

1 Introduction
The polar bear (Ursus maritimus) lives in the Arctic Circle (Fig. 1), where its preferred habitat is the sea ice along the coast and on the islands [1]. Like other mammals of the region, such as the Arctic fox (Vulpes lagopus) and Arctic hare (Lepus arcticus), the polar bear developed adaptations specific to sea ice habitat that distinguish it from its congeners living in milder conditions, the brown bear (U. arctos) and black bear (U. americanus). A thick layer of fat covered with dense white fur ensures thermal insulation and a perfect camouflage. The ears are small, which reduce heat loss due to cold temperatures. But unlike most other Arctic taxa, which have a more compact body compared to closely related species of lower latitudes (Allen's rule), the polar bear is more slender in appearance, with a longer neck and a smaller head [2]. These physical characteristics have probably been selected because of its semi-aquatic life, its survival greatly depending on its ability to hunt both on land and in water.
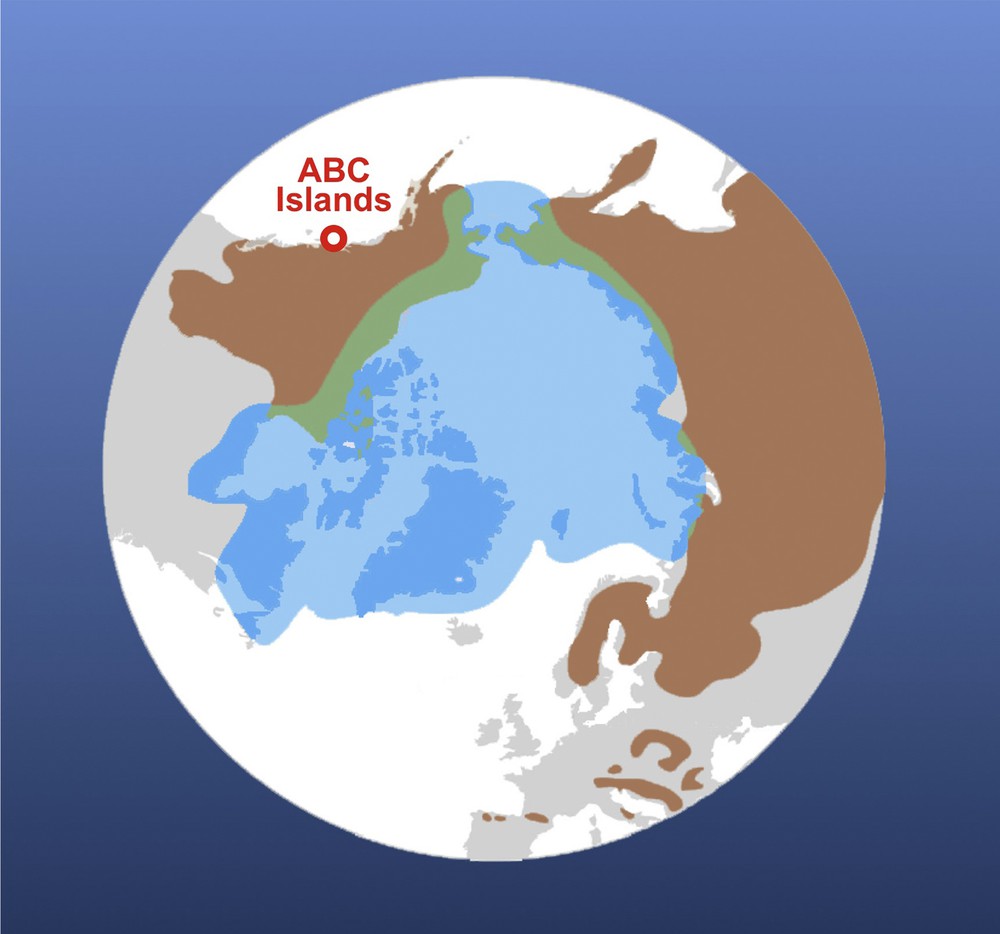
Current geographical distributions of the polar bear (Ursus maritimus, in light blue) and brown bear (Ursus arctos, in brown). The map was modified from Liu et al. [20]. The position of ABC islands is indicated in red. Both species can meet occasionally on the lands of the Arctic Circle, in western North America and eastern Siberia (in green). For interpretation of the references to colour in this figure caption, please see the online version of this article.
