

1 Introduction
Glucosinolates (GSLs) are sulfur- and nitrogen-enriched major secondary metabolites found in the Brassicaceae crop plants. The Brassicaceae crops include Chinese cabbage, broccoli, turnip, cabbage, cauliflower, and several other cruciferous vegetables possessing high GSL content [1]. Among these crops, the Chinese cabbage (Brassica rapa. ssp. pekinensis) is one of the most consumed vegetables in Southeast Asia. The outcome of several valuable properties, such as resistance against pathogens, auxin maintenance in plants, and cancer prevention in humans from GSL catabolism makes them one of the major groups among plant secondary metabolites [1,2]. For example, sulforaphane (SF) and indole-3-carbinol (I3C) are the derivatized catabolites of glucoraphanin and glucobrassicin, respectively, which govern cancer protection [3]. There are three different types of GSLs depending on their intake of amino acids for biosynthesis, namely, aliphatic GSL (AGSL), benzoic GSL (BGSL), and indole specific GSL (IGSL) [4].
Several previous reports suggest a positive role of R2R3 MYB (myeloblastosis) transcription factors in GSL biosynthesis [4]. According to their regulatory function in GSL biosynthesis, the MYB transcription factors have been classified into AGSL and IGSL regulators. For example, in Arabidopsis MYB34, MYB51 and MYB122 are involved in the regulation of IGSL biosynthesis [5,6], whereas MYB28, MYB29, and MYB76 are involved in the regulation of AGSL biosynthesis [7–10]. All these GSL regulators belong to subgroup 12 of the R2R3-type MYB TFs, and they are further classified into Clade I (IGSL regulators) and Clade II (AGSL regulators) [6,9,10].
The present study aims at exploring the molecular characterization of the B. rapa ssp. pekinensis AGSL regulatory genes BrMYB28 and BrMYB29. B. rapa is a polyploid and consists of multiple paralogous genes, which is one of the major obstacles that hinders the study of the role of its individual genes. The whole genome analysis of B. rapa reveals the presence of three paralogous genes, similar to the Arabidopsis MYB28 and MYB29 genes [11]. The nomenclature of all the paralogs is as follows: BrMYB28.1 (Bra012961), BrMYB28.2 (Bra035929), BrMYB28.3 (Bra029311), BrMYB29.1 (Bra005949), BrMYB29.2 (Bra009245), and BrMYB29.3 (FJ584292). To analyze the expression of BrMYB paralogous genes unambiguously, the BrMYB29.2 gene is excluded from the expression studies due to its high sequence identity (99%) with BrMYB29.1. We have also analyzed the putative cis regulatory motifs from the upstream (UPS) regulatory regions of the respective BrMYB TF genes and their DSGs to determine the distribution patterns of the evolutionarily conserved motifs. Differential expressions of BrMYBs have been carried out at different developmental stages, as well as in response to different stresses. In addition, differential expression of the DSGs was also performed in different developmental stages of B. rapa ssp. pekinensis.
2 Materials and methods
2.1 Plant material and growth conditions
In vitro grown B. rapa ssp. pekinensis (Chinese cabbage) variety Seoul (Kyoungshin seed, Korea) seedlings were transplanted into pots (25 × 25 cm) containing a sterile mixture of soil, sand, and Biopeat (Seminis Asia Ltd., Korea). The plants were then raised under controlled glasshouse conditions until they reached maturity. Plant samples were collected from seven different developmental stages, such as five-day-old seedling (SDL), one-month-old root (R), shoot (S), and leaf (L), two-month-old roots (2R), inner leaves (IL), and outer leaves (OL). All samples were collected and stored at –80 °C, immediately after sampling, for further analysis. For stress treatments, surface sterilized B. rapa seeds were grown on half strength Murashige and Skoog (MS Duchefa, Germany) medium with 0.8% agar and 0.5% sucrose for 10 days at 21 °C, under a 16/8-h photoperiod, and then the plantlets were shifted to a liquid MS medium supplemented with 0.5% sucrose. Different stress treatments, such as 100 μM of indole-3-acetic acid (IAA), gibberellic acid (GA), methyl jasmonate (MeJA), salycilic acid (SA), abscicic acid (ABA), and 3% glucose (MS basal medium devoid of sucrose) were given to seedlings and incubated for 24 h. MeJA and SA treatments were given to both shoots and roots simultaneously, whereas other stresses were applied through the root system. Low- and high-temperature treatments were given by incubating the plantlets at 4 °C for 24 h and 37 °C for 2 h. Wound stress was induced by cutting the leaves with a scalpel, and the samples were collected after five minutes of wounding. Root and shoot tissue samples were collected from all stress treatments, with the exception of high-temperature treatments, in which the whole plant was taken for quantitative real-time PCR (qRT-PCR) gene expression analysis.
