

1 Introduction
Mitochondrial DNA has been increasingly used for comparative genomic and phylogenetic studies in the past decade. It has various interesting properties such as abundance in animal tissues, small size, simple genomic structure, fast rate of evolution (in animals) and exclusive maternal inheritance with low level of recombination [1,2]. Until recently, the sequencing of complete mitochondrial genomes has been relying on long range Polymerase Chain Reactions (PCR), which are difficult to perform and time consuming. The immense yield now provided by Next Generation Sequencing (NGS) allows alleviating this necessity. The high copy number of mitochondrial genomes per cell allows the use of a relatively low sequencing capacity compare to what is necessary to recover nuclear genomes. This “genome skimming” approach, originally developed for plant organelles assembly [3] has been successfully used to assemble a wide variety of animal mitochondrial genomes [4–7]. However, mitochondrial studies have been unbalanced among taxa, and the amount of available data for insects is still small compared to that for vertebrates [8,9].
In most metazoans, including insects, the mitogenome is a circular double-stranded molecule of 14–20 kb in size, which contains a set of 13 protein-coding genes (PCGs), 22 transfer RNA genes (tRNAs) and two ribosomal RNA genes (rRNAs) [10,11]. However, variations in gene content exist [8,12–14]. It also exhibits a large non-coding region known as the control region (CR), which is implicated in the initiation of transcription and replication processes [15–18].
Cimicomorpha is the largest infraorder of Heteroptera with more than 20,000 species currently placed in 17 families [19]. Tingidae is a family of Cimicomorpha distributed worldwide. They are sister group of the Miridae into the super-family Miroidea, according to the last phylogenetical analysis [20,21]. They are commonly named “lace bugs” because of the highly reticulate lace-like patterning of the pronotum and forewings of adults. All Tingidae are phytophagous, and can be destructive to plants. Some of them are important economical pests in agriculture and forestry [22]. Tingidae is currently comprised of approximately 2500 species [23] for which only one mitochondrial genome has been described so far (the sycamore lace bug, Corythucha ciliata; [24]). The avocado lace bug, Pseudacysta perseae (Heidemann, 1908) was considered a minor pest of avocado for several years [25] but since the mid-1990s, population outbreaks of P. perseae have been observed in Florida and the Caribbean. It is now considered as a serious threat for avocado in these regions [26] and several studies have focused on biological and chemical solutions for its management [27–31]. The known geographic distribution of P. perseae includes southern states of the USA (Florida, Georgia, Louisiana, Texas and, more recently, California) as well as various locations in the Caribbean, Mexico and Guatemala [30]. In South America, it has recently been reported in Venezuela and French Guiana [32,33].
In this paper, we use a genome skimming approach based on Illumina technology to assemble the complete mitochondrial genome of P. perseae. Its organization and features are compared to the mitogenome of C. ciliata. A set of 55 heteropteran mitochondrial genomes was used to perform a phylogenetic analysis.
2 Material and method
2.1 Specimen, DNA extraction and sequencing
Specimens of P. perseae were collected on an avocado in Remire-Montjoly, French Guiana on May 6th 2009. Total genomic DNA was extracted from leg muscle tissues using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA) following a protocol adapted from the manufacturer's instructions. The quality and quantity of extracted genomic DNA was evaluated using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and the PicoGreen double stranded DNA quantitation assay kit (Life Technologies, Carlsbad, CA, USA).
The genomic DNA was sent for library construction and sequencing to the GeT-PlaGe core facilities of Genotoul (Toulouse, France). One hundred and sixty-two ng of DNA were used for library construction using the Illumina TruSeq Nano DNA Sample Prep Kit (Illumina Inc. San Diego, CA) following the instructions of the supplier. After shearing by ultrasonication with a Covaris M220 (Covaris Inc, Woburn, MA), purified fragments were A-tailed and ligated to sequencing indexed adapters. Fragments with an insert size around 450 bp were selected with Agencourt Ampure XP beads (Beckman Coulter, Inc.), and enriched with 8 cycles of PCR before library quantification and validation. The library was multiplexed with 23 other libraries (generated in other projects). The pool of libraries was then hybridized on one lane of Illumina Hiseq 2000 flow cell using the Illumina TruSeq PE Cluster Kit v.3, and paired-end reads of 100 nucleotides were collected on the HiSeq 2000 sequencer using the Illumina TruSeq SBS Kit v.3 (200 cycles). Quality filtering was performed by the Consensus Assessment of Sequence and Variation (CASAVA) pipeline (Illumina Inc., San Diego, CA, USA). Sequence data were stored on the NG6 platform [34] and all the computations were performed on the computer cluster of the Genotoul bioinformatic platform (Toulouse, France).
2.2 Sequence assembly
The mitochondrial genome was assembled using a previously used strategy [35–37] in essence similar to that of Hahn et al. [38]. Reads aligning with the mitochondrial protein sequences of C. ciliata [24] were retrieved using the PLAST program [39]. Reads with a match of at least 90% were assembled into contigs using the Velvet assembler [40] with a k-mer length of 69 and all the remaining parameters left at their default values. The resulting contigs were used as seeds to initiate the genome walking approach using the extractreads2 program (included in the Obitools package; http://metabarcoding.org/obitools). Reads sharing at least 80 consecutive bp with the seeds were selected and subsequently used as seed to repeat the operation until no new read was identified. The newly selected reads were assembled with the Velvet assembler. The few resulting contigs were assembled using Geneious 6.0.6 Pro (Biomatters, Auckland, New Zealand). Additionally, reads aligning with the 28S and 18S rRNA genes of Eurydema maracandica [41] were identified and the sequence of the complete nuclear ribosomal gene segment was recovered with the same method.
2.3 Genome annotation and sequence analysis
Coverage statistics were computed with Geneious 6.0.6 Pro (Biomatters, Auckland, New Zealand) by mapping the Illumina reads to the assembled genome. Reads were set as paired and mapped to the reference sequence using a custom sensitivity. The following mapping parameter were used: a minimum overlap of 10 bp, a minimum overlap identity of 90%, a maximum mismatch per read of 10%, a maximum ambiguity of four bases and no gaps allowed.
