

1 Introduction
Drug discovery, as its name indicates, aims at discovering new drugs against diseases. This process can be segmented into three steps: i) disease model provision, where experimental models are developed, ii) target identification, where therapeutic targets are proposed, and iii) target validation, where the proposed therapeutic targets are assessed. The present work focuses on the second step of drug discovery: target identification [1,2].
Given an organism suffering from a disease, target identification aims at finding where to act among its multitude of biomolecules in order to alleviate, or ultimately cure, the physiological consequences of the disease. These biomolecules on which perturbations should be applied are called targets and are targeted by drugs [3]. This raises two questions: which target should be therapeutically perturbed and what type of perturbation should be applied. Broadly, the functional perturbation of a target by a drug can be either activating or inactivating, regardless the way the drug achieves it.
One solution is to test all, or at least a large number of, biomolecules for activation or inactivation. Knowing that targeting several biomolecules is potentially more effective [4], the number of possibilities is consequently huge. This rather brute-force screening can be refined with knowledge about the pathophysiology by identifying potential targets based on the role they play in it [5]. Even with this knowledge, experimentally assessing the selected potential targets in vitro or in vivo is far from straightforward. Such experiments are costly in time and resources and exhibit a high risk of failure [6]. Fortunately, in silico experiments appear as valuable tools in improving the efficiency of therapeutic research [7] since they are less costly in time and resources than the traditional in vitro and in vivo ones. However, the stumbling block of in silico experiments is that they are built from the available knowledge: not all is known about everything.
Nevertheless, an impressive and ever increasing amount of biological knowledge is already available in the scientific literature, databases and knowledge bases such as, to name a few, DrugBank [8], KEGG [9], PharmGKB [10], Reactome [11] and TTD [12]. In addition to the complexity of integrating an increasing body of knowledge comes the inherent complexity of biological systems themselves [13]: this is where computational tools can help [14]. The interplay between experimental and computational biology is synergistic rather than competitive [15]. Since in vitro and in vivo experiments produce factual results, they are trustworthy sources of knowledge. Once these factual pieces of knowledge are obtained, computational tools can help to integrate them and infer new ones. This computationally obtained knowledge can be subsequently used to direct further in vitro or in vivo experiments, hence mutually potentiating the whole.
The goal of the present work is to propose a computational methodology implemented in an algorithm for target identification using Boolean network attractors. It assumes that Boolean network attractors correspond to phenotypes produced by the modeled biological network, an assumption successfully applied in several works [16–21] to cite a few. Assuming that a phenotype is an observable and hence a relatively stable state of a biological system and assuming that the state of a biological system results from its dynamics, a phenotype is likely to correspond to an attractor. This assumption can be stated for any dynamical model but, in the present work, only Boolean networks are considered. Reasons are that, in their most basic form, Boolean networks do not require parameter values [22] and that parameter values are not straightforward to estimate due to experimental limitations, particularly at the subcellular scale, the scale where drugs interact with their targets. Moreover, since synchronous Boolean networks are easier to compute than asynchronous ones [23], only synchronous Boolean networks are considered. This does not exclude the possibility, at a later stage, to extend the algorithm for both synchronous and asynchronous updating schemes.
For a biological network involved in a disease, two possible variants are considered: the physiological variant, exhibited by healthy organisms, which produces physiological phenotypes, and the pathological variant, exhibited by ill organisms, which produces pathological phenotypes or which fails to produce physiological ones. A physiological phenotype does not impair life quantity/quality while a pathological phenotype does. It should be noted that the loss of a physiological phenotype is also a pathological condition. The physiological and pathological variants differ in that the latter results from the occurrence of some alterations known to be responsible for disorders. With a pathological variant, there are two non-exclusive pathological scenarios: pathological phenotypes are gained or physiological phenotypes are lost.
The primary goal of the proposed algorithm is to identify, in a pathological variant, target combinations together with the perturbations to apply on them, here called bullets, which render it unable to exhibit pathological phenotypes. The secondary goal is to classify the obtained bullets according to their ability at rendering the pathological variant able to exhibit previously lost physiological phenotypes, if any.
2 Methods
This section briefly introduces some basic principles, namely biological networks [24,25] and Boolean networks [26], defines some concepts and then describes the proposed algorithm. An example network to illustrate it plus a case study to illustrate its intended applications are also described. Finally, some details about implementation and code availability are mentioned.
2.1 Basic principles
2.1.1 Biological networks
A network can be seen as a digraph G = (V, E) where
2.1.2 Boolean networks
A Boolean network is a network where nodes are Boolean variables xi and where edges (xi, xj) represent the binary is input of relation: xi is input of xj. Each xi has bi∈ 〚 0, n 〛 inputs
| 1 | fork∈ 〚 k0, kend − 1 〛 do |
| 2 | |
| 3 | … |
| 4 | |
| 5 | end for |
which can be written in a more concise form:
| 1 | fork∈ 〚 k0, kend − 1 〛 do |
| 2 | x(k + 1) = f(x(k)) |
| 3 | end for |
where f = (f1,…,fn) is the Boolean transition function and x = (x1,…,xn) is the state vector. In the present work, it is assumed that k0 = 1. The value x(k) = (x1(k),…,xn(k)) ∈ {0, 1}n of x at k belongs to the state space S = {0, 1}n which is the set of cardinality 2n containing exactly all the possible states. If the values of all the xi are updated simultaneously at each k then the network is synchronous, otherwise it is asynchronous. With synchronous Boolean networks, x(k) has a unique possible successor x(k + 1): synchronous Boolean networks are deterministic.
In the particular case where k = k0, x(k0) = x0 is the initial state and, in deterministic dynamical systems, determines entirely the trajectory
The set A = {a1,…,ap} of cardinality p containing exactly all the attractors ai is called the attractor set. Due to the determinism of synchronous Boolean networks, all the attractors are cycles. A cycle is a sequence (x1,…,xq) of length q such that ∀j ∈ 〚1, q 〛 , xj+1 = f(xj) and xq+1 = x1: once the system reaches a state xj belonging to a cycle, it successively visits its states xj+1,…,xq, x1,…,xj for infinity. In the particular case where q = 1, the cycle is called a point attractor. The set Bi ⊆ S containing exactly all the x ∈ S from which ai can be reached is called its basin of attraction. With deterministic dynamical systems, the family of sets (B1,…,Bp) constitutes a partition of S.