2.2 Sequence retrieval and bioinformatics analysis
All nucleotide and protein sequences of MYB and other genes used in this study were extracted from the Brassica database (BRAD) (http://brassicadb.org/brad), the National Centre for Biotechnology Information (NCBI) sequence database (http://www.ncbi.nlm.nih.gov/), and the Arabidopsis Information Resource (TAIR) Arabidopsis genome database (http://www.arabidopsis.org/) [11,12]. The putative orthologous MYB protein sequences from other crops were identified through the NCBI BLASTP program, using B. rapa ssp. pekinensis MYB28 and 29 amino acid sequences [11,12] as a query sequence, with a cut-off E-value of 1–10. The top 5–10 orthologous MYB proteins selected from each homology search were used for multiple sequence alignments and phylogenetic analysis. The sequence identity between the MYB proteins was analyzed with the ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2) program, by utilizing the default parameter [13]. The phylogenetic tree was built by the neighbour-joining method, using the Molecular Evolution Genetics Analysis (MEGA 4.02) program [14] to analyze the evolutionary relationships among the MYB proteins. The reliability of the tree was achieved with 1000 bootstrap replicates.
The putative structural domain identification in the B. rapa MYB28 and 29 paralogous proteins and Arabidopsis MYB28 and 29 proteins were analyzed by using the PROSITE (http://www.expasy.org/tools/scanprosite/) program [15]. For putative cis regulatory motif analysis, the promoter regions (–1 to –1.5 kb) from gene sequences of all the BrMYB28 and BrMYB29 paralogs and their DSGs, namely, methylthioalkylmalate synthase 1 (MAM1), MAM3, cytochrome P450 belonging to the CYP79 family gene (CYP79F1), CYP83 family gene (CYP83A1), C-S lyase superroot-1 (SUR1), sulfotransferase 5C (ST5C), flavin monooxygenase glucosinolate S-oxygenase 2 (FMO-GSOX2), and flavin monooxygenase glucosinolate S-oxygenase 5 (FMO-GSOX5) in the AGSL biosynthetic pathway, were extracted from the BRAD database. The distributions of putative cis regulatory motifs were identified with the help of the PLACE (http://www.dna.affrc.go.jp/PLACE/signalscan.html) and PlantPAN (http://plantpan.mbc.nctu.edu.tw/) software programs [16,17]. Apart from the common letters, such as A, G, C or T designated for nucleotides, the other letters used in the PLACE database were: K: G or T; N: A, C, G or T; W: A or T; Y: C or T. The flow chart of the work presented in the materials and method section was represented in Fig. 1.
Flow chart representation of the outline of the present study. Abbreviations: BRAD: Brassica database; NCBI: National Centre for Biotechnology Information; MEGA: Molecular Evolutionary Genetics Analysis.
2.3 Quantitative transcript variance analysis
Total RNA was isolated from seven different developmental stages (SDL, R, S, L, 2R, IL, and OL) and from various stress-treated B. rapa plants, using TRI reagent (Sigma–Aldrich, Saint-Louis, USA), followed by treatment with DNase I (Promega, MA, USA). cDNA synthesis was performed with 3 μg of total RNA, oligodT primer, and Super Script-II RT, according to the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). The B. rapa actin (BrAct – FJ969844) gene was used as an internal control for relative quantification of BrMYBs and their DSGs transcripts. The nucleotide multiple sequence similarities between the BrMYB amplicons were analyzed by using ClustalW2. qRT-PCR was performed in the CFX 96 Real-Time PCR system (Bio-Rad, Hercules CA, USA) using the SYBR PCR kit (Bio-Rad, USA). The PCR amplification reactions were performed as follows: 95 °C for 5 min, followed by 35 cycles at 94 °C for 30 s, annealing temperature for 30 s and an extension at 72 °C for 2 min using the primers listed in Table S1. The specificity of amplification was confirmed by the melt curve analysis and agarose gel electrophoresis. The relative gene expression was quantified by 2−ΔΔCt method [18]. Comparative threshold (Ct) values were normalized to actin control and compared to quantify the relative transcript levels.
2.4 Statistical analysis
Each experiment was carried out independently, at least thrice, with three replicates. The resulting data from different experiments were analyzed using Origin 8.1 (Origin lab corporation, Northampton MA USA) software program. Statistical analyses were done using one-way analysis of variance (ANOVA) followed by Tukey's multiple comparison tests, and the standard error (SE) was calculated using the n values of each experiment.