The mitochondrial genome was first annotated using the MITOS web server [42] applying the invertebrate mitochondrial genetic code (NCBI code table 5). The annotations of tRNA genes were kept unchanged. The annotations of protein-coding genes (PCGs) were refined by checking manually for consistent start/stop codons and reading frames. The annotations of rRNA genes were extended until adjacent tRNAs when existing, following the punctuation model of mtDNA transcription [43,44]. Annotation of srRNA's 3′ end was further adjusted by mapping the srRNA of C. ciliata for which the secondary structure was predicted and presented all expected domains [24]. We used Geneious 6.0.6 Pro with the following parameters: a word length of seven, maximum gap size of 15 and maximum mismatches of 40%. This approach conducted to extend lrRNA's annotation by 13 bp until tRNALeu at its 3′ end and by 194 bp until tRNAVal at its 5′ end. srRNA's annotation was extended by 1 bp until tRNAVal at its 3′ end and by 35 bp at its 5′ end. We further verified the consistency of these new annotations by mapping the rRNAs of C. ciliata as described above. The large remaining non-annotated sequence was annotated as the control region in concordance with other insect mitogenomes. The tandem repeats copy number was inferred using a coverage analysis.
The 18S and 28S rRNAs were annotated in comparison with that of E. maracandica [41]. The 5.8S rRNA was annotated in comparison with that of Triatoma dimidiata (accession number: KF142517).
2.4 Sequence analyses and phylogenetics
Base composition and codon usage where computed with MEGA6 [45]. AT-skew ([A−T]/[A + T]) and GC-skew ([G−C]/[G + C]) where used to measure nucleotide compositional differences between genes [46]. Relative synonymous codon usage (RSCU) where used to described bias in synonymous codon composition. Tandem repeats were identified using Tandem Repeat Finder web server [47]. tRNA's secondary structure were inferred via the MITOS web server pipeline. We looked for putative secondary structures within the 100 bp of the control region flanking tRNAIle where stem loops structures have already been reported in Tingidae and other heteropterans [24,36,48,49].
A set of 54 additional heteropteran mitogenomes was downloaded from Genbank (Table 1). Two mitogenomes of Auchenorrhyncha were used as outgroups. The 13 PCGs were used for the analysis to allow for comparison with previous studies [24,50]. They were first aligned separately based on amino-acids translation with translatorX [51]. Divergent regions were removed with Gblocks.0.91b before back-translation in order to conserve reading frames. All resulting alignments were then concatenated using FASconCAT [52]. The best partitioning scheme and substitution model were inferred with PartitionFinder.1.1.1 [53], using the greedy algorithm for scheme search and the Bayesian information criterion for scheme selection. A maximum-likelihood (ML) analysis was performed with RAxML 8.0 [54] and nodal support was estimated using a rapid bootstrap procedure [55] with the majority-rule tree based bootstopping criteria. A Bayesian analysis was conducted using Mr. Bayes 3.2 [56], starting from four random trees with ten Markov chains (nine heated chain and one cold chain), 2,000,000 generations and all other parameters set to default. Each set was sampled every 200 generations with a burn-in of 25% of the sampled trees. At the end of the analysis, the average standard deviation of split frequencies was below the recommended 0.01.
Complete or near complete mitochondrial genomes used in this study.
| Suborder | Infra-order/Superfamily | Family | Species | Accession num. | Reference |
| Auchenorryncha | Fulgoroidea | Fulgoridae | Lycorma delicatula | EU909203 | [83] |
| Flatidae | Geisha distinctissima | NC_012617 | [70] | ||
| Heteroptera | Cimicomorpha | ||||
| Cimicoidea | Anthocoridae | Orius niger | EU427341 | [74] | |
| Miroidea | Miridae | Adelphocoris fasciaticollis | NC_023796 | [84] | |
| Apolygus lucorum | NC_023083 | [85] | |||
| Lygus lineolaris | NC_021975 | Unpublished | |||
| Nesidiocoris tenuis | NC_022677 | [86] | |||
| Tingidae | Corythucha ciliata | NC_022922 | [24] | ||
