

1 Introduction
Around one billion years old, Nematoda (round worms) constitute the most ancient and diverse animal phylum [1], with estimates of the number of extant species ranging from 25,000 to certainly more than 100 million [2]. As free living organisms as well as parasites of animals and plants, these species represent an ecologically diverse assemblage of roundworms distributed in nearly every environment, including soil and sediments, fresh and sea water [3]. Five main lineages were identified using molecular data, and all diverged from a shared ancestor several hundred million years ago [4]. Today parasitic nematodes are recognized to cause significant impacts on agriculture, domestic animals and human health [5].
Endoparasitic root-knot nematodes (Meloidogyne spp.) are foremost economically damaging genera of plant parasitic nematodes [6]. Meloidogyne spp. belongs to the class Secernentea, order Tylenchida, suborder Hoplolaimina, superfamily Hoplolaimoidea, family Meloidogynidae, subfamily Meloidogyninae. Recent phylogenies indicated that Meloidogyne is a monophyletic group but these analyses also pointed to poorly resolved paraphyletic relationships with the sister lineage that includes genera Pratylenchus, Hirschmanniella and Zygotylenchus [7,8]. The Meloidogyne genus includes more than 60 species, while some of the members even have several races. Meloidogyne species are major pests worldwide with very large plant host ranges (e.g. M. javanica, M. arenaria, M. incognita and M. hapla) and cause important crop losses locally, as it has been reported for M. graminicola in rice fields [9].
Meloidogyne species use various reproduction strategies (i.e. amphimixis, facultative meiotic parthenogenesis or obligatory mitotic parthenogenesis [10,11]). For instance, M. incognita, M. javanica and M. arenaria have a mitotic partenogenetic reproduction mode whereas M. hapla and M. graminicola reproduce usually by a facultative meiotic parthenogenetic mode and less so frequently by cross fertilization [10]. Although these nematodes are particularly well studied because of their economic importance and their ecological widespread adaptation, their characterization faces constraints. Species and race identification of Meloidogyne based on phenotypic characterization is time-consuming because adult forms are hardly accessible since males are usually rare and females are distorted (pear-shaped due to their settlement inside roots). Therefore, taxonomists use routinely morphometrics of second stage juveniles (e.g. tapering tail, body and stylet size) and female perineal patterns [12,13]. Consequently, species identification is long and requires trained nematologists. For these reasons, biochemical and molecular tools have been developed to assist the identification of taxa [13–15] as well as to investigate the phylogeography and population dynamics of nematodes (for a recent review on this topic, see [16]). However, the molecular investigation of nematodes still has to face other challenges caused by the difficulty to isolate individuals or colonies for extracting enough protein or DNA. Despite of this, isozymes (esterase) as well as ribosomal and mitochondrial DNA sequences are generally used for routine diagnostics of nematodes [12,15,17]. Polymorphic DNA markers (microsatellites) have been also developed to study the genetic structure of the potato cyst nematode Globodera pallida in a whole country [18], or to detect and locate rare sexual events in predominantly asexual Xiphinema spp. [19]. Lastly, it is worth to mention that the nuclear genome of Meloidogyne spp. is usually small (e.g., M. graminicola exhibits the smallest animal genome referenced to date, C-value = ca. 0.03 pg; [20]) and recent advances about the complete genome sequence of these organisms should provide new insights in nematode diagnostics and ecology [21].
Mitochondrial DNA (mtDNA) is usually maternally inherited in nematodes [22,23], and polymorphism of this genome is thus useful to follow maternal lineages. For instance, the cox1 mitochondrial gene has been used to study the genetic structure of the plant parasitic nematode Xiphinema spp. [24]. No reference mitochondrial genome (or mitogenome) is presently available for root-knot nematodes, although the structure of the M. javanica mtDNA has been described more than 20 years ago, and was partially sequenced (X57625/X57626; ca. 20% of the genome; [25]). Today, in the superfamily Hoplolaimoidea, only three complete mitogenomes have been deposited in the public DNA databases [26–28] for the distantly related species Heterodera glycines (Heteroderidae), Radopholus similis, and Pratylenchus vulnus (Pratylenchidae). Thus, little is known about the variations in mitogenomes of this important phytoparasitic lineage. This also represents a limitation to develop genetic tools based on mtDNA sequence polymorphism for various purposes from species or race identification to phylogeography or population genetics. In addition, frequent mitogenome reorganizations have been reported in Hoplolaimoidea [28], rendering fastidious the development of PCR-based methods to sequence long mtDNA regions. Lastly, the gene annotation of nematode mitochondrial genomes remains a challenge, especially the typical truncated tRNA (transfer RNA) genes are hard to detect with current tools.