3 Results
3.1 Bioinformatics analysis of putative structural domains in BrMYBs
Identification of putative structural domains in BrMYBs and Arabidopsis MYB28 and MYB29 proteins, using the PROSITE tool (http://www.expasy.org/tools/scanprosite/), indicated the presence of a specific DNA-binding domain (HTH) in all the BrMYBs, except in BrMYB29.3 (Fig. 2). In addition to the DNA-binding domain (DBD), the BrMYB28.3 paralog had a putative transmembrane domain between the amino acid region 332 and 354. BrMYB29.2 possessed only a single HTH motif along with an MYB-like domain and did not contain the regulatory protein–protein interaction domain (Fig. 2).
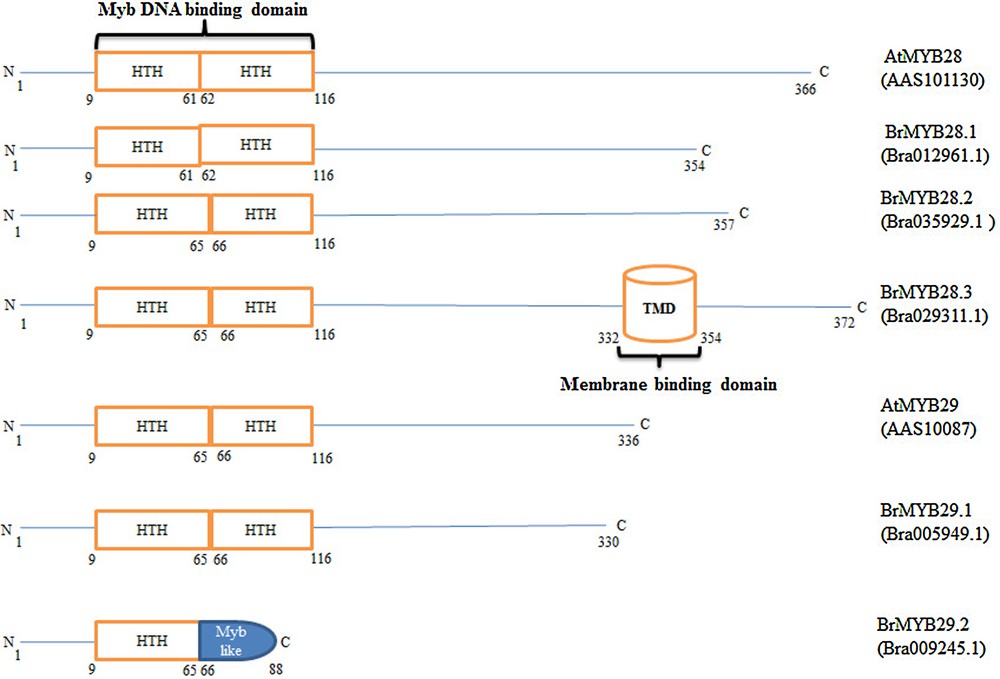
Schematic structural representation of Brassica rapa ssp. pekinensis R2R3 MYB28 and 29 paralogous proteins and Arabidopsis thaliana MYB28 and 29 proteins. All paralogs of B. rapa MYB28 and 29, except B. rapa MYB29.3, contain MYB DNA-binding domains (HTH). A transmembrane domain (TMD) is also observed in the C-terminal region of B. rapa MYB28.3 along with a DNA-binding domain. The positions of the amino acids are indicated numerically. The Genbank accession numbers are as follows: B. rapa ssp. pekinensis: BrMYB28.1 (Bra012961.1), BrMYB28.2 (Bra035929.1), BrMYB28.3 (Bra029311.1), BrMYB29.1 (Bra005949.1) and BrMYB29.2 (Bra009245.1). Arabidopsis thaliana: AtMYB28 (AAS10113), AtMYB29 (AAS10087).
3.2 Protein homology between paralogs of BrMYB28, 29 and AtMYB28, 29
The multiple sequence alignment results revealed that B. rapa MYB28 paralogs, such as BrMYB28.1, 28.2, and 28.3 showed 83, 80, and 73% sequence identity, respectively, with AtMYB28 (AAS10113), at the protein level. Similarly, Brassica MYB29 paralogs, such as BrMYB29.1, 29.2, and 29.3 amino acid sequences showed 73, 98, and 72% sequence identity, respectively, with AtMYB29 (AAS10087) (Fig. S1). Furthermore, in the phylogenetic analysis, all the IGSL and AGSL regulators had formed distinct clusters (Fig. S2).