The first molecular phylogenies, based on mitochondrial DNA (mtDNA) sequences of the cytochrome b gene, indicated that U. maritimus and U. arctos are closely related species [3,4], in agreement with the paleontological scenario of Kurtén [5] and subsequent phylogenetic studies of the family Ursidae based on nuclear data [6–8]. However, the mitochondrial paraphyly of U. arctos, due to the inclusive position of U. maritimus (Fig. 2), had suggested from the beginning that the mitochondrial phylogeny does not reflect the true species tree [3]. Indeed, the mtDNA sequences of brown bears of Alaska's ABC Islands (Admiralty, Baranof and Chichagof; see Fig. 1) were found to be very similar to those of polar bears, but more distant from those of other brown bears [3,4,9,10].
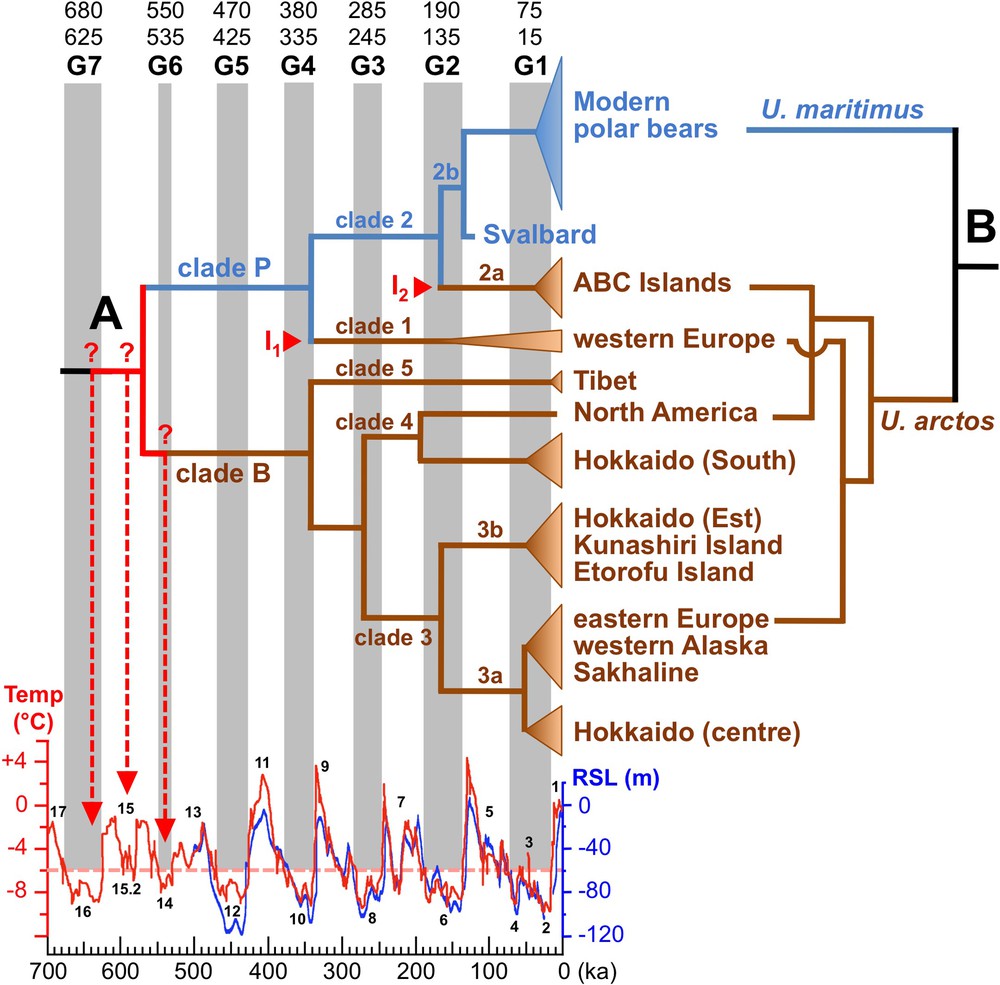
Mitochondrial and nuclear phylogenies of bears. A. The Bayesian chronogram at the left was modified from Hirata et al. [10]. It was constructed from an alignment of complete mitochondrial genomes, and divergence times were estimated using the sequence of two fossils, a cave bear (Ursus spelaeus; not shown in the tree) [22] and the oldest polar bear fossil, a specimen from Svalbard, dated at 120 ± 10 ka [9]. Conventional naming for bear mtDNA haplogroups is indicated on the tree (clades 1–5). Mitochondrial introgressions (I1 and I2) from female polar bears into brown bear populations are indicated in red. The seven major glacial periods since 700 ka, here named G1–G7, were interpreted from Antarctic temperature data (in red) [25] and Red Sea radiometrically dated relative sea-level (RSL) records (in blue) [26]; their lower and upper bounds were defined at 6 °C below late Holocene temperature (dashed pink line). Marine isotope stages (MIS) 1 to 17 are written in black. B. The nuclear tree at the right is from Hailer et al. [14]. For interpretation of the references to colour in this figure caption, please see the online version of this article.
