioxidants, sequentially or simultaneously with gene augmentation therapy.
2.2 Current challenges
The outcomes of the clinical trials collectively point to the safety of AAV in gene delivery. They also highlight the importance of dosage, early intervention, immune privilege of the subretinal space, the unfavorable properties of the fovea for subretinal administration and the importance of implementing survival factors for better rescue. These constitute the challenges for the development of the next series of clinical trials. Vector requirements for gene replacement and correction strategies require controlled amounts of gene expression in the outer retinal cells as depicted in Fig. 2. The retina can be targeted by two routes of injection: intravitreal and subretinal [10]. The performance of all vectors depends on their administration route. Most viral and non-viral vectors are able to transduce the RPE when injected into the subretinal space, whereas only AAV has thus far been successful in gene delivery after vitreal administration in the adult mammalian retina [11,12]. Many studies over the past years have provided evidence of the higher efficiency of viral versus non-viral vehicles [13–15]; however, recent reports on nanoparticle-mediated retinal gene therapy showed an improvement compared with the previous studies with non-viral agents [16,17]. The vectors most studied and used for retinal gene transfer are those derived from adenoviruses [18], lentiviruses [19], and adeno-associated viruses [20], which are capable of infecting and transducing non-dividing cells, such as photoreceptors and RPE.
Intravitreal injection delivers the therapeutic agent into the vitreous, while the subretinal injection detaches photoreceptors from their supporting epithelium, creating a dome-shape bleb where therapeutic substances can be injected (Fig. 3). The intravitreal injection is less invasive than the subretinal, but the diffusion of the therapeutic agent to the photoreceptors and RPE is limited by significant physical barriers, such as the vitreous, the inner limiting membrane, and the inner retina with tightly filled extracellular matrix and cells [21]. Thus, the subretinal injections have been the preferred route used to date to target RPE and photoreceptors by all vectors. An additional advantage of the subretinal space compared to the vitreous is the immune privilege of this compartment in respect to the vitreous. The eye is considered to be an immunologically protected space [22]. The origin of this immune privilege is complex and is generated by multiple layers and mechanisms, including the blood-retina barrier and other physical barriers, an immunosuppressive microenvironment, and the existence of deviant systemic immunity that limits the production of proinflammatory effector cells. These mechanisms provide the eye with a degree of immune protection that lacks acute, destructive inflammation, thus, sparing the delicate visual axes, which is incapable of regeneration after early development.
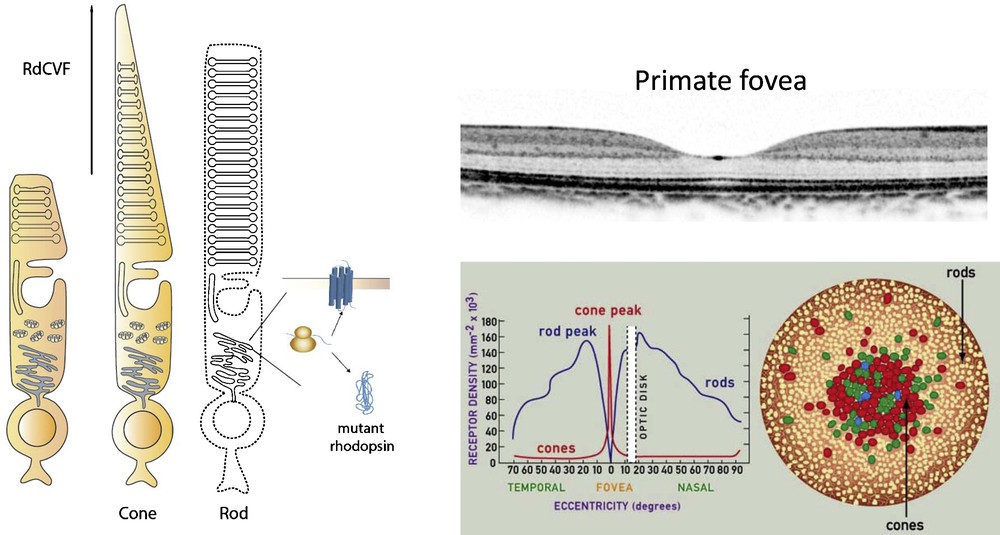
(Color online) Schematic drawing representing loss of cone outer segment in the absence of rods and therapeutic effect of RdCVF (left) and the anatomy of primate fovea, with cross section and rod and cone density in the fovea (right).