| Pseudacysta perseae | This study | ||||
| Naboidea | Nabidae | Alloeorhynchus bakeri | HM235722 | [87] | |
| Gorpis annulatus | JF907591 | [87] | |||
| Gorpis humeralis | JF927830 | [87] | |||
| Himacerus apterus | JF927831 | [87] | |||
| Himacerus nodipes | JF927832 | [87] | |||
| Nabis apicalis | JF907590 | [87] | |||
| Reduvioidea | Reduviidae | Agriosphodrus dohrni | NC 015842 | [50] | |
| Oncocephalus breviscutum | NC 022816 | [88] | |||
| Sirthenea flavipes | HQ645959 | [49] | |||
| Triatoma dimidiata | NC 002609 | [48] | |||
| Valentia hoffmanni | NC 012823 | [69] | |||
| Enicocephalomorpha | |||||
| Enicocephaloidea | Enicocephalidae | Stenopirates sp. | NC_016017 | [89] | |
| Gerromorpha | |||||
| Gerroidea | Gerridae | Aquarius paludum | NC_012841 | [69] | |
| Hydrometroidea | Hydrometridae | Hydrometra sp. | NC_012842 | [69] | |
| Leptopodomorpha | |||||
| Leptopodoidea | Leptopodidae | Leptopus sp. | FJ456946 | [69] | |
| Saldoidae | Saldidae | Saldula arsenjevi | EU427345 | [74] | |
| Nepomorpha | |||||
| Corixoidea | Corixidae | Sigara septemlineata | FJ456941 | [69] | |
| Naucoroidea | Aphelocheiridae | Aphelocheirus ellipsoideus | FJ456939 | [69] | |
| Naucoridae | Ilyocoris cimicoides | NC_012845 | [69] | ||
| Nepoidea | Belostomatidae | Diplonychus rusticus | FJ456940 | [69] | |
| Nepidae | Laccotrephes robustus | NC_012817 | [69] | ||
| Notonectoidea | Notonectidae | Enithares tibialis | NC_012819 | [69] | |
| Ochteroidea | Gelastocoridae | Nerthra sp. | NC_012838 | [69] | |
| Ochteridae | Ochterus marginatus | NC_012820 | [69] | ||
| Pleoidea | Helotrephidae | Helotrephes sp. | FJ456951 | [69] | |
| Pentatomomorpha | |||||
| Aradoidea | Aradidae | Aradacanthia heissi | HQ441233 | [90] | |
| Brachyrhynchus hsiaoi | NC_022670 | [91] | |||
| Neuroctenus parus | EU427340 | [74] | |||
| Coreoidea | Alydidae | Riptortus pedestris | EU427344 | [74] | |
| Coreidae | Hydaropsis longirostris | EU427337 | [74] | ||
| Rhopalidae | Aeschyntelus notatus | EU427333 | [74] | ||
| Stictopleurus subviridis | NC_012888 | [69] | |||
| Lygaeoidea | Berytidae | Yemmalysus parallelus | EU427346 | [74] | |
| Colobathristidae | Phaenacantha marcida | EU427342 | [74] | ||
| Geocoridae | Geocoris pallidipennis | EU427336 | [74] | ||
| Malcidae | Chauliops fallax | NC_020772 | [74] | ||
| Malcus inconspicuus | EU427339 | [74] | |||
| Pentatomoidea | Cydnidae | Macroscytus gibbulus | EU427338 | [74] | |
| Dinidoridae | Coridius chinensis | JQ739179 | [92] | ||
| Pentatomidae | Dolycoris baccarum | NC_020373 | [93] | ||
| Halyomorpha halys | NC_013272 | [94] | |||
| Nezara viridula | NC_011755 | [74] | |||
| Plataspidae | Coptosoma bifaria | EU427334 | [74] | ||
| Megacopta cribraria | NC_015842 | [73] | |||
| Tessaratomidae | Eusthenes cupreus | NC_022449 | [95] | ||
| Urostylididae | Urochela quadrinotata | NC_020144 | [96] | ||
| Pyrrhocoroidea | Largidae | Physopelta gutta | EU427343 | [74] | |
| Pyrrhocoridae | Dysdercus cingulatus | EU427335 | [74] |
3 Results and discussion
3.1 Genome sequencing, assembly and annotation
After filtering 5.08% of the initial reads, raw sequence data represent a total of 6,965,443 paired-end reads (13,930,886 reads in total). Fifty thousands and eight hundred reads were assembled into a 15,850-bp circular sequence (Genbank accession number: KM278221), representing the complete mitochondrial genome with an average sequencing depth of 320.5. A circular map of the mitogenome and the assembly coverage are presented on Fig. 1. The typical set of 37 genes (13 PCGs, 22 tRNAs, two rRNAs) and one control region were identified. Twenty-three genes are encoded on the majority strand and the others located on the minority strand. Eight gene overlaps where observed, the longest being a 14-bp region between ATP6 and COX3 (Table 2). Apart from the control region, nine non-coding regions ranging from 1 bp to 19 bp where identified.
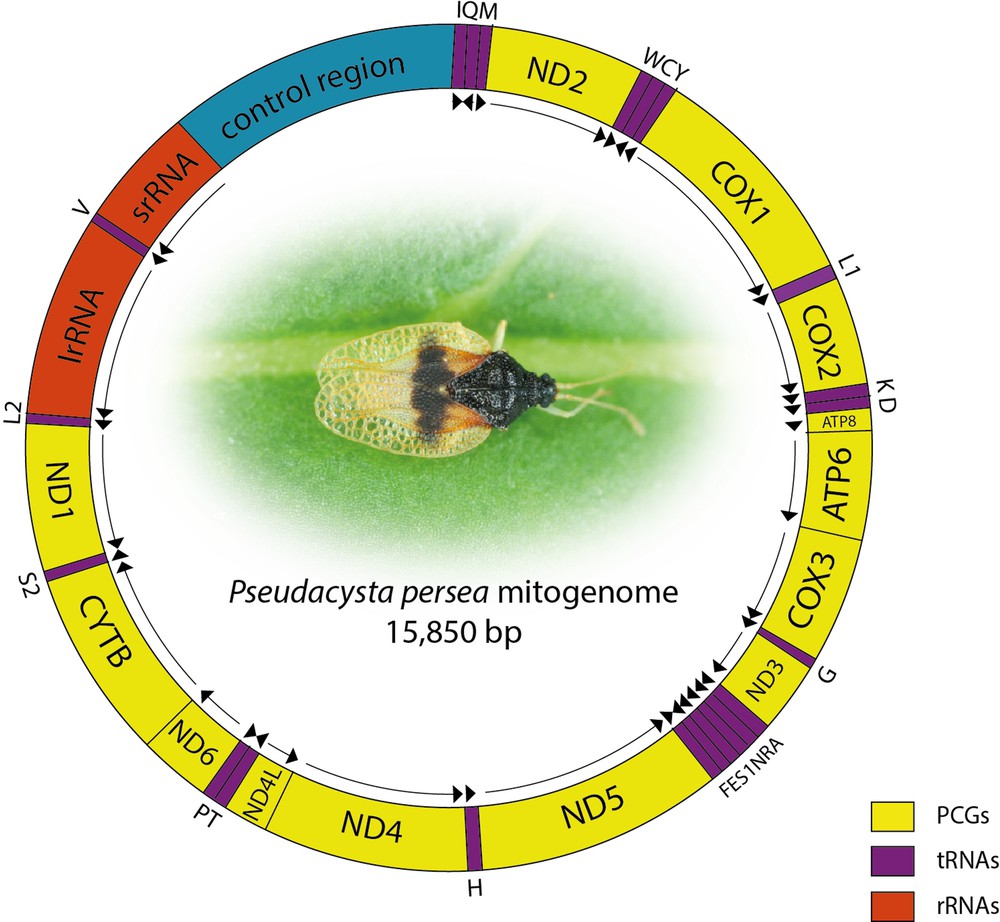
(Color online.) Schematic representation of Pseudacysta perseae mitogenome. tRNAs are labelled according to the IUPAC-IUB single-letter amino acid codes. Arrows indicate directions of genes (clockwise = majority strand).