In the past five years, multiple molecular studies have led to various interpretations of the evolutionary origin of the polar bear. In this paper, all recent hypotheses are reviewed, and a new scenario is proposed in the light of available molecular and eco-ethological data.
2 Molecular hypotheses proposed for the origin of the polar bear
2.1 A rapid and recent morphological differentiation of U. maritimus?
In 2010, Lindqvist et al. [9] sequenced the mitochondrial genome of the oldest polar bear fossil, which is a specimen from the Svalbard Archipelago in Norway, dated at 120 ± 10 ka. Using this ancient sequence, it was possible to date more precisely the timing of divergence events in the mitochondrial tree of bears [9–11] (Fig. 2, tree A): 566–490 ka for the most recent common ancestor (MRCA) of brown and polar bears, 336–310 ka for the MRCA of “clade P”, which is composed of clade 1 (brown bears from western Europe) and clade 2 (polar bears and ABC islands brown bears); 162–152 ka for the MRCA of clade 2; 136–134 ka for the divergence between the fossil from Svalbard and modern polar bears (clade 2b); and 52–41 ka for the MRCA of extant polar bears. Based on these time estimates, Lindqvist et al. [9] concluded that there was a rapid and recent morphological differentiation of the polar bear, i.e., between 162–152 and 136–134 ka. Davison et al. [12] agreed with such a recent split between U. maritimus and U. arctos, and made the ad hoc assumption that the mitochondrial paraphyly of U. arctos is due to incomplete lineage sorting of mtDNA haplotypes. However, all these assumptions imply that the mitochondrial history was not too much distorted by possible hybridization events between polar and brown bears.
2.2 An Irish origin for the modern polar bear matriline?
In 2011, Edwards et al. [13] sequenced a D-loop fragment of ∼ 300 bp in brown and polar bears from various geographic and temporal origins (38 ka to present). Their analyses suggested that the haplogroup of modern polar bears was more closely related to extinct Irish brown bears than to the polar bear fossil from Svalbard. Based on the apparent paraphyly of extinct Irish brown bears with respect to modern polar bears, Edwards et al. [13] concluded in favour of the hypothesis involving mitochondrial introgression from Irish brown bears into polar bears. However, this conclusion was poorly supported by the data, as most nodes of the tree, including the paraphyletic pattern, were found to be unstable (posterior probability < 0.85). Such a lack of robustness can be explained by three factors: (1) an alignment of very short DNA sequences (< 300 bp) containing only a few informative sites; (2) a large amount of missing data (the alignment of the 22 prehistoric sequences, accession numbers JF900154–JF900175, contains 32% of missing nucleotides on the total length of 295 bp); (3) a significant proportion of exclusive autapomorphies, which might suggest sequencing errors in the dataset.
Apparently, the only robust node was the one grouping modern and extinct polar bears, modern ABC islands brown bears, and extinct Irish brown bears.