3.3 Analysis of putative cis regulatory motif distributions
The analysis of the putative regulatory motifs from the promoter region of the DSGs from the key steps of the AGSL biosynthetic pathway, such as MAM1, MAM3 (chain elongation), CYP79F1, CYP83A1, SUR1, ST5C (core structure formation), FMO-GSOX2, and FMO-GSOX5 (secondary side chain modification) was carried out to analyze the motifs related to the binding sites of the MYB TF genes, and also the stress-specific and tissue-specific gene expressions. Similarly, putative regulatory motifs from the promoter regions of Brassica MYB TF genes (BrMYB28 and BrMYB29 paralogs) and Arabidopsis MYB28, 29 genes were also analyzed, to find the motifs related to the stress-specific and tissue-specific gene expressions. The proximal promoters (–101 to –1000) function as a docking site for TFs, and therefore, we selected the promoter region covering –1 to –1500 bp from the translational start site of the DSGs and all paralogs of the BrMYB28, 29 genes as well as the Arabidopsis MYB28, 29 genes for in silico cis motif analysis. The results showed the presence of highly conserved regulatory elements, such as, CCAAT, CGCG, TATA boxes, sulfur-response motifs, binding sites for MYB, as well as motifs related to abiotic and biotic stresses, hormones, glucose signalling, circadian, and organ-specific. Duplication frequency of putative cis regulatory motifs and their functions in the UPS region of BrMYB28, 29 paralogs and their DSGs as well as the Arabidopsis MYB28, 29 genes are presented in the supplementary table (Tables S1 and S2). The respective positions of the important putative cis motif clusters in the 5′ UPS region of BrMYB28, 29 paralogs and their DSGs are shown in the figure (Fig. S3a and S3b).
3.4 Differential expression of the BrMYB genes
In order to monitor the quantitative gene expression variation of BrMYB28 and BrMYB29 paralogous genes, qRT-PCR was performed at different developmental stages of B. rapa. The sequence similarities between the amplicons of BrMYBs were shown in Fig. S4. The gene expression data showed a profound variation in tissues of different developmental stages, such as seedlings, one-month-old root, shoot, and leaf, and two-month-old roots, inner leaves and outer leaves. BrMYB28.3 gene expression was ubiquitous and exceptionally high in all the stages compared to other closely related BrMYB genes, such as BrMYB28.1 and BrMYB28.2 genes (Fig. 3a). Among the different developmental stages, gene expression of all three paralogs of BrMYB28 was exclusively higher in the two-month-old outer leaf tissues. Similarly, among the BrMYB29 paralogs (BrMYB29.1 and BrMYB29.3) studied, BrMYB29.1 showed high transcript abundance in all the developmental stages compared to BrMYB29.3 (Fig. 3b). Both BrMYB29 paralogs were preferentially expressed in two-month-old root samples as well as in two-month-old outer leaf tissues.
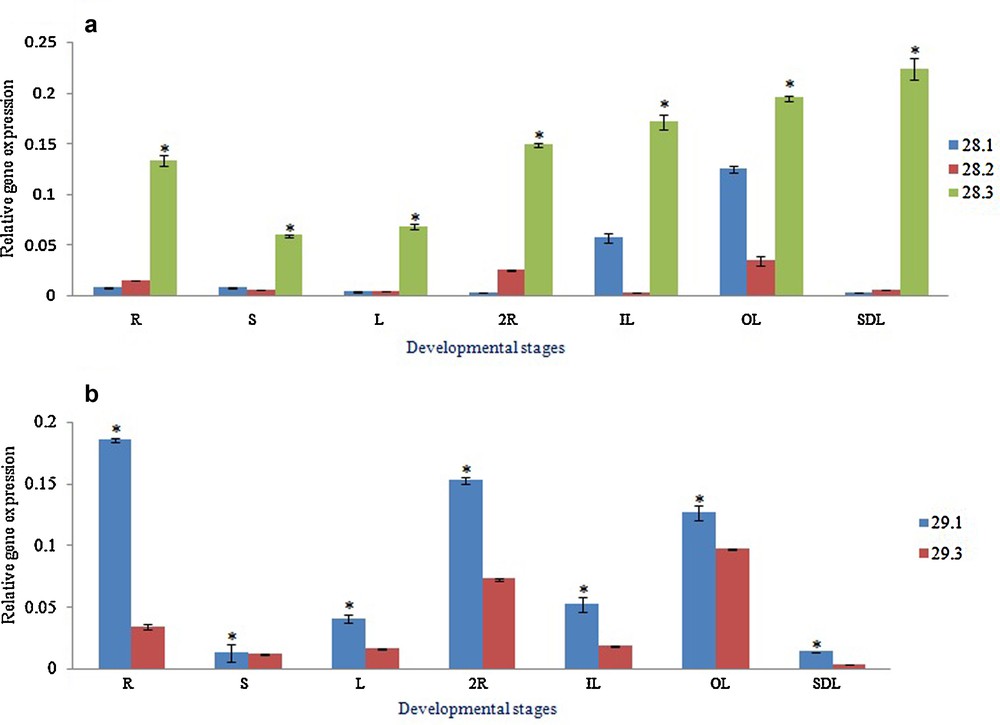
(Color online.) Quantitative real-time gene expression analysis of Brassica rapa. ssp. pekinensis: (a) MYB28 paralogous genes, 28.1 (Bra012961), 28.2 (Bra035929), 28.3 (Bra029311); (b) MYB29 paralogs, 29.1 (Bra005949) and 29.3 (FJ584292) in different developmental stages; one-month-old root (R), one-month-old shoot (S), one-month-old leaf (L), two-month-old roots (2R), two-month-old inner leaves (IL), two-month-old outer leaves (OL), five-day-old seedling (SDL) of B. rapa ssp. pekinensis. Values indicate mean ± SE of three replicates per treatment. The asterisk indicates significant differences (P ≤ 0.05) in relative gene expression between the different stages after Tukey's test (n = 3).