Recent advances in sequencing technologies, so called next generation sequencing or NGS, are bringing about a revolution for the sequencing of organellar genomes [29]. NGS has been used to generate mitochondrial genomes of various metazoan organisms, but the method is still usually based on the sequencing of long-range PCR products. The method however offers the possibility of fast assembly of the most represented DNA regions in a genome with high sequencing depths [29] but this has never been tested on nematodes. In particular, the Illumina technology (e.g. HiSeq 2000; [30]) allows generating tens of millions of short DNA segments (i.e. 100-bp reads) that can be used for this purpose.
Here, the Illumina technology was tested on M. graminicola in order to generate a reference mitogenome for root-knot nematodes. A complete mtDNA genome sequence was released and compared to the mtDNA gene map of M. javanica, and also to other species of the superfamily Hoplolaimoidea. Lastly, the utility of our approach for generating full mtDNA resources in nematodes is briefly discussed.
2 Material and methods
2.1 DNA extraction and DNA sequencing
An isolate of M. graminicola from a rice field in Laurel, Batangas, Philippines [31], was propagated in a hydroponic solution on rice cultivar IR64 during four weeks before egg extraction. Eggs were recovered from infested roots using the hypochlorite extraction method with minor modifications [32] and were collected in 0.8% hypochlorite solution with 0.1% Tween 20 then left for 30 min at room temperature. Eggs were rinsed several times with sterile ddH2O and placed on a strainer covered by three damp Kimwipe tissues on a 50-mL beaker filled with sterile ddH2O and streptomycin for a final concentration of 10 μg/mL. After 24 h in the dark at room temperature, water containing nematodes was discarded and replaced with new sterile ddH2O. Twenty-four hours later, the freshly hatched second stage juveniles (J2s) were collected by centrifugation and conserved at –80 °C. For 100 μL of J2, an equal volume of 2 × lysis buffer containing 20 mM Tris–HCl pH 8.4, 100 mM KCl, 3 mM MgCl2 and 0.9% Tween 20 was added. Fresh DTT (2 mM) and 20 μg of fresh proteinase K were added and the tube was placed in a water bath at 50 °C. After a 2-h treatment, 0.1 μg of RNase A (Qiagen Inc., GmbH, Hilden, Germany) was added and the tube was placed at 37 °C for an additional 30 min. Then, genomic DNA was recovered by a phenol–chloroform method [33]. DNA quality was inspected with a NanoDrop® spectrophotometer and 1 μL of genomic DNA (gDNA) was loaded on 1% agarose gel to control DNA integrity.
Three hundred nanograms of DNA were used for shotgun sequencing with the Illumina technology (e.g., HiSeq 2000; [30]). The library was constructed using the Illumina TruSeq DNA Sample Prep v2 kit and following the instructions of the supplier. Briefly, genomic DNA was sheared by sonication and the fragments were end-repaired. Purified fragments were A-tailed and ligated to sequencing indexed adapters. Fragments with an insert size of approximately 300 to 500 bp were gel-extracted and enriched with 10 cycles of PCR before library quantification and validation. This library was multiplexed with 23 other libraries (generated by other colleagues on various plant species). The pool of libraries was then hybridized to the Hiseq 2000 flow cell using the Illumina TruSeq PE Cluster Kit v3. Bridge amplification was performed to generate clusters, and paired reads of 100 nucleotides were collected on the HiSeq 2000 sequencer using the Illumina TruSeq SBS Kit v3 (200 cycles).
2.2 Read assembly, sequencing depth and gene annotation
The read assembly method is similar to the approach reported by [34] to generate a complete plastid genome in plants. We used the cox2 sequence of M. graminicola as a seed because it was already available in public DNA databases (JN241929; 530 bp). This sequence allowed initiating the global assembly process that involved two main steps. First, we used the program extractread2 (included in the OBITools package, http://metabarcoing.orf/obitools) to select sequence reads sharing at least 80 bases with the seed. The newly selected reads were subsequently used as probes. This process was iteratively repeated until no new read could be identified. Second, the set of selected reads were assembled into two large contigs using the Velvet program [35]. Long repeat elements in two regions (see below) hampered us to generate one contig of the full mitogenome. All computations were done on the computer cluster of the Genopole Bioinformatic Platform (Toulouse, France).