2.3 Are polar bears an old and distinct bear lineage, which has been recently introgressed by ABC islands brown bears?
In 2012, Hailer et al. [14] sequenced 14 independent introns (representing an alignment of 9116 nucleotides) in 45 individuals of polar, brown, and black bears. They found that brown bears constitute a monophyletic group that separated from polar bears at 603 ka (Fig. 2, tree B). This study was strongly criticized by Nakagome et al. [15,16], who revealed that, of the 13 loci analysed (one locus, LRGUK, was excluded due to high frequencies of recombinants), only one supported the monophyly of brown bears, four did not provide any resolution, and eight were in agreement with the paraphyly of brown bears. To this, Hailer et al. [17] responded that the analyses of Nakagome et al. [15,16] did not take into account the effect of incomplete lineage sorting, as well as the possible impact of gene flow (introgression) between polar and brown bears.
Miller et al. [11] performed deep sequencing of the nuclear genome for five bears, including one polar bear, three brown bears from Alaska (two from ABC islands and one from Kenai Peninsula), and one black bear. They also sequenced about 12 million nuclear genome SNP markers in 22 additional modern polar bears and the fossil from Svalbard. The results supported the monophyly of both U. maritimus and U. arctos; the Svalbard's fossil being divergent from extant polar bears. Miller et al. [11] estimated that polar bears diverged from brown bears between 4 and 5 million years (Ma), which is much older than all other studies (Table 1).
Molecular estimates of divergence time for the most recent common ancestor of brown and polar bears.
| Publication | Data | Methods | Parameters | Node age (ka) |
| Lindqvist et al. [9] | mtDNA genome 15 taxa Alignment length? |
BEAST v1.4.8 [42] | Three dated fossils: polar bear (120 ka) cave bears (31.87 ka and 44 ka) |
490 (620–360) |
| Hailer et al. [14] | 14 nuclear markers 45 taxa 9116 bp |
BEAST v1.6.2 [42] | d (giant panda) = 12 ± 4 Ma | 603 (934–338) |
| Miller et al. [11] | Nuclear genome 4 bears |
Pairwise sequentially Markovian Coalescent model [43] | g = 10 years and μ = 10−9/year/site |
5000–4000 |
| Hirata et al. [10] | mtDNA genome 74 taxa 15,863 bp |
BEAST v1.6.2 [42] | Two dated fossils: polar bear (120 ka) cave bear (31.87 ka) |
566 (944–251) |
| Bidon et al. [19] | Y chromosome 33 taxa 5197 bp |
BEAST v1.7.4 [42] | A. d (American black bear) = 0.95 ± 0.095 Ma | 770 (940–560) |
| B. d (spectacled bear) = 6 ± 0.6 Ma | 1120 (1610–730) | |||
| C. d (spectacled bear) = 10.91 ± 1.091 Ma | 2060 (2950–1310) | |||
| D. g = 10 years and μ = 3 × 10−9/year/site |
430 (610–290) | |||
| Kutschera et al. [24] | 15 nuclear markers Number of taxa? 13,898 bp |
*BEAST [44] | d (giant panda) ≥ 11.6 Ma and g = 7.2 years |
620 (890–380) |
| Liu et al. [20] | Nuclear genomes 79 polar bears 10 brown bears |
Identity by state [45] | d (giant panda) ≥ 14.1 Ma and g = 11.35 years |
479–343 |
Hailer et al. [14] and Miller et al. [11] both suggested that the mtDNA of polar bears was replaced by that of ABC islands brown bears at around 160 ka. In addition, Miller et al. [11] estimated that 5–10% of the nuclear genome of ABC islands brown bears is most closely related to polar bears, indicating ancient admixture between the two species.
2.4 Introgression of polar bears by male brown bears on the ABC islands?
In 2013, Cahill et al. [18] also performed genomic analyses, based on seven polar bears, two brown bears (from mainland Alaska and ABC islands), and one black bear. Simulated admixture revealed that ABC islands brown bears have polar bear sequences, up to 1% for the autosomal genome and 6.5% for the X chromosome. The authors proposed that ABC islands brown bears emerged from an isolated population of polar bears that has been gradually converted into brown bears via male-mediated dispersal and sex-biased gene flow.