3.5 Differential expression profiles of AGSL biosynthesis genes
The gene expression variations of B. rapa AGSL biosynthetic pathway genes (paralogs that are closely related to the Arabidopsis AGSL biosynthetic genes based on sequence homology) was analyzed at different developmental stages of B. rapa. The gene expression profile for the following AGSL biosynthetic genes, such as MAM1, MAM3, CYP79F1, CYP83A1, SUR1, ST5C, FMO-GSOX2, and FMO-GSOX5 was analyzed in all the selected developmental stages (SDL, 1R, S, L, 2R, IL and OL) through qRT-PCR (Fig. 4). Among the genes studied, ST5C, CYP83A1, and MAM3 showed a ubiquitous and strong expression, irrespective of the developmental stages. However, the expressions of MAM1, CYP79F1, SUR1, FMO-GSOX2 and FMO-GSOX5 genes revealed to be of low level (Fig. 4). Most of the AGSL biosynthetic pathway genes were also found to be highly expressed in two-month-old outer leaf tissues as well as in the seedling stages.
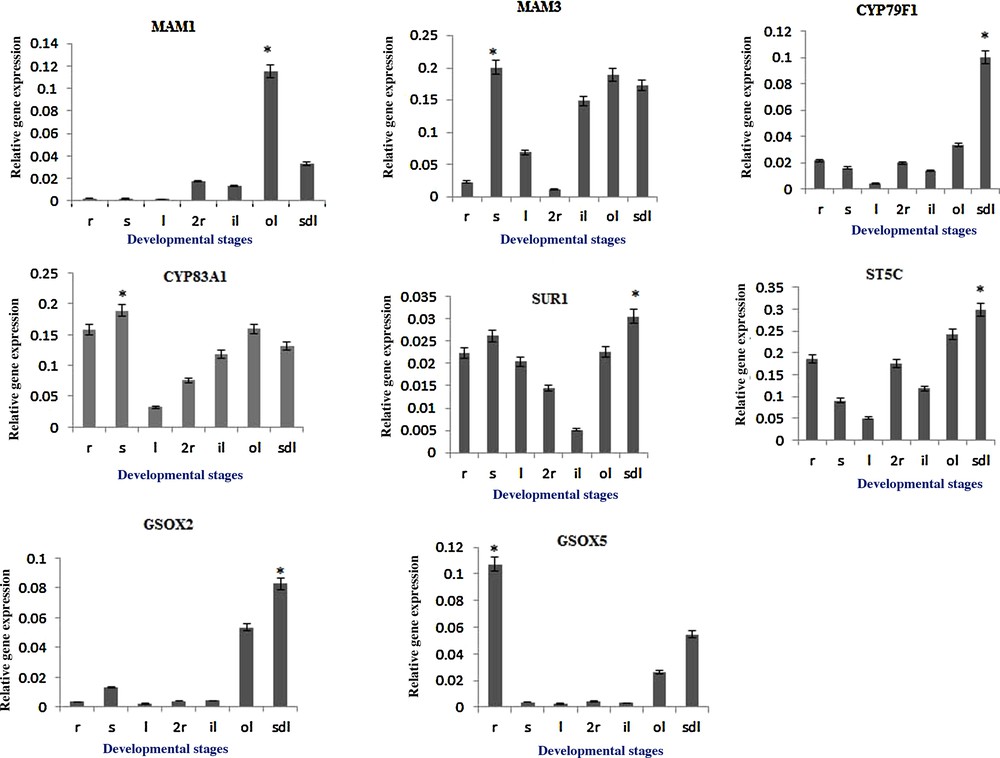
Relative gene expression of aliphatic glucosinolate biosynthesis genes, MAM1 (Bra013007), MAM3 (Bra013009), CYP79F1 (Bra026058), CYP83A1 (Bra032734), SUR1 (Bra036490), ST5C (Bra025668), GSOX2 (FMO-GSOX2, Bra027035) and GSOX5 (FMO-GSOX5, Bra026988) in different developmental stages; one-month-old root (R), one-month-old shoot (S), one-month-old leaf (L), two-month-old roots (2R), two-month-old inner leaves (IL), two-month-old outer leaves (OL), five-day-old seedling (SDL) of B. rapa ssp. pekinensis. Values indicate mean ± SE of three replicates per treatment. The asterisk indicates significant differences (P ≤ 0.05) in relative gene expression between the different stages after Tukey's test (n = 3).