Two regions, namely 111R and 94R, are composed of 111-bp and 94-bp repeats and could not be assembled with our approach (see below). While region 111R includes about 30 repeats and is difficult to sequence with current methodologies, 94R is relatively short (inferior to 1000 bp). This later region was generated by PCR with the following primers (forward: GAATGAAAACGATCAGAGACATG; reverse: CAACTAATATTTTTGATAGCACTCC). The 94R region was amplified by PCR in a final volume of 50 μL containing 10 ng of gDNA, 500 nM of each PCR primer, 200 μM of each dNTP, 10 μL of 5 × Phusion buffer and one unit of the proof-reading enzyme “Phusion High-Fidelity DNA Polymerase” (Thermo Fisher Scientific Inc., France). The following PCR program was used: 98 °C for 30 s; 35× (98 °C, 10 s; 55 °C, 30 s; 72 °C, 70 s); 72 °C for 10 min. PCR products were gel-purified and eluted in 30 μL ddH2O using a gel purification kit (Qiagen Inc., Germany). Approximately 200 ng of purified amplicon were sent for sequencing with a classical Sanger approach (Cogenics, UK). The generated sequence allowed merging both contigs. Finally, reads were mapped onto the obtained mtDNA sequence using Geneious v6.0.5 (Biomatters Ltd.) to assess the depth of sequencing and the number of repeats in the region 111R.
We annotated the consensus mtDNA sequence using the protein prediction pipeline of Mitos with the invertebrate mitochondrial code [36], but could not detect all common mitochondrial genes, especially rRNA (ribosomal RNA) and tRNA genes. We thus used a semi-automated procedure: all ORFs of the mitochondrial genome of M. graminicola were extracted and translated with the help of Getorf from a local installation of the Emboss package v6.4.0.0 [37]. Candidate genes were aligned to known nematode mitochondrial proteins extracted from RefSeq 58 using a local installation of Mafft v7.058b [38], and identified manually by sequence similarity. All tRNA genes could be detected with MiTFi [39], using Infernal v.1.0 [40] and covariance models as described by [41]. The applied models additionally contained truncated tRNAs of nematodes of the class Enoplea, which are partially experimentally validated [42]. Using the nematode models without the Enoplea tRNAs, some tRNAs could not be found in M. graminicola. All tRNAs have been checked manually, the start/stop positions of trnG, trnH and trnS1 have been improved. Both rRNA genes were cut using alignments of the Tylenchida (Bursaphelenchus mucronatus, Pratylenchus vulnus, Radopholus similis) extracted from RefSeq 58. For these sequences the 5′ ends of the adjacent tRNAs were used as start position, and the 3′ end of the rrnL gene was trimmed manually to fit with the nad3 gene. The structures have been cleaned manually and all wrongly predicted base pairs, resulting from the Infernal output, were removed.
The newly generated sequence was deposited within GenBank (HG529223). A physical map was finally drawn with Mtviz (http://pacosy.informatik.uni-leipzig.de/mtviz/) and colors were modified. Gene order in the M. graminicola mtDNA was compared with the three available complete mitochondrial genomes of the chromadorean nematode clade (i.e. Heterodera glycines, Radopholus similis and Pratylenchus vulnus; [28]). In order to check the phylogenetic position of M. graminicola in the superfamily Hoplolaimoidea, the 12 protein encoding genes were extracted and aligned with the complete mitochondrial gene sequences of 41 other nematodes as used by [28]. A Maximum Likelihood analysis was conducted with PhyML 3.0 [43]. Statistical support for the different internal branches was assessed through bootstrap resampling (1000 replicates).