By analysing Y-chromosomal datasets of 3.1 and 5.3 kb for at least 45 brown bears, 15 polar bears and 4 black bears, Bidon et al. [19] found three distant species-specific groups of haplotypes. The lack of Y-chromosomal haplotype shared among species supports the absence of recent Y-chromosomal introgression between brown and polar bears. Based on the scenario of Cahill et al. [18], which implies an initial polar bear ancestry on the ABC islands followed by extensive male-biased immigration of mainland brown bears, Bidon et al. [19] proposed that the absence of Y chromosome of polar bear on the ABC islands indicates a complete replacement of the original polar bear Y chromosome.
2.5 Gene flow from polar bears into North American brown bears?
Most previous molecular studies have postulated that introgression occurred from brown bears into polar bears. This is only recently that Liu et al. [20] made the opposite assumption, that is to say, the one involving introgression from polar bears into brown bears. They came to this conclusion after sequencing the complete nuclear genome in 89 bears, including 79 polar bears and 10 brown bears from North America and Fennoscandia (Sweden and Finland). Based on the identity by state (IBS) tract method, they found that admixture took place between 319 ka and 148 ka from polar bears into North American brown bears, the strongest evidence of gene flow being found with ABC islands brown bears. They also estimated that polar bears diverged from brown bears at 479–343 ka, and suggested that the divergence between polar and brown bears coincides with MIS 11, a long warm period, which spanned 424–374 ka, characterized by a substantial decrease in Greenland ice sheet volume.
3 Interpreting bear evolutionary history: what can be inferred from previous molecular studies?
3.1 Brown and polar bears evolved during glacial periods of the Pleistocene
Molecular clocks can be more accurately calibrated using temporal information from ancient DNA sequences, because they allow for a direct estimation of substitution rates. In a recent study using 320 modern and 26 ancient human mtDNA genomes, timescale estimates based on individual dated tips (tip calibration) were shown to be more accurate and reliable than those based on internal node calibration [21]. In the three recent mtDNA studies on bears [9–11], divergence times were calculated using the complete mitochondrial genome from at least two fossil specimens, including the polar bear from Svalbard dated at 120 ± 10 ka [9], and one or two cave bears (U. spelaeus) dated at 31.87 ka [22] and 44 ka [23]. Although based on different taxa samplings, these studies have provided very similar divergence time estimates: 566–490 ka for the MRCA of brown and polar bears; 336–310 ka for clade P; 162–152 ka for clade 2; 136–134 ka for clade 2b; and 52–41 ka for the MRCA of modern polar bears. The accuracy of divergence times estimated on the basis of mitochondrial genomes relies on three other arguments. Firstly, the age inferred for the MRCA of brown and polar bears (566–490 ka) fits well with the fossil record, as Kurtén [5] suggested that polar bears branched off from brown bears during the early Middle Pleistocene. Secondly, similar date estimates were found using three different nuclear datasets: 603 ka with 14 autosomal introns [14]; 620 ka with a concatenation of autosomal and Y-chromosomal datasets [24]; and 479 ka with complete nuclear genomes [20] (Table 1). Thirdly, when compared with records on temperature and sea-level variations over the last 700 ka [25–27], it is astonishing to note that all divergence times estimated by Hirata et al. [10] on the basis of mitochondrial genomes can be linked to glacial periods of the Pleistocene (Fig. 2A). Such correlations were expected because glacial and interglacial cycles of the Pleistocene have had profound impacts on the geographic distribution of both brown and polar bears. During glacials, most populations of U. arctos, which is a temperate-adapted species, moved to southern refugia of the Northern Hemisphere, although some populations may have been isolated in a few northern refugia, such as those located on the ABC islands [28] and Ireland [29] during the Last Glacial Maximum. In parallel, populations of U. maritimus, which is a cold-adapted species, occupied a wider southern distribution, due to the extension of the sea ice and polar climate, from the Arctic Circle to the South. By contrast, during interglacials, the polar bear was isolated to its Arctic refugia, as it is today, whereas populations of brown bears were more prone to move towards northern latitudes.