2.2 Definitions
Some concepts used in the present work should be formally defined.physiological phenotype
A phenotype which does not impair life quantity/quality of the organism which exhibits it.
pathological phenotypeA phenotype which impairs life quantity/quality of the organism which exhibits it.
variant (of a biological network)Given a biological network of interest, a variant of it is one of its versions, namely the network plus eventually some modifications. It should be noted that this does not exclude the possibility that a variant can be the network of interest as is.
physiological variantA variant which produces only physiological phenotypes. It is the biological network of interest as it should be, namely the one of healthy organisms.
pathological variantA variant which produces at least one pathological phenotype. It is a dysfunctional version of the biological network of interest, namely a version found in ill organisms.
physiological attractor setThe attractor set Aphysio of the physiological variant.
pathological attractor setThe attractor set Apatho of the pathological variant.
physiological Boolean transition functionThe Boolean transition function fphysio of the physiological variant.
pathological Boolean transition functionThe Boolean transition function fpatho of the pathological variant.
runAn iterative computation of x(k) starting from an x0 until an ai is reached. It returns
An ai such that ai ∈ Aphysio.
pathological attractorAn ai such that ai ∉ Aphysio.
modalityThe functional perturbation modai applied on a node
A node targi ∈ V of the network on which a modai is applied.
bulletA couple (ctarg, cmoda) where ctarg = (targ1,…,targr) is a combination without repetition of targi and where cmoda = (moda1,…,modar) is an arrangement with repetition of modai, r∈ 〚1, n 〛 being the number of targets in the bullet. Here, modai is intended to be applied on targi.
therapeutic bulletA bullet which makes Apatho ⊆ Aphysio.
silver bulletA therapeutic bullet which makes Apatho ⊊ Aphysio.
golden bulletA therapeutic bullet which makes Apatho = Aphysio.
The assumed link between phenotypes and attractors is the reason why attractors are qualified as either physiological or pathological according to the phenotype they produce. This is also the reason why, in the present work, target identification aims at manipulating attractor sets of pathological variants.
2.3 Steps of the algorithm
The algorithm has two goals: i) finding therapeutic bullets and ii) classifying them as either golden or silver. A therapeutic bullet makes the pathological variant unable at reaching pathological attractors, that is Apatho ⊆ Aphysio. If such a bullet is applied on a pathological variant, the organism bearing it no longer exhibits the associated pathological phenotypes. However, a therapeutic bullet does not necessarily preserve/restore the physiological attractors. If a therapeutic bullet preserves/restores the physiological attractors, namely if Apatho = Aphysio, then it is a golden one but if Apatho ⊊ Aphysio then it is a silver one.
Given a physiological and a pathological variant, that is fphysio and fpatho, the algorithm follows five steps:
- 1. with fphysio it computes the control attractor set Aphysio
- 2. it generates bullets and, for each of them, it performs the three following steps
- 3. with fpatho plus the bullet, it computes the variant attractor set Apatho
- 4. it assesses the therapeutic potential of the bullet by comparing Aphysio and Apatho to detect pathological attractors
- 5. if the bullet is therapeutic then it is classified as either golden or silver by comparing Aphysio and Apatho for equality.
These steps can be written in pseudocode as:
| 1 | with fphysio compute Aphysio |
| 2 | generate bullet _ set |
| 3 | forbullet ∈ bullet _ set do |
| 4 | with fpatho plus bullet compute Apatho |
| 5 | if Apatho ⊆ Aphysio then |
| 6 | bullet is therapeutic |
| 7 | if Apatho = Aphysio then |
| 8 | bullet is golden |
| 9 | else |
| 10 | bullet is silver |
| 11 | end if |
| 12 | end if |
| 13 | end for |
2.3.1 Step 1: computing Aphysio
First of all, Aphysio has to be computed since it is the control and, as such, determines what is pathological. To do so, runs are performed with fphysio and the reached ai are stored in Aphysio. However, x0 ∈ S and card S increases exponentially with n. Even for reasonable values of n, card S explodes: more than 1 000 000 possible x0 for n = 20. One solution ensuring that all the ai are reached is to start a run from each of the possible x0, that is from each of the x ∈ S. Practically, this is unfeasible for an arbitrary value of n since the required computational resources can be too demanding. For example, assuming that a run requires 1 millisecond and that n = 50, performing a run from each of the 250 x ∈ S requires nearly 36 000 years.
Given that with deterministic dynamical systems (B1,…,Bp) is a partition of S, a solution is to select a subset D ⊆ S of a reasonable cardinality containing the x0 to start from. In the present work, D is selected randomly from a uniform distribution. The stumbling block of this solution is that it does not ensure that at least one x0 per Bi is selected and then does not ensure that all the ai are reached. This stumbling block holds only if card D < card S.
Again given that synchronous Boolean networks are deterministic, if a run visits a state already visited in a previous run then its destination, that is the reached attractor, is already found. If so, the run can be stopped and the algorithm can jump to the next one. To implement this, previous trajectories are stored in a set H, the history, and at each k the algorithm checks if
Since, with deterministic dynamical systems, attractors are cycles, the algorithm checks at each k if x(k + 1) is an already visited state of the current run, namely if ∃k′ ∈ 〚1, k 〛 : x(k + 1) = x(k′). If this check is positive then ai = (x(k′),…,x(k)).
This step can be written in pseudocode as:
| 1 | prompt card D |
| 2 | card D = min(card D, 2n) |
| 3 | generate D ⊆ S |
| 4 | H = {} |
| 5 | Aphysio = {} |
| 6 | forx0 ∈ D do |
| 7 | k = 1 |
| 8 | x(k) = x0 |
| 9 | while true do |
| 10 | if |
| 11 | break |
| 12 | end if |
| 13 | x(k + 1) = fphysio(x(k)) |
| 14 | if ∃k′ ∈ 〚1, k 〛 : x(k + 1) = x(k′) then |
| 15 | Aphysio = Aphysio ∪ {(x(k′),…,x(k))} |
| 16 | break |
| 17 | end if |
| 18 | k = k + 1 |
| 19 | end while |
| 20 | H = H ∪ {(x(1),…,x(k))} |
| 21 | end for |
| 22 | returnAphysio |
| 23 | do step 2 |
It should be noted that the purpose of the present work is not to propose an algorithm for finding Boolean network attractors since advanced algorithms for such tasks are already published [30,31]. The purpose is to introduce a methodology exploiting Boolean network attractors for target identification, a methodology which requires de facto these attractors to be found.