3 Results and discussion
3.1 De novo assembly of a root-knot nematode mitochondrial genome
We generated 6,012,732 paired-end 100-bp reads. Using extractread2, we selected 35,597 paired-end reads from the mitochondrial genome (ca. 0.6% of initial sequences). The first assembly generated two contigs of 15,109 and 1813 bp. These contigs were separated by regions with long repeats (namely 111R and 94R) that were not possible to properly assemble. These two repeats are located in the same regions than 102R and 63R in M. javanica ([25]; see below). Sequence comparisons did not show homology between repeats from the different species (see also [44] for 102R in M. hapla). The sequencing depth was estimated on the non-repeated regions at 318.5× ± 121.7 (min. 68×, max. 651×). Based on this value, we estimated that 94R is composed of three repeats of 94 bp that was confirmed by a classical Sanger sequencing. Similarly, sequencing depth of region 111R (9,077× ± 1830) suggested that about 29 tandem repeats of 111 bp composed this region. A consensus sequence of an 111R repeat was generated considering all mismatches: TATTTTTAATTATTRYYATTTTTGATGATTTTTCAGAATTTTGYTTAATTTAAYTTTATTTTTAARCCAATTTTTTGTAAAATATRAATYATAWAGGTAGTTACCTTTTYA. Considering 29 repeats in 111R, the mitochondrial genome size of M. graminicola was estimated at 20,030 bp, a size similar to the mtDNA size reported for M. javanica and M. incognita (ca. 20.5 kb and 19.5 kb, respectively; [25]). Lastly, the mtDNA of M. graminicola was especially AT-rich (84.3%) as previously reported in M. javanica and M. hapla [25,44].
Our complete sequence was also compared to mitochondrial DNA sequences of M. graminicola available in GenBank. Polymorphism was found in both genes cob and cox2: 16 substitutions/531 bp (3%) were found in cob compared to sequences of American isolates (AY757884 and AY757885) and three to five substitutions/530 bp (ca. 1%) were found in cox2 compared to six sequences of Asian isolates (from India, China and Bangladesh; JN241926 to JN241929, JN241939, GU187309, GQ865513). This result indicates that intraspecific variation is present in coding regions and potentially useful for population genetic analyses or race identification, but non-coding regions – such as 94R – may potentially be more variable (due to relaxed selective pressures compared to protein encoding genes) but their informativeness still needs to be assessed.
3.2 Mitochondrial genome organisation
Thirty-six genes were detected in the M. graminicola mtDNA sequence (Table S1, Supplementary data) as in other available mitogenomes of the suborder Hoplolaimina [28]: 12 protein encoding genes, 22 transfer RNA genes (trn) and two ribosomal RNA genes (rrn). Among the typical mitochondrial genes of metazoans, only atp8 is missing as already reported in other nematode mitogenomes [28,45]. An annotated physical map of the mtDNA genome was reconstructed and is shown in Fig. 1. We found the same gene order than in the incomplete gene map of M. Javanica [25], suggesting no important gene reorganisation between these two Meloidogyne species. In contrast, the comparison of the M. graminicola mtDNA with those of the chromadorean nematode clade reveals a deep reorganisation (Fig. 2). The gene order between rrnS and cox2 in the M. graminicola mtDNA is, however, similar to the mitogenome of Pratylenchus vulnus. In this genomic region of about 5000 bp (that includes 16 genes; Table S1, Supplementary data), only one gene is not located at the same position in these two species: trnF, which is located between trnN and trnG in M. graminicola, while it is located between trnC and nad6 in P. vulnus. Additionally, we detected in the mitogenomes of M. graminicola and P. vulnus the same three pairs of genes nad4-trnD, atp6-nad5 and trnQ-trnA plus another region with three genes (rrnL-nad3-cob). In contrast, a comparison of the mtDNA gene orders between M. graminicola and Heterodera glycines or Radopholus similis – two other members of the superfamily Hoplolaimoidea – show deep genome rearrangements. Only small regions with the same gene arrangements were detected between M. graminicola and R. similis [from cox2 to nad3 (four genes), from nad2 to trnN (four genes), from atp6 to trnA (four genes), from trnY to nad1 (three genes) and nad6-nad4L; Fig. 2], and between M. graminicola and H. glycines (from atp6 to trnA (four genes), plus three pairs of genes: trnE-trnS1, trnI-cox3 and rrnL-nad3; Fig. 2). Only four pairs of genes are conserved between the four mitogenomes of Hoplolaimoidea (i.e. atp6-nad5, trnQ-trnA, trnI-cox3 and rrnL-nad3). Note that Pratylenchus is the most related species to Meloidogyne for which a complete mitogenome is available ([7,8]; Supplementary data, Fig. S1) and the genome arrangement may reflect the phylogenetic distance between taxa [45]. The timing of mitogenome rearrangements needs to be investigated on a larger taxon sample in a phylogenetic framework. M. graminicola and P. vulnus also share a large non-coding region (that harbours only one tRNA gene). The length of this region is ca. 7.5 and 7.8 kb in M. graminicola and P. vulnus, respectively. To date, such a long non-coding region has not been reported in other mitogenomes of Hoplolaimoidea. This region is reported to contain the replication origin (the control region) that varies from a few hundred bp to tens of kb in metazoans [28].