Adapted from www.webexhibits.org/causesofcolor/1G.html
It is commonly assumed that pre-exposure to AAV may not pose significant problems with regard to the performance of AAV vectors in the eye because of this ocular immune privilege. Immune responses have been shown to vary by different routes of ocular administration and re-administration of AAV vectors [23]. The effect of previous exposure to AAV vector in one eye has an effect on subsequent intraocular AAV-mediated gene delivery to the partner eye. Intravitreal administration of AAV vectors has been shown to generate a humoral immune response against AAV capsid that blocked expression from re-administered intravitreal vector in the partner eye. However, intravitreal vector in the first eye has no effect on re-administering vector into the subretinal space of the partner eye, which suggests greater immune privilege in the subretinal compartment. Moreover, it has been shown that the initially treated eye received subretinal vector, no humoral immune response against AAV capsid was elicited, and no effect on subsequent administration of the AAV vector either intravitreally or subretinally ensued. These findings are in line with the clinical data showing no complications in eyes treated with a second subretinal administration in the LCA2 studies [6]. Nevertheless, subretinal injections are technically much more challenging with higher risks of leading to surgical complications as suggested by the finding that the retinas of patients with LCA2 do not tolerate subretinal injections at the macula [7].
Adeno-associated viral vectors have been the vector of choice for gene delivery in most inherited retinal degenerations. The safety profile that has now been demonstrated in the clinic is a strong argument for further development of these vectors. In mice, recent developments at the viral capsid engineering level has now produced AAV variants that are able to transduce all retinal cells, pan-retinally, after intravitreal administration [24,25]. Such vectors provide opportunities to test gene therapeutic strategies without the bias of subretinal injections in rodents. Namely in studies where efficacy of trophic factor delivery is measured, the subretinal injection procedure can be very confounding as the mere fact of performing subretinal detachment leads to the release of a number of trophic factors which lead to significant rescue effect in control eyes. The practical aspect of intravitreal injections compared to subretinal are also a reason why these vectors will find more and more applications in the laboratory gene therapy setting.
Unfortunately, these developments in AAV technology do not translate easily to the non-human primate and currently efforts are being invested in this direction. Reasons for this is the significantly different anatomy of the mouse and human retinas as well as the differences in the volume and composition of the vitreous, and inner limiting membrane. Earliest AAV transduction studies in the non-human primates aiming at inner retinal gene delivery have shown that because of these differences, a simple AAV2 administration that leads to satisfactory pan-retinal ganglion cell transduction in the mouse retina, at a pharmacologically equivalent dose only produces transduction in a small ring of ganglion cells around the fovea. This study and the need to non-invasively deliver genes to the fovea have motivated researchers to investigate better vector technologies. It has since been shown that tyrosine mutant AAV2 and 8 based vectors as well as artificial variants designed for intravitreal gene delivery to deeper retina in mice can mediate strong gene expression in foveal cones [25,26]. The ability to target the ganglion cells around the macula [27] as well as foveal photoreceptors opens up possibilities for intravitreally delivered gene therapies in the clinic while further developments in our understanding of retinal barriers to transduction are needed to find an ideal AAVs for gene delivery to primate retinas.