Summary of the mitochondrial genome of Pseudacysta perseae. Strand + –refers to major/minor strand. Negative values of IGS (intergenic spacer) refer to overlapping nucleotides.
| Locus | Strand | Location (bp) | Size (bp) | Anticodon | Start codon | Stop codon | IGS |
| tRNAIle | + | 1–63 | 63 | GAT | 0 | ||
| tRNAGln | – | 65–132 | 68 | TTG | 1 | ||
| tRNAMet | + | 134–196 | 63 | CAT | 1 | ||
| ND2 | + | 197–1178 | 982 | ATA | T−− | 0 | |
| tRNATrp | + | 1179–1245 | 67 | TCA | 0 | ||
| tRNACys | – | 1238–1301 | 64 | GCA | –8 | ||
| tRNATyr | – | 1304–1367 | 64 | GTA | 2 | ||
| COX1 | + | 1368–2903 | 1536 | ATG | TAA | 0 | |
| tRNALeu (UUR) | + | 2906–2969 | 64 | TAA | 2 | ||
| COX2 | + | 2970–3648 | 679 | ATT | T−− | 0 | |
| tRNALys | + | 3649–3720 | 72 | CTT | 0 | ||
| tRNAAsp | + | 3721–3781 | 61 | GTC | 0 | ||
| ATP8 | + | 3782–3940 | 159 | ATA | TAA | 0 | |
| ATP6 | + | 3934–4599 | 666 | ATG | TAA | –7 | |
| COX3 | + | 4586–5372 | 787 | ATT | T−− | –14 | |
| tRNAGly | + | 5373–5435 | 63 | TCC | 0 | ||
| ND3 | + | 5436–5789 | 354 | ATT | TAA | 0 | |
| tRNAAla | + | 5793–5853 | 61 | TGC | 3 | ||
| tRNAArg | + | 5854–5914 | 61 | TCG | 0 | ||
| tRNAAsn | + | 5915–5979 | 65 | GTT | 0 | ||
| tRNASer (AGN) | + | 5979–6047 | 69 | GCT | –1 | ||
| tRNAGlu | + | 6048–6112 | 65 | TTC | 0 | ||
| tRNAPhe | R | 6111–6174 | 64 | GAA | –2 | ||
| ND5 | – | 6175–7853 | 1679 | ATT | TA− | 0 | |
| tRNAHis | – | 7854–7918 | 65 | GTG | 0 | ||
| ND4 | – | 7921–9240 | 1320 | ATG | TAA | 2 | |
| ND4L | – | 9234–9515 | 282 | ATA | TAA | –7 | |
| tRNAThr | + | 9517–9578 | 62 | TGT | 1 | ||
| tRNAPro | – | 9578–9643 | 66 | TGG | –1 | ||
| ND6 | + | 9645–10,124 | 480 | ATT | TAA | 1 | |
| CYTB | + | 10,124–11,264 | 1141 | ATT | T−− | –1 | |
| tRNASer (TCN) | + | 11,265–11,333 | 69 | TGA | 0 | ||
| ND1 | – | 11,353–12,279 | 927 | ATA | TAA | 19 | |
| tRNALeu (CUN) | – | 12,281–12,344 | 64 | TAG | 0 | ||
| lrRNA | – | 12,345–13,564 | 1220 | 0 | |||
| tRNAVal | – | 13,565–13,632 | 68 | TAC | 0 | ||
| srRNA | – | 13,633–14,424 | 792 | 0 | |||
| Control region | 14,425–15,850 | 1426 | 0 |
The complete nuclear ribosomal sequence was recovered. A total of 45,600 reads were assembled into a 7588-bp sequence comprising of 18S rRNA (1899 bp), ITS1 (232 bp), 5.8S rRNA (155 bp), ITS2 (1220 bp) and 28S rRNA (4082 bp). The sequence was deposited on Genbank with the accession number KM278220.
P. perseae mitogenome shares the same architecture and orientation as C. ciliata. This gene arrangement is shared with many insects [57,58]. It is also found in crustaceans and was the first to be determined differing by a single tRNA translocation from that of the Chelicerata Limulus polyphemus, considered as ancestral for Arthropoda [59,60]. Therefore, this mitogenome organisation is thought to be ancestral for the insect-crustacean clade [10,61]. P. perseae mitogenome is 593 longer than C. ciliata mitogenome which is almost entirely due to a variation in the size of the control region (1426 bp in P. perseae vs 818 in C. ciliata). More generally, variations in insect mitogenomes size are mainly due to the control region, which ranges from 70 bp in Ruspolia dubia (Orthoptera) [62] to 4599 bp in Drosophila melanogaster (Diptera) [62,63].
3.2 Protein-coding genes
The total length of the 13 PCGs was 10,992 bp. Their nucleotide composition is strongly biased toward AT with an overall AT content of 76.4% (Table 3). All PCGs have an ATN start codon (Table 2). Six PCGs initiated with ATT (COX2, COX3, ND3, ND5, ND6 and CYTB), four initiated with ATA (ND2, ATP8, ND4L and ND1) and three initiated with ATG (COX1, ATP6 and ND4). Six genes initiated with the same ATG start codon between both lace bugs mitogenomes (COX1, ATP8, ATP6, ND4, ND6 and ND1). Other unconventional start codons were found in insects such as TTG in heteropterans [24], CGA and TTAG in lepidopterans [64,65], or ATAA, GTAA and TTAA in dipterans [57,66] but that was not the case in P. perseae.