3.2 When did polar and brown bears split?
All recent nuclear studies agree to support mutual monophyly of the two species U. arctos and U. maritimus [11,14,19,24], but they have yielded a wide range of divergence time estimates for the split between polar and brown bears, i.e., ranging from 343 ka to 5 Ma (Table 1). The results were found to be highly dependent on the data and model parameters used as priors, such as the selection of internal node calibration(s) (e.g., the divergence between the giant panda and other bears ranges from 11.6 to 14.1 Ma), the generation time of bears, which ranges from 7.2 to 11.35 years, and the rates of mutation, which were generally extrapolated from human genomic data (10−9/year/site for autosomal sequences, and 3.10−9/year/site for Y-chromosomal sequences). As shown in Bidon et al. [19], the use of different priors on the same Y-chromosomal dataset resulted in a strong variation of mean estimates for the MRCA of polar and brown and bears, i.e., from 430 ka to 2.06 Ma (Table 1). Since calibration point(s), generation time(s), and mutational rate(s) cannot be chosen objectively for inferring divergence times within bears, I consider that nuclear estimates are necessarily less reliable than mitochondrial estimates, which were generated using DNA sequences from fossil specimens (see paragraph 3.1 for details).
Mitochondrial estimates for the time to the MRCA of brown and polar bears range from 566 to 490 ka. In Hirata et al. [10] and Lindqvist et al. [9], the 95% confidence intervals for the ages of this node were, however, much larger than those calculated for more recent divergences within bears: 944–251/620–360 ka versus 545–174/400–240 ka for clade P, and 216–125/177–131 ka for clade 2. Two methodological aspects suggest that the age of the MRCA of brown and polar bears was less reliably estimated than that of more recent nodes. Firstly, only two or three ancient specimens were sequenced to calibrate the molecular clock of bears, and they represent two distant lineages in the tree, i.e., polar bear (clade 2b) and cave bear (U. spelaeus), which was used as an outgroup species. As the common ancestor of brown and polar bears is the most distant node from these two or three dated tips, it is the most sensitive to all types of biases potentially encountered with molecular dating based on tip calibration(s), such as uncertainties associated with tip-dating (radiometric dating and biostratigraphy), sequencing errors in ancient samples, and high deviations from the molecular clock [21,30,31]. Secondly, the mitochondrial estimates for the MRCA of brown and polar bears may have been underestimated because several geographically isolated populations of U. arctos were not included in molecular dating analyses. Indeed, divergent mtDNA haplotypes have been detected in short D-loop or cytochrome b sequences of brown bears currently found in Iran, Pakistan, and Mongolia (Gobi Desert), as well as in extinct populations of North Africa [32–35].
Given that all splitting events within large bears occurred during glacial periods, three hypotheses can be proposed for the age of the MRCA of polar and brown bears (see dashed red arrows in Fig. 2A): (1) the first large ice sheet in the Atlantic during MIS 16 at ∼ 630 ka; (2) the short glacial MIS 15.2 at ∼ 590 ka; (3) glacial MIS 14 at ∼ 540 ka [25,36].
To distinguish among the three hypotheses, it will be necessary to improve the precision of divergence time estimates for the ancestor of polar and brown bears. I recommend, therefore, to perform further molecular dating analyses using an increased taxon sampling, including all divergent mtDNA lineages occurring in Asia, as well as the extinct Atlas bear (U. arctos crowtheri), which is the most interesting taxa, firstly, because it was found to be highly divergent from other populations [34], and secondly, because it can be used as an additional dated tip calibration in order to improve the accuracy of divergence time estimates.
3.3 Evidence for female-mediated gene flow from polar bears into brown bears during glacial periods
As detailed in Section 2, the discordance between mtDNA and nuDNA data has been variously interpreted in recent studies. My point of view is that brown bear populations have been introgressed by female polar bears during glacial periods of the Pleistocene. This scenario was deduced from molecular results published in the past five years, including divergence time estimates [9,10,14,20], data on global climate changes over the Pleistocene [25,26], and evidence from Canadian Arctic that the two bear species can produce viable and fertile hybrids in the wild [37].