3 Gene correction and interference strategies
Approximately one third of all retinal dystrophies are inherited in an autosomal dominant fashion (RetNet database: http://www.sph.uth.tmc.edu/retnet/sum-dis.htm), caused by gain-of-function mutant alleles producing gene products with dominant negative or abnormal functions. Dominant mutations pose bigger challenges for gene therapy than the recessive ones. Rhodopsin mutations have frequently been studied with respect to autosomal dominant retinitis pigmentosa [28]. In the case of dominant negative mutations, gain-of-function results in interference of wild-type protein processing. In some dominant disorders, however, the mutated gene product has a toxic impact on the cell, as a result, interfering adversely with its normal cellular functions (mutant rhodopsin can trigger an unfolded protein response). In these cases, supplementing the cell with a normal copy of the gene would not necessarily reduce the negative effect of the mutation although distracting the cellular machinery from making mutant protein might still prove beneficial [29]. Ablating the mutated gene's expression is essential for the treatment of gain-of-function in dominantly inherited disorders and if haploinsufficiency results after the silencing of the mutation, it can be treated with a replacement gene therapy approach. The term “suppression and replacement” has been used to describe this concept. The common genetic approaches to correct dominant conditions rely first of all on silencing gene expression. The main target of gene silencing strategies is the messenger RNA (mRNA) transcript, the function of which can be inhibited by antisense oligonucleatide-based, ribozyme-based or RNA interference-based strategies. Other emerging technologies exist for doing genome editing, meaning the correction of the mutation at the DNA level. Zinc finger nucleases, TALE nucleases and the newly emerging CRISPR technology can have a large impact in future therapeutic prospects in dominantly inherited disorders. However, as most of these technologies are still largely in experimental development, these will likely be implemented much later for human gene therapy. Nevertheless, the vector technologies developed for gene replacement strategies in the outer retina will be applicable to gene correction and interference strategies to be developed in the years to come.
4 Neuroprotection through gene therapy (anti-apoptotic and trophic factors)
One broadly applicable gene therapy approach to treat IRDs is the expression of compounds, which treat the most damaging symptoms of disease. In most IRDs, neuronal cell death, namely the loss of the primary retinal neurons responsible for light capture, is the main issue. Neuroprotection focuses on prolonging the lifespan of neurons in spite of their genetic abnormality, and it can be a straightforward way to deal with photoreceptor cell loss in IRD. Neuroprotection is typically accomplished through the expression of naturally occurring low molecular weight “survival factors”, which when present at sufficient concentration elicits a neuroprotective effect. A distinct advantage of this approach is that such factors are highly diffusible, and so, the therapeutic gene can be delivered to any tissue, typically the RPE or ganglion cell layer for ease, from where the factor is secreted resulting in a paracrine effect. Additionally, most of these factors do not have an adverse affect when secreted in large quantities so, the precise control overexpression levels and cell-to-cell variation of expression is not a concern in this treatment option. Several neurotrophic factors have been demonstrated to have efficacy in the treatment of dominant RP arising from rhodopsin mutations, including ciliaryneurotrophic factor (CNTF) [30], brain-derived neurotrophic factor (BDNF) [31] and glial-cell derived neurotrophic factor (GDNF) [32–34]. Neuroprotective approaches have also been used successfully in models of recessive retinal diseases both in vitro and in vivo, and present one of the only approaches for the treatment of inherited retinal disease which do not have identified genes [32–35]. Inhibition of apoptosis follows a similar approach as neuroprotection, in that gene defects are not corrected, but the effect they have on the cell are instead counteracted. The functional mechanism of anti-apoptotic proteins require that they are expressed intracellularly and so, in contrast to neuroprotection, the therapeutic gene must be delivered directly to the mutation harboring cell. X-linked inhibitor of apoptosis (XIAP) has shown considerable promise in attenuating the degeneration of photoreceptors in models of ischemic injury and in models of RP [36–39].