2.3.2 Step 2: generating bullets
Bullets are candidate perturbations to apply on the pathological variant to make it unable at reaching pathological attractors and hence unable at producing pathological phenotypes. Generating a bullet requires a choice of targi ∈ V and associated modai ∈ {0, 1}. In the present work, there is no time sequencing in target engagement nor in modality application. This means that, given a bullet and during a run, all the targi are engaged simultaneously and constantly and the modai do not change. As a consequence, for a given bullet, choosing the same targi more than once is senseless, while it is possible to choose the same modai for more than one targi. Therefore, a bullet is a combination ctarg without repetition of targi together with an arrangement cmoda with repetition of modai.
If bullets containing r targets have to be generated then there are n !/(r ! · (n − r) !) possible ctarg and, for each of them, there are 2r possible cmoda. This raises the same difficulty than with state space explosion since there are (n ! ·2r)/(r ! · (n − r) !) possible bullets. For example, with n = 50 and r = 3, there are more than 150 000 possible bullets. Knowing that the algorithm, as explained below, computes one attractor set per bullet, the computation time becomes practically unfeasible.
To overcome this barrier, the algorithm asks for r as an interval 〚rmin, rmax〛, asks for a maximum number
The result is the bullets: per r∈ 〚 rmin, rmax 〛, a Ctarg together with a Cmoda. As with state space explosion, the stumbling block of this method is that it does not ensure that all the possible ctarg together with all the possible cmoda are tested. This stumbling block holds only if
This step can be written in pseudocode as:
| 1 | prompt |
| 2 | rmax = min(rmax, n) |
| 3 | golden _ set = {} |
| 4 | silver _ set = {} |
| 5 | forr∈ 〚 rmin, rmax 〛 do |
| 6 | |
| 7 | |
| 8 | Ctarg = {} |
| 9 | Cmoda = {} |
| 10 | while |
| 11 | generate ctarg ∉ Ctarg |
| 12 | Ctarg = Ctarg ∪ {ctarg} |
| 13 | end while |
| 14 | while |
| 15 | generate cmoda ∉ Cmoda |
| 16 | Cmoda = Cmoda ∪ {cmoda} |
| 17 | end while |
| 18 | do steps 3 to 5 |
| 19 | end for |
| 20 | returngolden _ set, silver _ set |
2.3.3 Step 3: computing Apatho
Having the control attractor set Aphysio and a bullet (ctarg, cmoda) ∈ Ctarg × Cmoda, the algorithm computes the variant attractor set Apatho under the bullet by almost the same way Aphysio is computed in step 1. However, fpatho is used instead of fphysio and the bullet is applied: at each k, fj(x(k)) is overwritten by modai ∈ cmoda, that is xj(k + 1) = modai, provided that
In order to apply all the generated bullets, the algorithm uses two nested for loops. For each ctarg ∈ Ctarg it uses successively all the cmoda ∈ Cmoda. For each (ctarg, cmoda), the algorithm computes the corresponding Apatho and does steps 4 and 5.
This step can be written in pseudocode as:
| 1 | forctarg ∈ Ctarg do |
| 2 | for cmoda ∈ Cmoda do |
| 3 | H = {} |
| 4 | Apatho = {} |
| 5 | for x0 ∈ D do |
| 6 | k = 1 |
| 7 | x(k) = x0 |
| 8 | while true do |
| 9 | if |
| 10 | break |
| 11 | end if |
| 12 | x(k + 1) = fpatho(x(k)) |
| 13 | for targi ∈ ctarg do |
| 14 | for |
| 15 | if |
| 16 | xj(k + 1) = modai |
| 17 | end if |
| 18 | end for |
| 19 | end for |
| 20 | if ∃k′ ∈ 〚1, k 〛 : x(k + 1) = x(k′) then |
| 21 | Apatho = Apatho ∪ {(x(k′),…,x(k))} |
| 22 | break |
| 23 | end if |
| 24 | k = k + 1 |
| 25 | end while |
| 26 | H = H ∪ {(x(1),…,x(k))} |
| 27 | end for |
| 28 | do step 4 and 5 |
| 29 | end for |
| 30 | end for |
2.3.4 Step 4: identifying therapeutic bullets
To identify therapeutic bullets among the generated ones, for each (ctarg, cmoda) tested in step 3 and once the corresponding Apatho is obtained, the algorithm compares it with Aphysio to check if Apatho ⊆ Aphysio. This check ensures that, under the bullet, all the pathological attractors are removed and if new attractors appear then they are physiological ones. If this check is positive, then the bullet is therapeutic and the algorithm pursues with step 5.
This step can be written in pseudocode as:
| 1 | ifApatho ⊆ Aphysio then |
| 2 | do step 5 |
| 3 | end if |
2.3.5 Step 5: assessing therapeutic bullets
Therapeutic bullets are qualified as either golden or silver according to their ability at making the pathological variant reaching the physiological attractors. All therapeutic bullets, being golden or silver, remove the pathological attractors without creating new ones, that is Apatho ⊆ Aphysio. However, this does not imply that therapeutic bullets preserve/restore the physiological attractors. A golden bullet preserves/restores the physiological attractors: Apatho = Aphysio while a silver bullet does not: Apatho ⊊ Aphysio.
In this setting, golden bullets are perfect therapies while silver bullets are not. However, since precious things are rare and just as gold is rarer than silver, finding golden bullets is less likely than finding silver ones. Indeed, given that more constraints are required for a therapeutic bullet to be a golden one, it is more likely that the found therapeutic bullets are silver ones, except in one case: card Aphysio = 1.
Theorem 1
If card Aphysio = 1 then all therapeutic bullets are golden.
Proof
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
Practically, in the present setting, an organism bearing a pathological variant treated with a therapeutic bullet no longer exhibits the associated pathological phenotypes. Moreover, if the therapeutic bullet is golden then the organism exhibits the same phenotypes than its healthy counterpart. However, if the therapeutic bullet is silver then the organism fails to exhibit at least one physiological phenotype. With a silver bullet this is a matter of choice: what is the less detrimental between a silver bullet and no therapeutic bullet at all.