Based on the chronogram made by Hirata et al. [10] (Fig. 2, tree A), I propose that two mitochondrial introgression events occurred from U. maritimus into U. arctos: the most recent involved ABC islands brown bears, whereas the most ancient involved brown bears from western Europe. According to this scenario, mtDNA haplogroups can be divided into two deep lineages: the “polar bear” mtDNA lineage (named “clade P”, blue line in tree A of Fig. 2) that includes clades 1 and 2; and the “brown bear” mtDNA lineage (named “clade B”, brown line in tree A of Fig. 2), which contains clades 3, 4 and 5. Molecular dating estimates indicate that both introgression events took place during glacial periods, at 155 ± 5 ka for ABC islands brown bears, and at 340 ± 10 ka for western European brown bears.
In ABC islands brown bears, the Y chromosome is very similar to that found in other populations of U. arctos [19], whereas 6.5% of the X chromosome and 1% of the autosomes are represented by polar bear sequences [18]. Based on genomic analyses, Liu et al. [20] found strong evidence of gene flow from polar bears into ABC islands brown bears. It seems, therefore, that the introgression of mtDNA from U. maritimus was also accompanied by nuclear gene flow in the X chromosome and autosomes. All these data clearly indicate that the brown bear population found on the ABC islands has been introgressed by female polar bears. An extinct mitochondrial subclade 2c, as detected by control region and cytochrome b sequences, has been found in three permafrost-preserved brown bear specimens: one from Yukon (Sixty Mile) dated at 36.5 ka [38]; one from Alaska (Colville River) dated at 48.2 ka [39]; and another one from northeast Siberia (Duvanny Yar, undated) [39]. Since the subclade 2c was found to be equally distant from subclades 2a (ABC islands brown bears) and 2b (polar bears) [39], it can be hypothesized that brown bears from both eastern and western Beringia have been introgressed by female polar bears at the same time than ABC islands brown bears, i.e., at 155 ± 5 ka.
In western European brown bears, the hypothesis involving a similar introgressive hybridization by female polar bears, but this time at 340 ± 10 ka, was interpreted from the mtDNA tree of Fig. 2 (tree A), which shows the existence of two mtDNA lineages, named B and P (for brown bears and polar bears, respectively). This hypothesis is also supported by two other arguments.
Firstly, the recent demographic history analyses of Liu et al. [20] have suggested that nuclear gene flow from U. maritimus to U. arctos occurred several times between 319 and 148 ka. Since the study of Liu et al. [20] included ABC islands brown bears, as well as one brown bear from southern Sweden, which is supposed to have a mtDNA haplotype of clade 1 (western Europe) [40] (Fig. 3), I suggest that the introgression of nuclear genes happened at the same times as that of the mitochondrial genome, i.e., during the two glacial periods corresponding to G4 (at 340 ± 10 ka) and G2 (at 155 ± 5 ka) (Fig. 2A). Indeed, divergence times estimated by Liu et al. [20], on the basis of nuclear genomes, are very similar to those estimated by Hirata et al. [10], on the basis of mitochondrial genomes: 479 vs 566 ka for the MRCA of brown and polar bears; 319 vs 336 ka for the most ancient introgression event; and 148 vs 161 ka for the last introgression event. The time to the MRCA of brown and polar bears inferred from nuDNA genomes is younger than that estimated with mtDNA genomes, probably because Liu et al. [20] used an overestimated value for the generation time of brown and polar bears (11.35 years, whereas previous authors used 10 or 7.2 years [11,19,24]), and/or because they sequenced only a small fraction of the extant biodiversity of brown bears (populations from southern Europe and Asia were not sampled in their analyses).