Nucleotide composition of the mitogenomes of Tingidae: Pseudacysta perseae (P) and Corythucha ciliata (C).
| A + T% | AT skew | GC skew | ||||
| P | C | P | C | P | C | |
| Whole genome | 79.8 | 77.0 | 0.05 | 0.13 | –0.23 | –0.16 |
| Protein coding genes | 76.4 | 76.4 | –0.12 | 0.16 | –0.01 | –0.17 |
| 1st codon position | 72.7 | 74.2 | 0.18 | 0.36 | –0.07 | –0.02 |
| 2nd codon position | 69.8 | 73.5 | –0.03 | 0.46 | –0.15 | –0.22 |
| 3rd codon position | 86.6 | 80.7 | 0.20 | 0.24 | –0.54 | –0.33 |
| Protein coding genes-Ma | 75.0 | 75.3 | 0.00 | 0.44 | –0.16 | –0.15 |
| 1st codon position | 70.6 | 78.9 | 0.16 | 0.06 | 0.09 | –0.30 |
| 2nd codon position | 68.6 | 76.5 | –0.35 | 0.16 | –0.23 | –0.07 |
| 3rd codon position | 85.7 | 70.5 | 0.16 | –0.10 | –0.53 | –0.11 |
| Protein coding genes-m | 78.6 | 78.0 | 0.31 | 0.35 | –0.26 | –0.21 |
| 1st codon position | 76.1 | 77.4 | 0.21 | 0.33 | –0.39 | –0.19 |
| 2nd codon position | 71.6 | 79.8 | 0.45 | 0.37 | –0.03 | –0.23 |
| 3rd codon position | 88.0 | 77.2 | 0.27 | 0.33 | –0.55 | –0.23 |
| tRNAs | 80.0 | 78.3 | 0.04 | 0.08 | –0.11 | –0.11 |
| tRNAs-M | 79.9 | 78.2 | 0.05 | 0.66 | 0.03 | 0.01 |
| tRNAs-m | 80.1 | 78.5 | 0.03 | 0.11 | –0.37 | –0.31 |
| rRNAs | 80.5 | 79.2 | 0.05 | 0.11 | –0.27 | –0.25 |
| Control region | 82.2 | 74.0 | –0.07 | –0.07 | –0.01 | 0.10 |
a M/m: genes encoded on the major/minor strand.
The majority of PCGs have a usual TAA stop codon, but four T and one TA stop codons were identified (ND2, COX2, COX3, CYTB and ND5 respectively). These incomplete stop codons are immediately adjacent to tRNA genes encoded on the same strand which is consistent with the punctuation model for primary transcripts processing followed by 3′ polyadenylation of mature mRNA that will allow the completion of termination codons [43,44,67]. Incomplete stop codons can also be found in C. ciliata and are shared with many arthropods [68].
3.3 Ribosomal and transfer RNA genes
rRNA genes locations and lengths are similar to those of other insect mitogenomes. lrRNA is located between tRNALeu(CUN) and tRNAVal and is 1220 bp-long. srRNA is located between tRNAVal and the control region and is 792 bp-long. Their AT content is respectively 81.0% and 79.8%.
The classical set of 22 tRNAs found in arthropods is present in P. perseae. Their lengths vary between 61 bp (tRNAAla and tRNAArg) and 72 bp (tRNALys). Secondary structures of tRNAs are schematized in Fig. 2. The classical clover leaf structure was observed for each of them, except for tRNASer(AGN), in which the D arm is reduced to a simple loop, as in many insects, and more generally, in most bilaterians [11,15]. The unusual TTC anticodon observed in C. ciliata for tRNASer(AGN) was not found in P. perseae which exhibits the classical GCT anticodon.
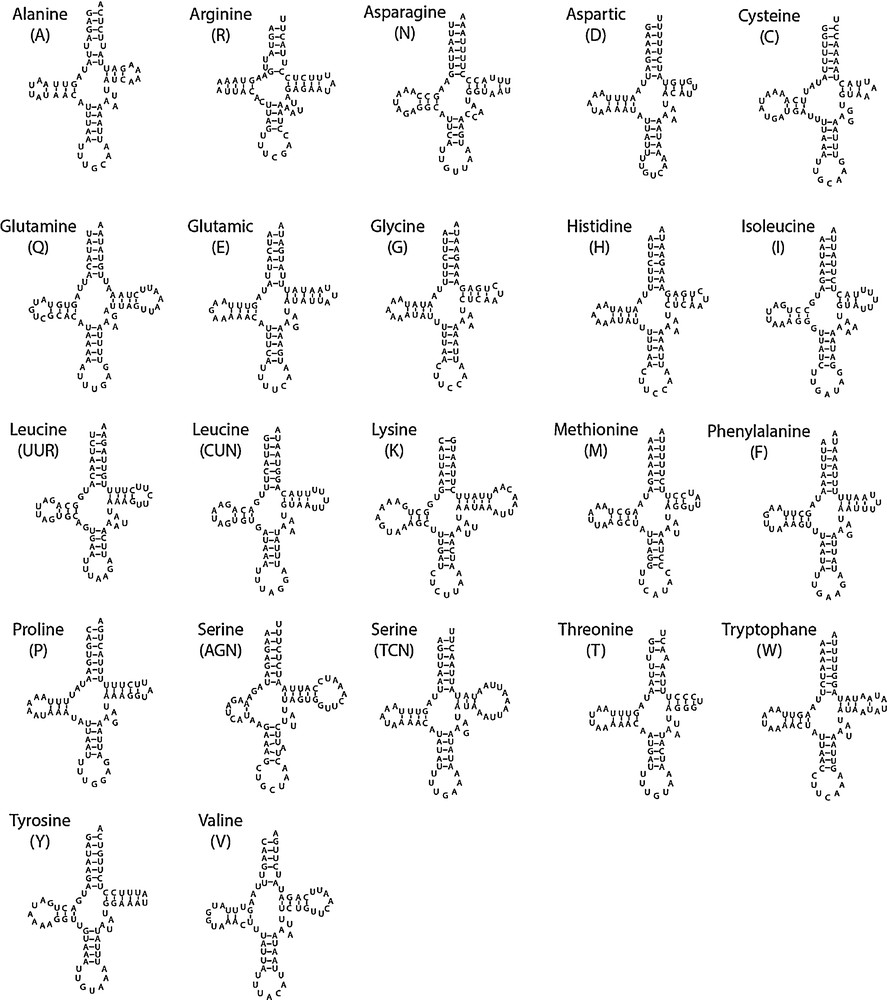
Secondary structure of Pseudacysta perseae mitochondrial tRNAs.