This step can be written in pseudocode as:
| 1 | ifApatho = Aphysio then |
| 2 | golden _ set = golden _ set ∪ {(ctarg, cmoda)} |
| 3 | else |
| 4 | silver _ set = silver _ set ∪ {(ctarg, cmoda)} |
| 5 | end if |
2.4 Example network
To illustrate the algorithm, it is used on a Boolean model of the mammalian cell cycle published by Faure et al. [18]. This model is chosen for several reasons: i) synchronous updating is performed: to date, the algorithm focuses on synchronous Boolean networks, ii) a mammalian biological system is modeled: the closer to human physiology the model is, the better it illustrates the intended applications, iii) the cell cycle is a at the heart of cancer: this gives relevancy to the example network, iv) the network comprises ten nodes: easily computable in face of its state space and v) attractors are already computed: useful to validate the algorithm in finding them.
Below are the Boolean functions of the example network where, for the sake of readability, xi stands for xi(k) and xi+ stands for xi(k + 1).
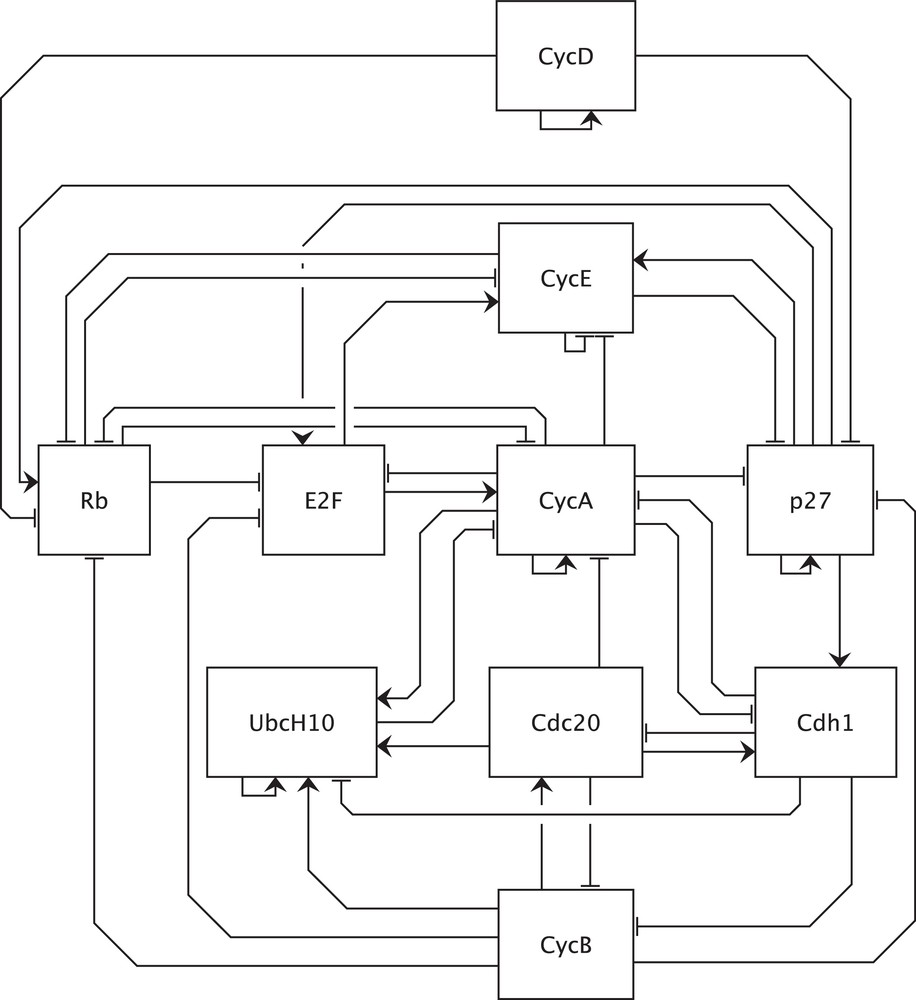
Graphical representation of the example network adapted from [18]. CDKs (cyclin-dependent kinases) are the catalytic partners of cyclins and, in this model, are not explicitly shown since the activity of CDK-cyclin complexes essentially depends on cyclins. Furthermore, inhibition of E2F by Rb is modeled by opposing Rb to the effects of E2F on its targets. The same applies to inhibition of CycE and CycA by p27. For a complete description of the model, please see [18]. CycD: CDK4/6-cyclin D complex, input of the model, initiates the cell cycle, activated by positive signals such as growth factors; CycE: CDK2-cyclin E complex; CycA: CDK2-cyclin A complex; CycB: CDK1-cyclin B complex; Rb: retinoblastoma protein, a tumor suppressor; E2F: a family of transcription factors divided into activator and repressor members, in this model E2F represents activator members; p27: p27/Kip1, a CKI (CDK inhibitor); Cdc20: an APC (Anaphase Promoting Complex, an E3 ubiquitin ligase) activator; Cdh1: an APC activator; UbcH10: an E2 ubiquitin conjugating enzyme. Masquer
Graphical representation of the example network adapted from [18]. CDKs (cyclin-dependent kinases) are the catalytic partners of cyclins and, in this model, are not explicitly shown since the activity of CDK-cyclin complexes essentially depends on cyclins. Furthermore, inhibition of ... Lire la suite
Having the example network, two variants of it are needed: the physiological one and the pathological one. The physiological variant is the network as is while the pathological variant is the network plus a constitutive activation/inactivation of at least one of its nodes. For simplicity, and given the relatively small number of entities, only one is chosen: the retinoblastoma protein Rb, for which a constitutive inactivation is applied. To implement this, the corresponding fi becomes:
2.5 Case study
To illustrate the intended usage of the proposed methodology, the algorithm is used on a Boolean model of the Fanconi Anemia/Breast Cancer (FA/BRCA) pathway published by Rodriguez et al. [33]. This model is chosen for several reasons: i) two pathological conditions are studied: required for a case study of an in silico target identification, ii) the physiological and pathological variants are clearly described: required by the algorithm iii) it is nearly three times bigger than the example network: representative of a more comprehensive biological model while remaining computationally tractable, iv) synchronous updating is used: to date, the algorithm focuses on synchronous Boolean networks and v) attractors are already interpreted in terms of phenotypes.
The FA/BRCA pathway is dedicated to DNA repair and more precisely to interstrand cross-links (ICLs) removal. As expected with any DNA repair impairment, individuals suffering from FA/BRCA pathway malfunction are subjected to increased risk of cancer, such as in Fanconi anemia, a rare genetic disorder causing bone marrow failure, congenital abnormalities and increased risk of cancer [34–36].