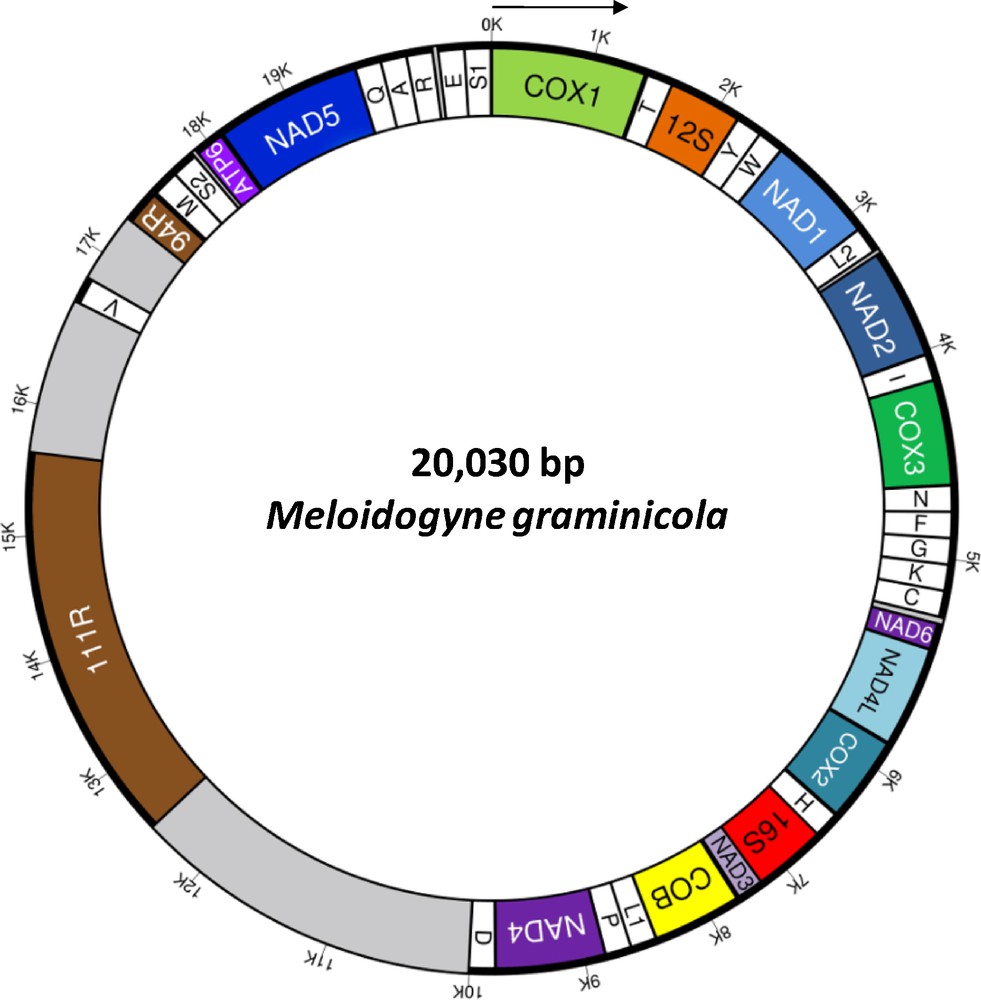
(Color online). Circular gene map of the complete mitochondrial genome of Meloidogyne graminicola. Genes and non-coding regions are represented as boxes. The direction of transcription of all genes is the same and indicated by the arrow. Abbreviations of protein coding and rRNA genes are: NADi = subunit i of NADH dehydrogenase; COXi = subunit i of cytochrome c oxydase; COB = cytochrome b; ATP6 = subunit 6 of ATP-synthase, 12S = 12S ribosomal RNA, 16S = 16S ribosomal RNA. tRNA genes are named with single-letter amino acid abbreviations except for those coding for leucine and serine, which are named as L1 (anticodon uag), L2 (uaa), S1 (ucu), and S2 (uga). Two regions, namely 111R and 94R, are composed of 111-bp and 94-bp repeats. These elements are located in a large AT-rich non-coding region (that harbours only one tRNA gene, trnV) of 7.5 kb (in grey).