3.4 Non-coding regions
Nine short intergenic spacers (IGS) and a long control region were identified, matching the usual organisation of insect mitogenomes. Most of IGS are very short, with less than four base pairs, and eight overlapping sequences are found (Table 2). The longest IGS are the 19 bp found between ND1 and tRNASer2. The control region is 1426 bp-long and is located between srRNA and tRNAIle genes. In P. persea, it exhibits one of the highest A + T content of the genome. The alternative name “A + T rich region” that has been used for insect control region could therefore be appropriate in this case [18]. However, this designation is hardly generalizable for heteropterans. Indeed, in C. ciliata and other bugs such as T. dimidiata [48] and Valentia hoffmanni [69], the control region exhibits a lower A + T content compared to that the whole mitogenome. In P. persea and C. ciliata, the control regions can be divided in a similar way (Fig. 3a):
- • a region with a G + C content higher than the average;
- • a region with a high A + T content;
- • a region with a G + C content higher than the average, and;
- • a region containing tandem repeats.
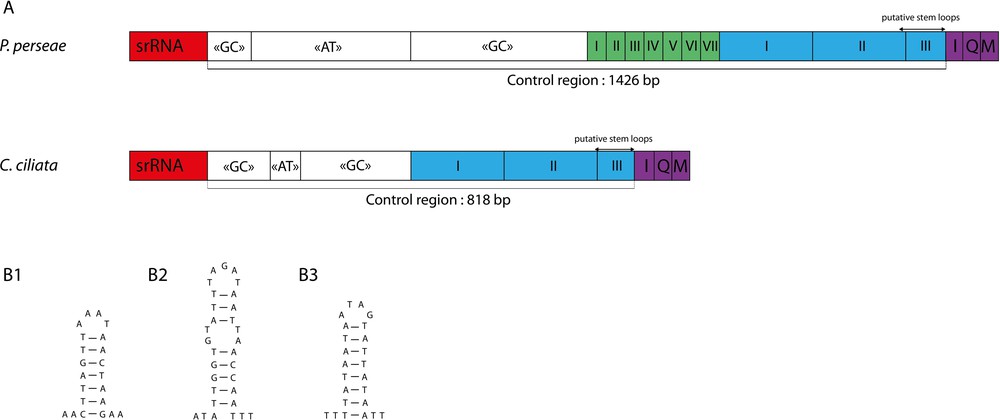
(Color online.) A. Structural organization of the mitochondrial control region of Tingidae. The boxes with roman numerals indicate tandem repeat units; B. Putative stem loop structures found in the mitochondrial control region of Pseudacysta perseae.
Tandem repeats in P. persea control region consisted of six 38-bp repeated units and one 31-bp unit (corresponding to a partial copy of the previous ones) followed by two 180 bp repeated units and one 84-bp partial copy. In C. ciliata, it consisted of two 189-bp repeated units followed by a 48-bp partial copy. However, the sequencing technology used in this study does not allow inferring repeats copy numbers with certainty. We identified three putative stem loop structures in the control region of P. perseae (Fig. 3b). Such features may be involved in the mitochondrial DNA replication mechanism [70,71], but further studies are needed to assess this hypothesis.
In Drosophila and other insect species, a conserved string of about 20 Ts was identified in the control region and suspected to be involved in the initiation of replication [17]. Similarly, a conserved sequence block including a “G element” was observed in Reduviidae [49]. None of these were found in P. persea. The alignment of the two lace bugs mitogenomes revealed various identical segments all along the control region, but more data would be necessary to know if some of them are well conserved among Tingidae.
3.5 Nucleotide content and codon usage
The nucleotide composition is strongly biased toward adenine and thymine in P. perseae mitogenome, with A + T representing 79.8% of the whole sequence and ranging from 76.4% in the protein-coding genes, 80.0% in tRNA genes, and 80.5% in rRNA genes to 82.2% in the control region. The nucleotide biais is reflected in the codon usage. AT-rich codons are predominant in protein-coding genes, with the most prevalent being in order TTA (Leu), ATT (Ile), ATA (Met) and TTT (Phe). The relative synonymous codon usage (RSCU) clearly indicates that AT rich codons are favoured among synonymous codons (Table 4). At the third codon position, AT content is particularly high (86.6%), and G nucleotides are under-represented (GC skew = −0.54). AT content, as well as A–T and G–C skews patterns are similar between the two lace bugs mitochondrial genomes (Table 3).
Codon usage of Pseudacysta perseae mitogenome protein-coding genes.
| Amino Acida | Codon | N b | RSCUc | Amino Acid | Codon | N | RSCU |
| F | UUU | 291 | 1.69 | Y | UAU | 150 | 1.72 |
| UUC | 54 | 0.31 | UAC | 24 | 0.28 | ||
| L | UUA | 363 | 4.02 | H | CAU | 51 | 1.59 |
| UUG | 61 | 0.68 | CAC | 13 | 0.41 | ||
| CUU | 52 | 0.58 | Q | CAA | 45 | 1.7 | |
| CUC | 8 | 0.09 | CAG | 8 | 0.3 | ||
| CUA | 54 | 0.6 | N | AAU | 159 | 1.58 | |
| CUG | 4 | 0.04 | AAC | 42 | 0.42 | ||
| I | AUU | 313 | 1.67 | K | AAA | 108 | 1.74 |
| AUC | 62 | 0.33 | AAG | 16 | 0.26 | ||
| M | AUA | 304 | 1.77 | D | GAU | 51 | 1.55 |
| AUG | 39 | 0.23 | GAC | 15 | 0.45 | ||
| V | GUU | 81 | 1.89 | E | GAA | 77 | 1.75 |
| GUC | 10 | 0.23 | GAG | 11 | 0.25 | ||
| GUA | 70 | 1.64 | C | UGU | 43 | 1.91 | |
| GUG | 10 | 0.23 | UGC | 2 | 0.09 | ||
| S | UCU | 115 | 2.67 | W | UGA | 80 | 1.72 |
| UCC | 14 | 0.33 | UGG | 13 | 0.28 | ||
| UCA | 85 | 1.98 | R | CGU | 13 | 1.04 | |
| UCG | 10 | 0.23 | CGC | 1 | 0.08 | ||
| P | CCU | 69 | 2.28 | CGA | 33 | 2.64 | |
| CCC | 10 | 0.33 | CGG | 3 | 0.24 | ||
| CCA | 39 | 1.29 | S | AGU | 36 | 0.84 | |
| CCG | 3 | 0.1 | AGC | 1 | 0.02 | ||
| T | ACU | 64 | 1.39 | AGA | 83 | 1.93 | |
| ACC | 15 | 0.33 | AGG | 0 | 0 | ||
| ACA | 103 | 2.24 | G | GGU | 71 | 1.65 | |
| ACG | 2 | 0.04 | GGC | 4 | 0.09 | ||
| A | GCU | 47 | 1.9 | GGA | 74 | 1.72 | |
| GCC | 10 | 0.4 | GGG | 23 | 0.53 | ||
| GCA | 39 | 1.58 | |||||
| GCG | 3 | 0.12 |
a Amino acids are labeled according to the IUPAC-IUB single-letter codes.