Rodriguez et al. propose a Boolean model comprising the FA/BRCA pathway and three types of DNA damages commonly observed in Fanconi anemia, namely ICLs, double-strand breaks (DSBs) and DNA adducts (ADDs). DSBs and ADDs can be created during ICLs repair before being removed, therefore leaving an undamaged DNA ready for the cell cycle. For a complete description of the model, please see [33]. The Boolean functions can be found in Appendix B.
The physiological variant is the FA/BRCA pathway model as is. To it, Rodriguez et al. propose two pathological variants, here called patho1 and patho2, modeling two mutations involving genes of the FA/BRCA pathway. These mutations are observed in patients suffering from Fanconi anemia [37]. The first one involves the FANCA gene, corresponding to the FAcore variable, and the second one involves the FANCD1/BRCA2 or FANCN/PALB2 gene, corresponding to the FANCD1N variable. These mutations are of loss-of-function kind: to simulate them the corresponding fi becomes
2.6 Implementation
The algorithm is implemented in Fortran 95 compiled with GFortran.1 The code is available on GitHub2 at https://github.com/arnaudporet/kali-targ under a BSD 3-Clause License.3
3 Results
This section exposes results produced with the algorithm on the example network to illustrate how it works. Next, results produced with the algorithm on the case study are exposed to illustrate its intended applications in target identification.
3.1 Results of step 1
Owing to the relatively small size of the example network, card D is set to card S = 1024. Since card D = card S, all the attractors are found. The algorithm returns the following attractors:
Attractors are presented as matrices where, for an attractor of length q, lines correspond to the xi(k), k∈ 〚1, q 〛 and columns to x(k). Aphysio = {a1, a2}, which corresponds to results obtained by Faure et al.. By the way, a1 and a2 are the two physiological attractors. In terms of phenotypes, a1 corresponds to the cell cycle while a2 corresponds to quiescence.
3.2 Results of steps 2 to 5
Results of steps 2 to 5 are grouped since only therapeutic bullets found in step 4 and classified in step 5 are returned. The algorithm is launched with rmin = 1 and rmax = 2. Again due to the relatively small size of the example network,
To better illustrate what is performed to obtain these therapeutic bullets, below is Apatho without any bullet:
It should be noted that a4 = a1 ∈ Aphysio: a4 is a physiological attractor. It is possible that the pathological variant produces physiological attractors: Apatho is not the set containing exactly all the pathological attractors, it is the attractor set of the pathological variant. As a consequence, Aphysio∩ Apatho ≠ ∅ is possible. However, a3 ∉ Aphysio: it is a pathological attractor and is what a therapeutic bullet, being golden or silver, is intended to avoid.
Again to better illustrate what is performed to obtain these therapeutic bullets, below is Apatho under the third bullet:
3.3 Results on the case study
With the case study, card S = 228 = 268 435 456: computing attractors from all the x ∈ S becomes too demanding. Indeed, it should be recalled that the algorithm computes one attractor set per bullet, namely Apatho under the tested bullet. As a consequence, card D is set to a more reasonable value: card D = 10 000. Despite that card D < card S, it seems sufficient for the algorithm to find all the attractors, just as Rodriguez et al. whose computation covers the whole state space. Below are the computed attractors:
- • Aphysio = {a1}
- • Apatho1 = {a1}
- • Apatho2 = {a1, a2}, a1 and a2 attracting respectively 29.5% and 70.5% of the x ∈ D under fpatho2
- • a1: cell cycle progression
- • a2: cell cycle arrest
In physiological conditions, in case of damaged DNA, cells repair it before performing the cell cycle, or die if repair fails. Such checkpoints enable cells to ensure genomic integrity by preventing damaged DNA to be replicated and then propagated [38,39]. Otherwise, genetic instability may appear, potentially leading to cancer [40]. The results show that the physiological variant is able to ensure genomic integrity since its unique attractor is a1, where ICL = DSB = ADD = 0: DNA damages are repaired, if any, and the cell cycle can safely occur. Interestingly, the same physiological phenotype is computed for patho1 where Apatho1 = Aphysio. This suggests that cells bearing FANCA gene null mutation are nonetheless able to repair DNA.
With patho2, a pathological attractor appears: a2, where DSB = 1. This suggests that cells bearing FANCD1/BRCA2 or FANCN/PALB2 gene null mutation are unable to repair DSBs, explaining why a2 corresponds to cell cycle arrest: DNA remains damaged. It should be noted that a1 ∈ Apatho2, suggesting that from certain x0, that is under certain conditions, such cells could be able to repair DNA. However, a1 attracts only 29.5% of the x ∈ D under fpatho2, indicating that the pathological phenotype associated with a2 is more likely to occur.
Altogether, according to computed attractors and their phenotypic interpretation, and limited to the scope studied by the model of Rodriguez et al., FANCA gene null mutation may not induce pathological phenotypes. However, with FANCD1/BRCA2 or FANCN/PALB2 gene null mutation, two phenotypes are predicted: a physiological one and a pathological one, the latter being the most likely to be exhibited. As a consequence, the algorithm has to operate on patho2 to find bullets able to remove the pathological attractor a2.
By comprehensively testing all bullets made of 1 to 3 targets, the algorithm returns the following results:
| number of all possible bullets | number of therapeutic bullets | |
| r = 1 | 56 | 1 (1.786%) |
| r = 2 | 1 512 | 20 (1.323%) |
| r = 3 | 26 208 | 191 (0.729%) |
Occurrence of each node in the found therapeutic bullets, in percentage of the total number of tested bullets, can be found in Appendix E. Below is the top five:
| ATM | 87.736% |
| ICL | 22.170% |
| BRCA1 | 18.396% |
| DSB | 11.792% |
| MRN | 10.377% |
In the present case study, DNA damages such as ICLs and DSBs are the pathological events. Unsurprisingly, the algorithm suggests them to be targeted: this is a logical consequence. However, DNA damages are not biomolecules in themselves and directly targeting them by means of drugs appears senseless. What is relevant are the biomolecules of the FA/BRCA pathway suggested as therapeutic targets. Interestingly, ATM dominates all the other candidates, predicting ATM to be a pivotal therapeutic target for the patho2 condition, namely the FA/BRCA pathway bearing FANCD1/BRCA2 or FANCN/PALB2 gene null mutation, as observed in Fanconi anemia.