3.6 Differential expression of BrMYBs in response to various stresses
The stress-specific gene expression variations of BrMYBs were analyzed under various stress treatments (Fig. 5a and b). Among the various stresses given to plants, a strong induction in most of the BrMYBs was observed in the wounding, IAA, and low- and high-temperature treatments. To investigate the possible involvement of MYB TF genes in phytohormone-mediated signaling in B. rapa ssp. pekinensis, we examined the expression of BrMYB genes in response to IAA, GA, ABA, MeJA and SA treatments. The phytohormone IAA strongly induced the upregulation of all BrMYBs in the aerial parts, whereas the expression of all genes was found to be the least in the root tissues. In contrast, GA triggered the expression of BrMYB29.1 in root tissues and BrMyb28.3 in shoot tissues (Fig. 5a). In the aerial parts of Brassica, MeJA induced the upregulation of BrMYB29.3 and BrMYB28.1 genes, whereas in the roots it induced the upregulation of BrMYB28.3 and BrMYB29.1 genes. In response to SA treatment, the expression of most of the BrMYBs increased significantly in roots except BrMYB29.3 gene. However, there were some differences in the ABA-induced expression levels of BrMYBs. The ABA-induced expression of BrMYB28.2 and BrMYB29.1 was observed in shoot and root tissues, respectively, and the expression of BrMYB28.1 was least expressed among the BrMYBs, in response to the ABA treatment (Fig. 5a).
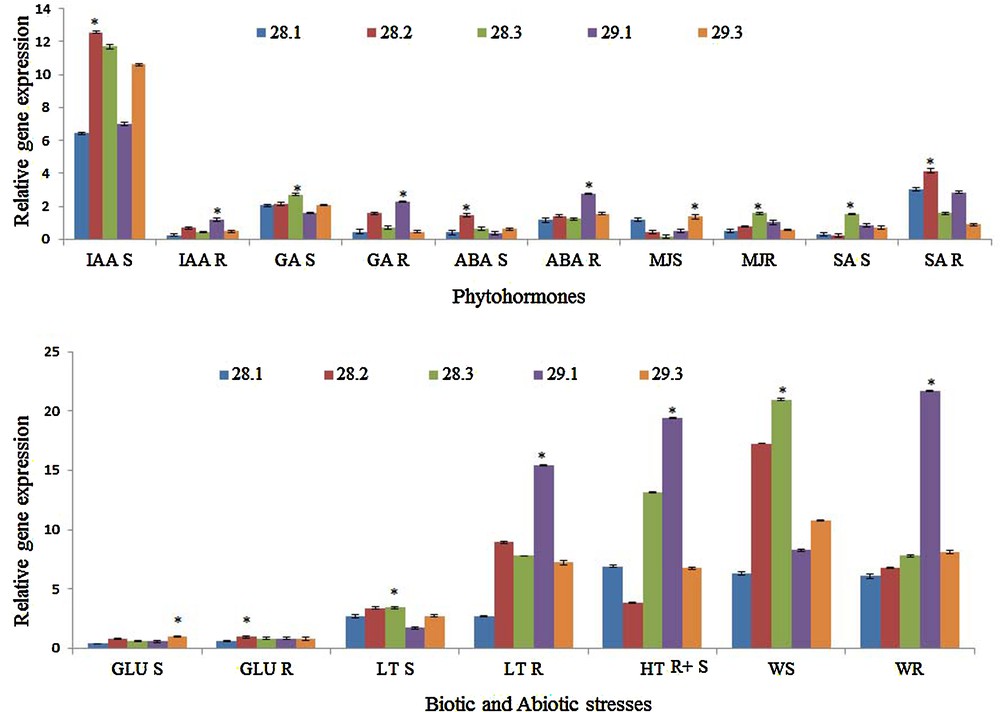
(Color online.) a: quantitative real-time gene expression of B. rapa ssp. pekinensis MYB28, 29 paralogous genes, 28.1 (Bra012961), 28.2 (Bra035929), 28.3 (Bra029311), 29.1 (Bra005949) and 29.3 (FJ584292) in Shoots (S) and roots (R) of B. rapa. ssp. pekinensis in response to various phytohormones, such as indole-3-acetic acid (IAA), gibberellic acid (GA), abscisic acid (ABA), methyl jasmonate (MJ), salicylic acid (SA); b: differential gene expression analysis of B. rapa ssp. pekinensis MYB 28, 29 paralogous genes, 28.1 (Bra012961), 28.2 (Bra035929), 28.3 (Bra029311), 29.1 (Bra005949) and 29.3 (FJ584292) in shoots (S), roots (R) and root and shoot (R + S) of B. rapa ssp. pekinensis in response to biotic and abiotic stresses such as glucose (Glu), low-temperature (LT), high-temperature (HT) and wound stresses (W). Values indicate mean ± SE of three replicates per treatment. Asterisk indicates significant differences (P ≤ 0.05) in relative gene expression between the different stages after Tukey's test (n = 3).