b N: total number in all PCGs.
c RSCU: relative synonymous codon usage.
3.6 Phylogenetic analysis
Bayesian inference and Maximum Likelihood analysis (ML) generated phylogenetic trees with very similar topologies (Figs. 4 and 5). P. perseae is placed as a sister group of C. ciliata. Miroidea is paraphyletic in our analysis, in contrast with previous results based on morphological and molecular data [20,72], but in accordance with the results of Tian et al. [73] inferred from mitochondrial and nuclear genes. The 18 other superfamilies represented in our dataset are monophyletic. At the infra-order level, Pentatomorpha is monophyletic with the following relationships between the five superfamilies: Aradoidea + (Pentatomoidea + [Lygaeoidea + [Pyrrhocoroidea + Coreoidea]]). These relationships are strongly supported in our analyses and do not confirm the results of previous studies based on a subset of the present mitogenomic data that placed Coreoidea and Lygaeoidea as sister groups [24,74]. On the tree generated by Bayesian inference, Nepomorpha is monophyletic with the following relationships between the six superfamilies: (Pleoidea + Corixoidea) + ([Notonectoidea + Naucoroidea] + [Nepoidea + Ochteroidea]). ML analysis differs by positioning Pleoidea as a sister group of the remaining Nepomorpha. These results are not consistent with those of a study based on molecular and morphological data [75]. Interestingly, they also contradict a previous analysis of a subset of the present mitogenomic dataset by confirming the monophyly of Nepomorpha including Pleoidea for which an infraordinal status was proposed [69]. Gerromorpha and Leptopodomorpha are also monophyletic but only two species of each were included in the study. Enicocephalomorpha was only represented by Stenopirates sp.
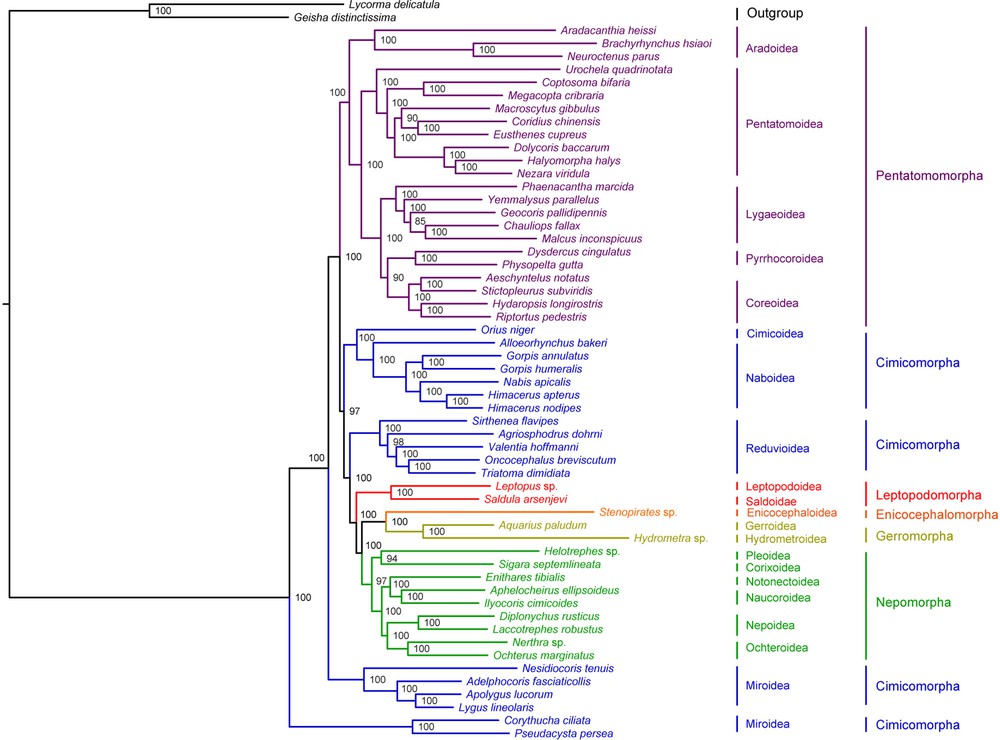
(Color online.) Phylogenetic tree inferred by Bayesian analysis from 55 heteropteran mitogenomes. Bayesian posterior probabilities expressed in percentages are indicated on the nodes.