4 Conclusion
Under the assumption that dynamical system attractors and biological network phenotypes are linked when the former models the latter, the results show that the algorithm succeeds in performing the proposed in silico target identification. It returns therapeutic bullets for a pathological variant of the mammalian cell cycle relevant in diseases such as cancer and for a pathological variant modeling Fanconi anemia. Consequently, the algorithm can be used on other synchronous Boolean models of biological networks involved in diseases for in silico target identification. However, both the physiological and pathological variants have to be known. This can constitute a limit of the proposed methodology since all the pathophysiologies are not known. On the other hand, this can constitute a motivation to unravel pathophysiologies of poorly understood diseases.
Target identification, whether performed in silico or not, is a step belonging to a wider process: drug discovery. Having demonstrated a potential target in silico, or even in vitro, is far from having a drug. Further work and many years are necessary before obtaining a drug which is effective in vivo. For example, and among other characteristics, such a drug has to be absorbed by the organism, has to reach its target and has to be non-toxic at therapeutic dosages. Furthermore, as with any in silico evidence, it should be validated in vitro and ultimately in vivo: there is a bridge to cross between theory and practice. For example, targeting ATM should restore a physiological running of the FA/BRCA pathway bearing FANCD1/BRCA2 or FANCN/PALB2 gene null mutation. However, if ATM operates in other pathways, targeting it may disrupt them, hence creating new pathological conditions. Nevertheless, it is expected that the algorithm is of interest for target identification.
While finding Boolean network attractors of biological networks is not the purpose of the present work, it is a necessary step which is in itself a challenging field of computational biology. As a consequence, incorporating advances made in this field could be a relevant improvement. Another possible improvement could be to extend the algorithm for asynchronous Boolean networks since such models are likely to more accurately describe the dynamics of biological systems. Indeed, in biological systems, events may be subjected to stochasticity, may not occur simultaneously or may not belong to the same time scale, three points that synchronous updating does not take into account. Yet another possible improvement could be to use finer logics, such as multivalued ones. Indeed, one of the main limitations of Boolean models is that their variables can take only two values. In reality, things are not necessarily binary and variables should be able to take more possible values. Multivalued logics enable it in a discrete manner where variables can take a finite number of values between 0 (false) and 1 (true). For example, one can state that Rb is partly impaired rather than totally. Such a statement is not implementable with Boolean models but is with multivalued ones such as, for example, a three-valued logic where true = 1, moderate = 0.5 and false = 0.
Finally, considering basin cardinalities of pathological attractors could be an interesting extension of the proposed criteria for selecting therapeutic bullets. In that case, the therapeutic potential of bullets could be assessed by estimating their ability at reducing the basin of pathological attractors, as performed by Fumia et al. with their Boolean model of cancer pathways [19]. Such a criterion enable to consider the particular case where pathological attractors are removed, that is where pathological basins are reduced to the empty set, but also the other cases where pathological basins are not necessarily reduced to the empty set. Such a less restrictive selection of therapeutic bullets would enable to consider more possibilities for counteracting diseases.
Appendix A
The algorithm in one block of pseudocode.
| 1 | prompt card D |
| 2 | card D = min(card D, 2n) |
| 3 | generate D ⊆ S |
| 4 | H = {} |
| 5 | Aphysio = {} |
| 6 | forx0 ∈ D do |
| 7 | k = 1 |
| 8 | x(k) = x0 |
| 9 | while true do |
| 10 | if |
| 11 | break |
| 12 | end if |
| 13 | x(k + 1) = fphysio(x(k)) |
| 14 | if ∃k′ ∈ 〚1, k 〛 : x(k + 1) = x(k′) then |
| 15 | Aphysio = Aphysio ∪ {(x(k′),…,x(k))} |
| 16 | break |
| 17 | end if |
| 18 | k = k + 1 |
| 19 | end while |
| 20 | H = H ∪ {(x(1),…,x(k))} |
| 21 | end for |
| 22 | return A physio |
| 23 |
prompt
|
| 24 | rmax = min(rmax, n) |
| 25 | golden _ set = {} |
| 26 | silver _ set = {} |
| 27 | forr∈ 〚 rmin, rmax 〛 do |
| 28 | |
| 29 | |
| 30 | Ctarg = {} |
| 31 | Cmoda = {} |
| 32 | while |
| 33 | generate ctarg ∉ Ctarg |
| 34 | Ctarg = Ctarg ∪ {ctarg} |
| 35 | end while |
| 36 |
while
|
| 37 | generate cmoda ∉ Cmoda |
| 38 | Cmoda = Cmoda ∪ {cmoda} |
| 39 | end while |
| 40 | forctarg ∈ Ctarg do |
| 41 | for cmoda ∈ Cmoda do |
| 42 | H = {} |
| 43 | Apatho = {} |
| 44 | for x0 ∈ D do |
| 45 | k = 1 |
| 46 | x(k) = x0 |
| 47 | while true do |
| 48 | if |
| 49 | break |
| 50 | end if |
| 51 | x(k + 1) = fpatho(x(k)) |
| 52 | for targi ∈ ctarg do |
| 53 | for |
| 54 | if |
| 55 | xj(k + 1) = modai |
| 56 | end if |
| 57 | end for |
| 58 | end for |
| 59 | if ∃k′ ∈ 〚1, k 〛 : x(k + 1) = x(k′) then |
| 60 | Apatho = Apatho ∪ {(x(k′),…,x(k))} |
| 61 | break |
| 62 | end if |
| 63 | k = k + 1 |
| 64 | end while |
| 65 | H = H ∪ {(x(1),…,x(k))} |
| 66 | end for |
| 67 | if Apatho ⊆ Aphysio then |
| 68 | if Apatho = Aphysio then |
| 69 | golden _ set = golden _ set ∪ {(ctarg, cmoda)} |
| 70 | else |
| 71 | silver _ set = silver _ set ∪ {(ctarg, cmoda)} |
| 72 | end if |
| 73 | end if |
| 74 | end for |
| 75 | end for |
| 76 | end for |
| 77 | returngolden _ set, silver _ set |
Appendix B
Boolean functions of the case study where, for the sake of readability, xi stands for xi(k) and xi+ stands for xi(k + 1).
Appendix C
Computed attractors for the case study.