In the context of rod-cone degenerations, one of the most potent neurotrophins is rod-derived cone viability factor (RdCVF) [40]. In most types of RP, mutations selectively affect rods (20 times more mutations are found in rods compared to cones). Nevertheless, cones still slowly degenerate in response to the death of rods. This secondary event takes years-to-decades after the initial loss of rod photoreceptors [41]. Considering that rods are only necessary for night vision, cone photoreceptors are much more important for day vision, in fine visual acuity and color vision that we use most. It is thus the secondary cone cell death that leads to progressive visual field constriction (known as tunnel vision) and to loss of central vision associated with complete blindness. This cascade of retinal degenerative events points to the importance of preserving the cones in human vision. It has been hypothesized and shown that rod cells produce a factor that helps the cone cells survive, which might have a large impact on the management of RP in humans [42]. It has been shown that cone viability is dependent upon the presence of viable rod photoreceptors (Fig. 4) which led to a high-throughput screen identifying a protein that leads to increased number of cone cells in rd1 mouse retina both in vitro and in vivo [43,44]. This trophic factor referred to as RdCVF is a truncated thioredoxin-like protein (encoded by the Nxnl1 gene) specifically expressed by photoreceptors (see article by Dr. Leveillard et al.). These studies have shown that the protective effect recorded by electroretinogram (ERG) was five orders of magnitude higher than the survival effect measured by the viability of the cones and that the outer segment of the cones, is significantly prePrimate fovea served by RdCVF.

(Color online) Schematic drawing representing a subretinal and an intravitreal injection of AAV particles into a primate eye.
Recently, very encouraging results with AAV-mediated delivery of RdCVF in rd10 mouse model of retinal degeneration has demonstrated the potential of RdCVF for retinal gene therapy [45]. Efficient expression of RdCVF has been shown to lead to functional rescue of the cone-mediated photopic ERG. RdCVF is now in translation into a possible therapeutic agent to save cones and treat a spectrum of degenerative eye diseases and its expression through AAV is likely to become a promising gene therapy option in rod-cone dystrophies. Importantly, this trophic factor can be combined with gene replacement or optogenetic therapies (described below) in order to add the benefit of cell survival. Dual vector systems permeating such expression are going to be key to the development in this mode of combinatorial gene therapy.
5 Optogenetics and restoring light sensitivity to blind retinas
Optogenetics refers to a neuromodulation technique employed in neuroscience that uses a combination of techniques from optics and genetics to control the activities of neurons in living tissue. The key elements used in optogenetics are light-sensitive proteins. Spatially precise neuronal control is achieved using optogenetic actuators, like channelrhodopsin, halorhodopsin, and archaerhodopsin. These proteins are able to modulate membrane potential and lead to a depolarization or hyperpolarization in neurons in which they are expressed. The key idea of optogenetic vision restoration is to target these genetically encoded light sensors to strategically important retinal cell types, thus, converting them into artificial photoreceptors [46]. If the artificial photoreceptors are connected to other cell types in the retinal circuit, light will also modulate the activity of these cells and travel across the visual pathway. Challenges in this strategy lie at many levels. First challenge is choosing the right optogenetic tool and targeting strategy, so that the light-evoked retinal activity will be similar to the activity of normal retinas stimulated through normal photoreceptive function [47–49]. The second challenge is the relatively high levels of light that are necessary to get light responses in retinal circuits using these light sensors [50]. Since these optogenetic tools do not benefit from amplification of the G-protein coupled cascade that normal retinal opsins do, high levels of optogenetic protein expression is needed to drive these systems alongside high light intensities.
Nevertheless, ectopic expression of photosensitive proteins is a promising strategy to restore light sensitivity and even sophisticated forms of vision to the visually impaired. The field of using optogenetics to restore vision started in 2006 with the publication by Bi and Pan where they show light responses in blind retinas by the expression of channelrhodopsin (ChR) [47] in the retinal ganglion cells (Fig. 5).

(Color online) Cell targets for optogenetic therapy in retinitis pigmentosa type retinal degeneration. Cone photoreceptors lose their outer segments but the dormant cell bodies remain as suitable targets for expression of light-responsive transporters such as halorhodopsin depicted on the right. The inner retinal circuitry (RGCs and ON bipolar cells shown in blue) can also be targeted with depolarizing proteins, such as channelrhodopsin.