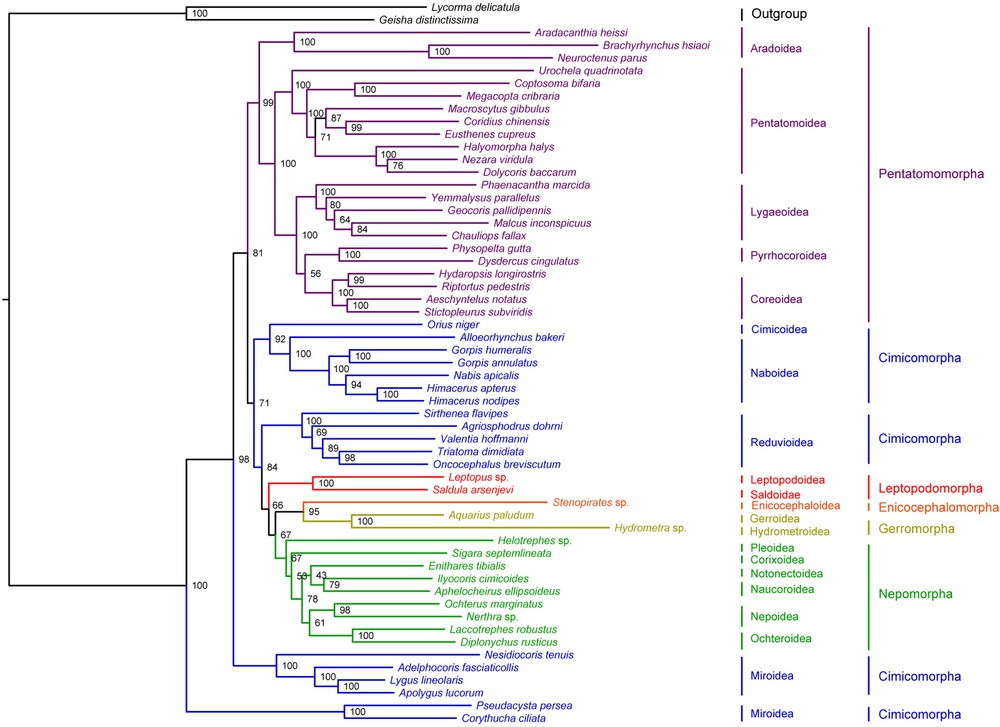
(Color online.) Phylogenetic tree inferred by ML analysis from 55 heteropteran mitogenomes. Bootstrap support values expressed in percentages are indicated on the nodes.
Our analysis supports the paraphyly of Cimicomorpha that consisted of four different clades: (Cimicoidea + Naboidea), Reduvioidea, Miridae and Tingidae. Tingidae is placed as a sister group to all remaining Heteroptera. However, the monophyly of Cimicomorpha has been widely accepted and is supported by various analyses. Schuh et al. [20] conducted a total-evidence phylogenetic analysis of Cimicomorpha based on 73 morphological characters, nuclear and mitochondrial DNA for 92 taxa including eight outgroups belonging to Pentatomomorpha, Leptopodomorpha and Nepomorpha. Their result confirmed the monophyly of the group except for the inclusion of Thaumastocoridae, which is not represented in our dataset. Similarly, Tian et al. [73] inferred the monophyly of Cimicomorpha from the analysis of nuclear and mitochondrial data on 46 taxa including three outgroups belonging to Pentatomomorpha and Leptopodomorpha. More recently, Li et al. [76] performed a phylogenetic analysis of Heteroptera with a dataset of nuclear and mitochondrial DNA for 83 species including 22 Cimicomorpha, which also confirmed the monophyly of the group.
Infraordinal relationships are conserved in both ML and Bayesian analyses: Tingidae + (Miridae + (Pentatomomorpha + ((Cimicoidea + Naboidea) + (Reduviodea + (Leptopodomorpha + ((Enicocephalomorpha + Gerromorpha) + Nepomorpha)))))). These results are in contrast with the general consensus that considers Enicocephalomorpha as the early infraorder of Heteroptera [19]. However, the relationships between the infraorders of Heteroptera remain controversial. Only few phylogenetic studies have addressed the question and those have only included a small number of taxa [77–79]. Mitogenomic data provide a new insight in this regard, but more taxa should be added to the current database, especially in the poorly represented infra-orders Enicocephalomorpha, Gerromorpha, Leptopodomorpha and Dipsocoromorpha.
Our results are incongruent with generally accepted hypothesis [19]. On the other hand, they are in accordance with previous studies based on a subset of the present mitogenomic data [24,50]. Analyses performed on individual genes by Tian et al. [73] and Schuh et al. [20] indicate that the monophyly of Cimicomorpha is only supported by nuclear rDNA partitions, among a dataset comprising 16 rDNA, 18S rDNA, 28S rDNA and COI. Incongruences of phylogenetic analyses among different genomic regions are a well-known issue that can have various biological causes such as incomplete lineage sorting or evolution rates variations among sites [80]. For instance, Kopp and True [81] reported that single-gene datasets of D. melanogaster species group produce several strongly supported conflicting clades. Lin and Danforth [82] studied the differences in the pattern of nucleotide substitution among nuclear and mitochondrial genes and concluded that insect phylogenetic studies should increasingly focus on nuclear data. This raises the limitations of phylogenetic inferences from complete mitochondrial genomes only. While reducing stochastic errors by providing large molecular datasets, this approach is susceptible to potential site-specific bias and would benefit from a conjoint analysis with other genes. Unfortunately, there is a lack of correspondence between publicly available mitogenomic and nuclear data. Currently, on Genbank, nuclear ribosomal sequences are available for only 14 species out of 55 represented in our mitochondrial dataset. Moreover, these are mainly partial sequences that do not necessarily overlap. The genome skimming-approach employed in the present study could provide an interesting improvement in this regard. In a single experimentation, it allows to recover the full mitogenome sequence as well as nuclear genes that are classically used for phylogenetic inference.
Acknowledgements
We would like to thank Maria Kamilari for help with DNA extraction and Eric Coissac for help with genome assembly. This work was supported by « Investissement d’avenir » grants managed by the Agence Nationale de la Recherche (CEBA, ref. ANR-10-LABX-25-01; TULIP, ANR-10-LABX-41, ANR-11-IDEX-0002-02) as well as project METABAR (ANR-11-BSV7-0020). We are grateful to the Genotoul bioinformatics platform Toulouse Midi-Pyrénées for providing computing and storage resources. The material studied was collected in the frame of the CAFOTROP-Muséum project.