Appendix D
Therapeutic bullets found for the case study.
| −ATM | golden | ||
| −ATM | −CHK2 | golden | |
| −HRR | −ATM | golden | |
| −ssDNARPA | −ATM | golden | |
| −BRCA1 | −ATM | golden | |
| −MRN | −ATM | golden | |
| −FAN1 | −ATM | golden | |
| −ICL | −DSB | golden | |
| −FAcore | −ATM | golden | |
| −USP1 | −ATM | golden | |
| −ATM | −H2AX | golden | |
| −ADD | −ATM | golden | |
| −RAD51 | −ATM | golden | |
| −XPF | −ATM | golden | |
| −FANCM | −ATM | golden | |
| −FANCD1N | −ATM | golden | |
| −ATM | −CHK1 | golden | |
| −ICL | −ATM | golden | |
| −ATM | −p53 | golden | |
| −FANCJBRCA1 | −ATM | golden | |
| −FANCD2I | −ATM | golden | |
| −ICL | −FANCD1N | −ATM | golden |
| −ICL | −FAcore | −DSB | golden |
| −BRCA1 | −USP1 | −ATM | golden |
| −BRCA1 | −ssDNARPA | −ATM | golden |
| −BRCA1 | −ATM | −CHK1 | golden |
| −ADD | −ATM | −H2AX | golden |
| −FAN1 | −MRN | −ATM | golden |
| −ATM | −CHK2 | −H2AX | golden |
| −ICL | −DSB | −MRN | golden |
| −XPF | −MRN | −ATM | golden |
| −FAcore | −FANCD2I | −ATM | golden |
| −FANCM | −ATM | −CHK2 | golden |
| −RAD51 | −ATM | −p53 | golden |
| −ICL | −ssDNARPA | −ATM | golden |
| −FANCM | −ATR | −ATM | golden |
| −RAD51 | −ATM | −H2AX | golden |
| −ADD | −FANCD1N | −ATM | golden |
| −ICL | −USP1 | −ATM | golden |
| −FANCM | −MRN | −ATR | golden |
| −MRN | −USP1 | −ATM | golden |
| −FAN1 | −HRR | −ATM | golden |
| −BRCA1 | −ATM | −H2AX | golden |
| −FANCJBRCA1 | −ADD | −ATM | golden |
| −MRN | −ssDNARPA | −ATM | golden |
| −FAcore | −ssDNARPA | −ATM | golden |
| −FAcore | −FANCD1N | −ATM | golden |
| −FANCD2I | −BRCA1 | −ATM | golden |
| −ADD | −MRN | −ATM | golden |
| −ATM | −p53 | −CHK2 | golden |
| −RAD51 | −ATM | −CHK2 | golden |
| −FANCM | −ATM | −H2AX | golden |
| −ADD | −PCNATLS | −ATM | golden |
| −FANCJBRCA1 | −ATM | −p53 | golden |
| −FANCM | −MRN | −ATM | golden |
| −FANCJBRCA1 | −ATM | −CHK2 | golden |
| −FANCD2I | −USP1 | −ATM | golden |
| −ADD | −ATM | −CHK2 | golden |
| −FANCD2I | −FANCD1N | −ATM | golden |
| −MRN | −HRR | −ATM | golden |
| −ICL | −DSB | −USP1 | golden |
| −FAN1 | −FANCD1N | −ATM | golden |
| −FAN1 | −ATM | −H2AX | golden |
| −FANCJBRCA1 | −FAN1 | −ATM | golden |
| −ssDNARPA | −ATM | −H2AX | golden |
| −ATM | −CHK1 | −CHK2 | golden |
| −ADD | −HRR | −ATM | golden |
| −ATM | −p53 | −CHK1 | golden |
| −FAcore | −ATM | −H2AX | golden |
| −FANCD2I | −ATM | −CHK2 | golden |
| −FAN1 | −RAD51 | −ATM | golden |
| −FANCD2I | −RAD51 | −ATM | golden |
| −FANCJBRCA1 | −XPF | −ATM | golden |
| −ICL | −FANCJBRCA1 | −DSB | golden |
| −ssDNARPA | −HRR | −ATM | golden |
| −MRN | −BRCA1 | −ATM | golden |
| −FANCM | −FAN1 | −ATM | golden |
| −ssDNARPA | −ATM | −p53 | golden |
| −FAN1 | −ATM | −CHK2 | golden |
| −FANCD2I | −ssDNARPA | −ATM | golden |
| −FANCD2I | −FAN1 | −ATM | golden |
| −XPF | −HRR | −ATM | golden |
| −FAN1 | −BRCA1 | −ATM | golden |
| −ADD | −ATM | −CHK1 | golden |
| −FAcore | −HRR | −ATM | golden |
| −XPF | −ATM | −CHK1 | golden |
| −ADD | −BRCA1 | −ATM | golden |
| −ICL | −FAN1 | −DSB | golden |
| −ADD | −ATM | −p53 | golden |
| −ICL | −MUS81 | −ATM | golden |
| −FAcore | −RAD51 | −ATM | golden |
| −ATM | −CHK1 | −H2AX | golden |
| −ICL | −MRN | −ATM | golden |
| −ssDNARPA | −ATM | −CHK2 | golden |
| −XPF | −RAD51 | −ATM | golden |
| −FANCM | −ATM | −CHK1 | golden |
| −ICL | −DSB | −KU | golden |
| −ICL | −MRN | −ATR | golden |
| −ssDNARPA | −RAD51 | −ATM | golden |
| −FANCJBRCA1 | −ssDNARPA | −ATM | golden |
| −XPF | −ATM | −p53 | golden |
| −FAcore | −MRN | −ATM | golden |
| −HRR | −ATM | −H2AX | golden |
| −HRR | −ATM | −p53 | golden |
| −FANCJBRCA1 | −FANCD1N | −ATM | golden |
| −FANCM | −ADD | −ATM | golden |
| −FAcore | −ATM | −CHK2 | golden |
| −ICL | −ATM | −CHK1 | golden |
| −MRN | −FANCD1N | −ATM | golden |
| −ADD | −ssDNARPA | −ATM | golden |
| −MRN | −RAD51 | −ATM | golden |
| −FANCD1N | −ATM | −p53 | golden |
| −FANCD1N | −RAD51 | −ATM | golden |
| −BRCA1 | −ATM | −CHK2 | golden |
| −ADD | −RAD51 | −ATM | golden |
| −ICL | −DSB | −FANCD1N | golden |
| −ICL | −RAD51 | −ATM | golden |