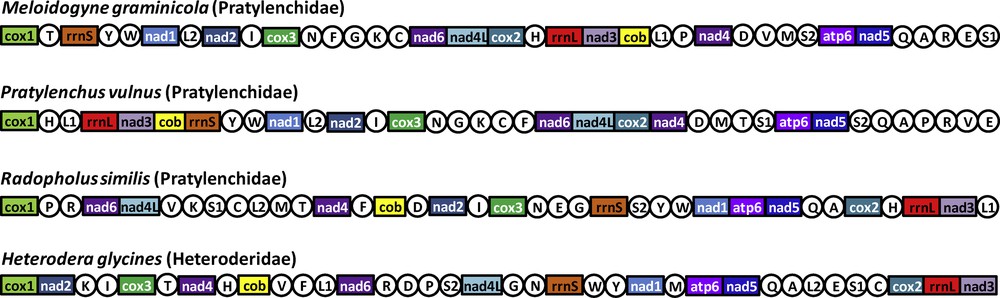
(Color online). Linearized representation of the mitochondrial gene arrangement in the four available mitogenomes of the superfamily Hoplolaimoidea. Gene and genome size are not to scale. All genes are transcribed in the same direction (from left to right). The tRNAs are designated by single-letter abbreviation (in circles). The AT-rich non-coding regions are not indicated.
3.3 Structures of tRNA and rRNA genes
The short sizes of tRNAs, ranging between 45 and 56 bp, and the fact that a part of them could only be detected by including truncated Enoplea sequences to the covariance models, indicates how minimal the mitochondrial tRNAs of M. graminicola are. Truncated mitochondrial tRNA sequences are found throughout nearly all metazoan phyla [41]. While nearly all nematodes share common truncated tRNA sequences missing either the D- or the T-stem, e.g., Caenorhabditis elegans and Ascaris suum [46], the most truncated sequences were found in Enoplea [41]. Here, tRNAs were found that lack both, the D- and T-stems. All tRNAs in M. graminicola share the typical nematode structure missing the T-stem, but forming a D-stem (data not shown). The only exceptions from this rule are serine tRNAs, which instead miss the D-stem in nematodes. This is also the case for trnS1 and trnS2 in M. graminicola. Both rRNA structures show characteristics that are common for nematodes, with the typical domains. Their short lengths (602/830 bp) again support the trend to minimal structures as related organisms encode larger rRNAs, e.g., Radopholus similis (692/840 bp). Both rRNA structures are shown on Supplementary data, Fig. S2.
The automated annotation of mitochondrial genomes is still a challenging problem. All highly conserved genes could be predicted by the Mitos pipeline [36]. However, the programs failed to predict the truncated non-coding RNAs correctly. We expect that our findings will help to improve these pipelines as most of predictive programs are based on former annotations, which is even more important for truncated sequences as it is the case in nematodes, and especially in the M. graminicola genome. Nice examples are the mitochondrial tRNAs of the American house dust mites. Here, an improvement in the detection methods led to the discovery of extremely short tRNAs [47].
4 Concluding remarks
Complete mitogenomes of nematodes that were recently released in DNA databases were generated with PCR-based methods [28], and this approach can be challenging due to numerous genome rearrangements. Our study indicates that NGS can overcome this difficulty and allows building assemblies of complete organellar genomes even when no sequence reference is available [29], and with a low amount of DNA (300 ng). In addition, our approach is fast and relatively cheap already today, and consequently can be applied to generate complete mitogenomes on a large panel of nematode lineages. In turn, such an accumulation of data will permit an improved automated gene annotation in nematodes. This may open new avenues to identify informative regions for developing genetic tools for race identification or phylogeographic investigations [48], to study the rearrangements of mitogenomes in nematodes [28], and may also allow the development of phylogenomics to depict the long evolutionary history of this still poorly known animal phylum [4].
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgments
We are grateful to Alexander Donath and Matthias Bernt for helpful analysis of protein coding genes and general discussions on gene annotations, and to Malyna Suong for nematode culture and DNA extraction. We also gladly acknowledge the Plateforme Génomique and Bioinformatics platform of Genopole Toulouse Midi-Pyrénées for providing support to analyse NGS data and a referee for his comments. Professor Joong-Ki Park kindly provided the alignment of mitochondrial protein coding genes used in this study. This work was funded by the fellowship ANR-12-AGRI-0002 (Arimnet 2011–PestOlive). This work has been conducted in the “Laboratoire Évolution & Diversité biologique” (EDB), part of the LABEX entitled TULIP managed by “Agence nationale de la recherché” (ANR-10-LABX-0041). Authors also wish to thank a Global Rice Science Partnership (GRiSP) Grant (Menergep Project) for financial support to SB.