Similarly, the other abiotic stresses (glucose, high- and low-temperature treatments) and biotic stress (wound stress) specific gene expressions of BrMYBs in B. rapa were analyzed by qRT-PCR. Glucose treatment slightly induced upregulation of the BrMYB28.2 paralog in both root and shoot tissues, while it slightly induced the BrMYB29.3 paralog in the shoot tissues. Low-temperature treatment induced the expression of most of the BrMYBs in the root tissues, whereas the expression of BrMYB28.1 was the lowest in this treatment. Among the BrMYB paralogs, BrMYB29.1 and BrMYB28.3 genes showed the strongest expression, whereas the BrMYB28.2 gene revealed the least expression in plants treated at high temperature (Fig. 5b). BrMYB paralogs, such as BrMYB28.2, 28.3, and BrMYB29.1 genes were predominantly expressed in wound stressed plants. In contrast, BrMYB28.1 showed least expression in shoots as well as in root tissues of wound stressed plants (Fig. 5b).
4 Discussion
Glucosinolates play important roles in plant–pathogen interactions and some of the glucosinolate-derived compounds are known to have potential human health benefits [2]. Although rapid progress has been made in recent years, there are many uncertainties and gaps in our knowledge of glucosinolate regulation in the Brassica crops. To better understand glucosinolate biosynthesis, in the present study, we have conducted a differential gene expression analysis of the GSL biosynthesis genes and their transcription factors, such as the BrMYB28 and BrMYB29 genes in B. rapa. B. rapa being a polyploid, it contains multiple copies of the same genes. Several paralogs of BrMYB28 and BrMYB29 TF genes involved in the regulation of AGSL biosynthesis have been reported previously in B. rapa ssp. pekinensis [11]. In agreement with this, we identified three paralogs for each of the BrMYB28 (BrMYB28.1, 28.2, and BrMYB28.3) and BrMYB29 (BrMYB29.1, 29.2, and BrMYB29.3) TF genes.
Bioinformatics analysis of the putative structural domain organizations of BrMYBs showed the presence of DBD in all BrMYBs, except in BrMYB29.3. Although BrMYB29.3 lacks the DBD, it may play an important role, similar to that of other BrMYBs, as is evident from its homology to AtMYB29 and varied gene expressions, similar to other paralogs of BrMYBs, in different developmental stages as well as under different stress treatments (Figs. 3b, 5a and b). Furthermore, TF proteins that lack DBD are also considered as TFs if they are able to interact with the DNA-binding protein, which in turn forms a transcriptional complex [19]. However, direct experimental evidences, such as reporter gene assays, DNA footprinting, and/or gel shift assays can only confirm their role. Similar to membrane-tethered transcription factors (MTTFs), BrMYB28.3 shows the presence of a putative TMD in addition to DBD, which indicates that this gene may act as an MTTF. MTTFs are a group of membrane-bound inactive transcription factors, activated by a proteolytic cleavage that leads to the release of a TF domain from the membrane-bound TMD [20].
In order to investigate the differential gene expression of BrMYB28 and BrMYB29 paralogous genes, we performed a gene expression analysis at different developmental stages and under different stress treatments. The qRT-PCR analysis of the Brassica MYB paralogs revealed that the BrMYB28.3 and BrMYB29.1 paralogs were dominantly expressed in most of the developmental stages, when compared with the others. These results were consistent with the previous report, which stated that the TF paralogs with a similar DBD could exhibit diversity in gene expression levels [21]. All paralogs of BrMYB28 showed an elevated expression level in the two-month-old matured OL and the lowest in the SDL stage. BrMYB29.1 and BrMYB29.3 showed a preferential expression in two-month-old root tissues followed by two-month-old matured OL tissues. The observed result was consistent with the previous study, which showed that MYB28 and MYB29 gene expressions were elevated in mature, young rosette leaves and trichomes, but no expression was detected in the seedling stage [10]. Recently, Kim et al. [22] studied the expression pattern of transcription factors involved in the regulation of glucosinolates in different stages of Chinese cabbage under light and dark conditions. They suggested that light favours the expression of glucosinolate transcription factors when compared to dark. Sonderby et al. [8] observed the upregulation of the transcript levels of AGSL biosynthetic genes in foliar tissues, via microarray analysis. Consistent with this, in the present investigation, most of the DSGs showed a prominent expression in the two-month-old OL and SDL tissues. Previous reports suggested a correlation between the gene expressions of MYB28 and AGSL biosynthetic genes in Arabidopsis [9]. A similar correlation in gene expression, between BrMYBs and AGSL biosynthesis genes, was also observed in the B. rapa ssp. pekinensis.