| −ICL | −ATM | −CHK2 | golden |
| −FANCD1N | −ATM | −H2AX | golden |
| −MRN | −ATM | −H2AX | golden |
| −FAcore | −FAN1 | −ATM | golden |
| −ICL | −XPF | −ATM | golden |
| −FANCD2I | −ADD | −ATM | golden |
| −FANCD2I | −ATM | −H2AX | golden |
| −ICL | −ATR | −ATM | golden |
| −FANCM | −HRR | −ATM | golden |
| −USP1 | −ATM | −H2AX | golden |
| −ICL | −DSB | −RAD51 | golden |
| −ICL | −ATM | −H2AX | golden |
| −FANCD1N | −USP1 | −ATM | golden |
| −FANCM | −FANCD2I | −ATM | golden |
| −FANCD2I | −MRN | −ATM | golden |
| −FAcore | −ADD | −ATM | golden |
| −ICL | −FAcore | −ATM | golden |
| −FANCM | −ssDNARPA | −ATM | golden |
| −XPF | −ATM | −H2AX | golden |
| −FAcore | −USP1 | −ATM | golden |
| −HRR | −ATM | −CHK1 | golden |
| −BRCA1 | −RAD51 | −ATM | golden |
| −FAN1 | −ADD | −ATM | golden |
| −FANCJBRCA1 | −MRN | −ATM | golden |
| −FANCM | −USP1 | −ATM | golden |
| −FANCJBRCA1 | −ATM | −H2AX | golden |
| −FANCM | −FAcore | −ATM | golden |
| −HRR | −USP1 | −ATM | golden |
| −ICL | −FANCM | −ATM | golden |
| −ICL | −DSB | −ssDNARPA | golden |
| −FAN1 | −USP1 | −ATM | golden |
| −FANCM | −FANCJBRCA1 | −ATM | golden |
| −ssDNARPA | −ATM | −CHK1 | golden |
| −FAcore | −FANCJBRCA1 | −ATM | golden |
| −FANCD2I | −HRR | −ATM | golden |
| −FANCD2I | −FANCJBRCA1 | −ATM | golden |
| −XPF | −ssDNARPA | −ATM | golden |
| −USP1 | −ATM | −CHK1 | golden |
| −ICL | −DSB | −ATM | golden |
| −ICL | −ADD | −DSB | golden |
| −USP1 | −ATM | −CHK2 | golden |
| −XPF | −BRCA1 | −ATM | golden |
| −RAD51 | −ATM | −CHK1 | golden |
| −FANCD1N | −ATM | −CHK2 | golden |
| −RAD51 | −HRR | −ATM | golden |
| −ICL | −ATM | −p53 | golden |
| −ICL | −DSB | −DNAPK | golden |
| −FANCM | −FANCD1N | −ATM | golden |
| −BRCA1 | −FANCD1N | −ATM | golden |
| −ICL | −HRR | −ATM | golden |
| −FANCJBRCA1 | −HRR | −ATM | golden |
| −USP1 | −ATM | −p53 | golden |
| −XPF | −ATM | −CHK2 | golden |
| −ICL | −DSB | −CHK2 | golden |
| −ICL | −XPF | −DSB | golden |
| −ssDNARPA | −FANCD1N | −ATM | golden |
| −FANCJBRCA1 | −RAD51 | −ATM | golden |
| −ICL | −DSB | −ATR | golden |
| −HRR | −ATM | −CHK2 | golden |
| −ADD | −USP1 | −ATM | golden |
| −FANCM | −RAD51 | −ATM | golden |
| −FANCJBRCA1 | −ATM | −CHK1 | golden |
| −FANCM | −ATM | −p53 | golden |
| −XPF | −FANCD1N | −ATM | golden |
| −FAcore | −BRCA1 | −ATM | golden |
| −ICL | −DSB | −NHEJ | golden |
| −BRCA1 | −ATM | −p53 | golden |
| −BRCA1 | −HRR | −ATM | golden |
| −FANCJBRCA1 | −USP1 | −ATM | golden |
| −ssDNARPA | −USP1 | −ATM | golden |
| −ICL | −DSB | −H2AX | golden |
| −FANCM | −BRCA1 | −ATM | golden |
| −MRN | −ATM | −CHK1 | golden |
| −ICL | −FANCJBRCA1 | −ATM | golden |
| −FANCD1N | −ATM | −CHK1 | golden |
| −ICL | −DSB | −BRCA1 | golden |
| −MRN | −ATM | −CHK2 | golden |
| −FANCJBRCA1 | −BRCA1 | −ATM | golden |
| −FAN1 | −ssDNARPA | −ATM | golden |
| −MRN | −ATM | −p53 | golden |
| −FANCD1N | −HRR | −ATM | golden |
| −ICL | −MUS81 | −DSB | golden |
| −ICL | −DSB | −p53 | golden |
| −XPF | −USP1 | −ATM | golden |
| −XPF | −ADD | −ATM | golden |
| −ATM | −p53 | −H2AX | golden |
| −ICL | −FANCM | −DSB | golden |
| −ICL | −DSB | −HRR | golden |
| −ICL | −BRCA1 | −ATM | golden |
| −RAD51 | −USP1 | −ATM | golden |
| −ICL | −FAN1 | −ATM | golden |
| −ICL | −ADD | −ATM | golden |
| −ICL | −DSB | −CHK1 | golden |
| −ICL | −FANCD2I | −DSB | golden |
| −ICL | −FANCD2I | −ATM | golden |
Appendix E
Occurrence of each node in the found therapeutic bullets, in percentage of the total number of tested bullets.
| ATM | 87.736% |
| ICL | 22.170% |
| BRCA1 | 18.396% |
| DSB | 11.792% |
| MRN | 10.377% |
| FANCM | 9.906% |
| ADD | 9.906% |
| FANCJBRCA1 | 9.434% |
| ssDNARPA | 9.434% |
| FANCD1N | 9.434% |
| RAD51 | 9.434% |
| HRR | 9.434% |
| USP1 | 9.434% |
| CHK2 | 9.434% |
| H2AX | 9.434% |
| FAcore | 8.019% |
| FANCD2I | 8.019% |
| FAN1 | 8.019% |
| p53 | 8.019% |
| CHK1 | 8.019% |
| XPF | 7.547% |
| ATR | 2.358% |
| MUS81 | 0.943% |
| PCNATLS | 0.472% |
| KU | 0.472% |
| DNAPK | 0.472% |
| NHEJ | 0.472% |
| CHKREC | 0% |
Vous devez vous connecter pour continuer.
S'authentifier