The inability to distinguish between the ON and OFF pathways and the lack of behavioral responses in the treated animals in this study underlined the importance of convergence and the ability to differentiate between ON and OFF pathways [51]. For this reason, the field has moved towards targeting the bipolar cells using the same optogenetic strategy. Lagali and Roska have shown that it is possible through the use of a ON bipolar cell-specific promoter to target channelrhodopsin expression to ON bipolar cells and thus gain in convergence and in behavioral responses that can be elicited through this kind of approach [48]. In this study, the authors used electroporation to gain entry into the bipolar cells. Another follow-up study used the same promoter but in combination with AAV to target optogenetic expression to on bipolar cells after subretinal delivery [52]. However, the ability to target bipolar cells within a given area of the retina poses the problem about acuity in the human translation.
A current strategy exploits the disease phenotype and chronology in RP in human patients [53]. Both clinical findings and model animals suggest that cone cell bodies remain alive long after the loss of photosensitivity in RP. Their number decreases, but about 25% of the cones still exist as long as 7 months after termination of rod-cone-mediated photosensitivity. Post-mortem examination of the retinas of RP patients has revealed at least one row of foveal cones remaining in the fovea [54]. Preservation of human cones has not been documented in the periphery. However, at later stages of RP, at least in rd1 mice, it is not easy to distinguish cone cell bodies from bipolar cells morphologically [49]. Based on these findings, when halorhodopsin – a hyperpolarizing chloride pump – was expressed in light-insensitive cones in mouse models of RP, the relevant retinal ON and OFF ganglion cells were activated; light-evoked activity was measured in the cortex and visually evoked behavior was documented [49]. Importantly, the result of sophisticated retinal processing, such as directional selective ganglion cell responses, has been measured in halorhodopsin-driven retinas. To show potential for human translation, the study was strengthened by testing halorhodopsin expression in cones of human retinal explants, which led to hyperpolarization of human cones upon light stimulation further demonstrating the potential for clinical application of this strategy.
Second generation microbial opsins are being developed rapidly. To overcome its principle downsides – the small single-channel conductance (especially in steady-state), the fixation on one optimal excitation wavelength (∼470 nm, blue) as well as the relatively long recovery time, not permitting controlled firing of neurons above 20–40 Hz – ChR2 has been optimized using genetic engineering. An important modification has been the mutation that created the ultra-light-sensitive version of ChR2 called CatCh [55]. More recently, variant of ChR, designated red-activatable ChR (ReaChR), that is optimally excited with orange to red light [56] and offers improved membrane trafficking, higher photocurrents and faster kinetics compared to existing red-shifted ChRs. Red light is less scattered by tissue allowing the use of safer light intensities for use in human retina. Photochemical damage spectra has a logarithmic dependency with photon energy, implying that even small changes in the wavelength will have a strong impact on the safety-threshold, increasing the safety and applicability of these second generation tools in vision restoration.
Further comparisons and studies in activating the retinal circuitry are needed to determine which optogenetic treatment is best suited for restorative treatment of which patient population. It is also important to back up the laboratory investigations by clinical investigations looking at patient populations and the state of their retinas with respect to the integrity of the retinal circuit and presence of different populations of cells [57]. The state of the art imaging techniques, such as optical coherence tomography and adaptive optics will be instrumental in these investigations. The development of vectors to accommodate the needs of these optogenetic strategies is an important step in translation to the clinic. An ideal vector should be able to deliver genes pan-retinally and trans-retinally across all retinal cell types and that through a non-invasive intravitreal injection. Such vector can then be combined with cell-specific promoters to fine tune expression and restrict to specific subsets of cells. The parallel development of cell-specific promoters for targeting sub-populations of neurons is particularly important for optogenetic vision restoration strategies as activation or inhibition of the neuronal circuit in undesired cell populations can inhibit responses and convolute the results obtained by this prosthetic strategy.
Disclosure of interest
Dalkara is a consultant for Gensight Biologics, José-Alain Sahel is a founder and consultant for Pixium Vision and GenSight Biologics, and a consultant for Sanofi-fovea and Genesignal.