Yanhui et al. [23] reported that GA, jasmonic acid (JA), and SA were shown to induce MYB28 and MYB29 genes in Arabidopsis by using northern gene expression analysis. Similarly, in the present investigation, both paralogs of BrMYB28 and BrMYB29 genes were induced by GA, MeJA and SA treatments in B. rapa. Moreover, in Arabidopsis, IAA and ABA treatments were shown to induce MYB28 gene, whereas, they did not affect the expression of the MYB29 gene [23]. However, in the present study, both paralogs of BrMYB28 and BrMYB29 genes were upregulated under IAA and ABA treatments. Furthermore, the presence of a high frequency of putative ABA-related and auxin-related regulatory motifs found in the regulatory regions of BrMYBs indicated that they also might be responsible for the induction of BrMYBs in response to ABA and IAA treatments.
In B. rapa ssp. rapa, high-temperature stress was reported to increase the GSL level as well as the gene expression of some of the BrMYB genes that regulated GSL biosynthesis [24]. Kim et al. [25] demonstrated that the correlation between the expression of MYB28 and the level of aliphatic glucosinolates in Chinese cabbage. Similarly, Justen [26] observed the correlation between the transcripts of BrMYB28 and significant increase of AGSLs, such as gluconapin and glucobrassicanapin in the tissues of high-temperature grown B. rapa subsp. rapa. Consistent with these reports, in the present study, the observed induction of BrMYBs under high-temperature treatment might be regulating the accumulation of AGSLs in B. rapa ssp pekinensis. In low-temperature stress treatment, the transcript levels of BrMYBs were slightly lower compared to those under high-temperature treatment. Similar results were observed by Justen and Fritz [24], who reported that the total GSL and transcript levels of BrMYBs were lower at low temperature, as compared to high temperature. It has been demonstrated that glucose activates the MYB28, MYB29, MYB34, MYB51, and CYP79F1, CYP79F2 genes in Arabidopsis [27,28], and furthermore, the MYB28 gene was characterized by the presence of glucose-regulated motifs in its promoter region [29]. Consistently with this report, in the present investigation, we observed that some of the paralogs of BrMYBs were upregulated by glucose treatment. Moreover, the present in silico analysis also showed the presence of putative glucose signalling related motifs in the regulatory regions of BrMYBs. The gene expression results showed that the BrMYB28 and BrMYB29 paralogs showed stronger induction in the tissues of wound stress-induced plants. The observed results also coincided with the previous reports, which stated that there was rapid induction of MYB28 and MYB29 gene expression in wound stress-treated plants [9,10]. Furthermore, putative regulatory cis motif analysis of the promoter regions of the BrMYBs indicated the presence of several motifs related to abiotic and biotic stresses, which were probably responsible for their stress-specific differential gene expression.
We also observed the different types of general putative MYB binding site motifs in the regulatory regions of BrMYBs and their DSGs. A recent study showed that MYC TFs could activate the GSL biosynthesis genes directly, as well as their interaction with the GSL-related MYB TF genes resulted in a change in the GSL level. MYC TFs could bind to the G-box motifs (CACGTG) found in the promoter regions of their target gene [30]. Putative G-box binding motifs were observed in the UPS region of BrMYB28.2, 29.1 and BrMYB29.2 genes, as well as the DSGs, such as MAM3, CYP79F1, and SUR1, which indicated that these genes might be regulated by MYC TFs (Tables S2 and S3). Taken together, the results of the in silico and expression analyses indicated that the dominantly expressed BrMYBs (BrMYB28.3 and BrMYB29.1) in both different developmental stages and in various stresses might be the putative right choice for plant GSL metabolic engineering studies, and furthermore their strong stress responsiveness suggested their possible role in stress tolerance. In addition, a detailed molecular characterization, using genetic engineering, is needed to articulate their role in stress tolerance.
Acknowledgements
This paper resulted from the Konkuk University research support program.