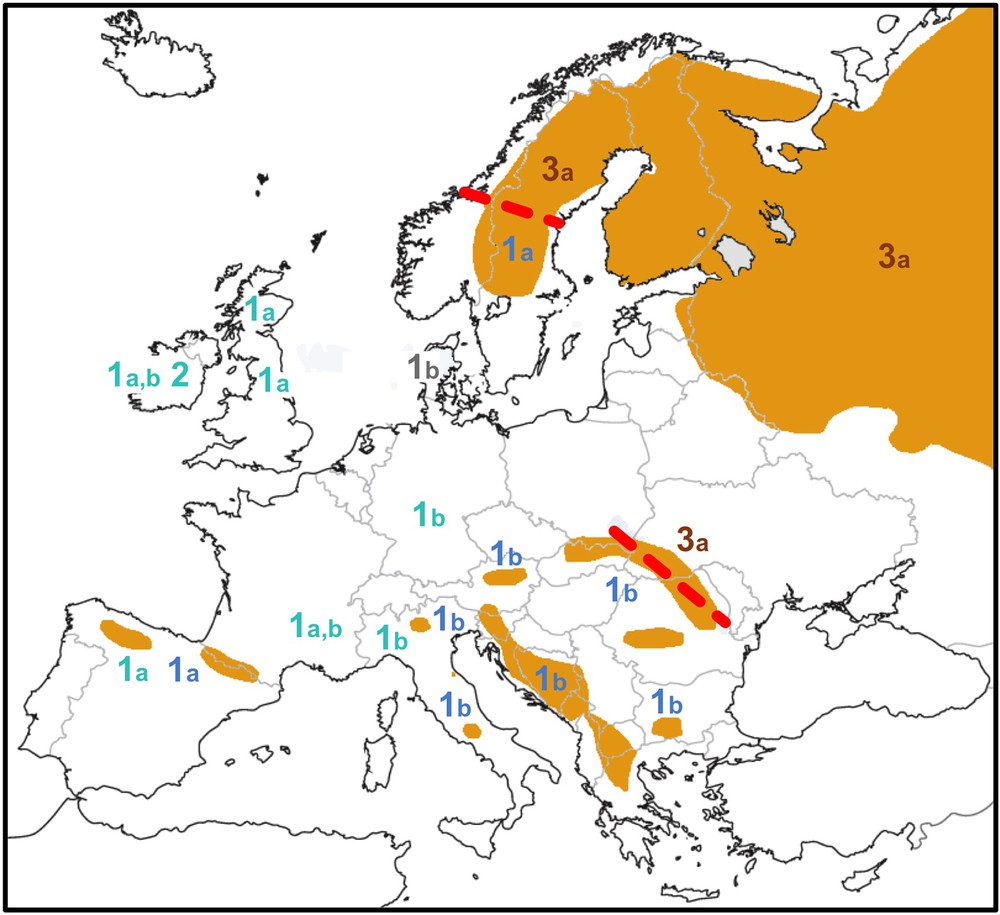
Distribution of brown bear mtDNA haplogroups (1a, 1b, 2, 3a) in Europe. The map was modified from Bray et al. [40]. The distribution of extant brown bear populations is shown in orange. Mitochondrial haplotypes of clades 1 and 2 are indicated in blue (fossil samples < 40 ka old in lighter blue), those of clade 3 in brown. Thick dashed red lines show current contact zones between divergent mtDNA haplogroups in eastern Europe (clade 1b/clade 3a) and Scandinavia (clade 1a/clade 3a). For interpretation of the references to colour in this figure caption, please see the online version of this article.
Secondly, more recent hybridization events from polar bears into European brown bears can be interpreted from previous mitochondrial studies. Indeed, two divergent mtDNA haplogroups, representing clades 1 (western Europe) and 2 (polar bears + ABC islands brown bears) were found in several Irish brown bear fossils dated between 38 and 3 ka [13] (Fig. 3). Such results showed that female polar bears have introgressed insular populations of western European brown bears, most probably during the glacial periods G2 (190–135 ka) and/or G1 (75–15 ka).
In theory, the introgression of mtDNA from a species to another related species should be favoured by two factors: (1) genetic drift in a small population; (2) selection, since mitochondria play a crucial role in ATP synthesis and heat production.
During Pleistocene glacials, several small populations of brown bears were likely isolated in a few northern glacial refugia, such as the ABC islands or southern Ireland. In these populations, polar bear mitochondria may have been positively selected during glacial periods, because of their better efficiency for heat production within brown adipose tissue. This hypothesis is supported by genomic evidence that shows that polar bears evolved under stronger positive selection than brown bears, in particular for genes associated with adipose tissue development and fatty acid metabolism [20], as well as biochemical evidence that suggests the major role of mitochondria in adipocyte differentiation and adipocyte homeostasis [41].
4 Conclusion and perspectives
The reinterpretation of published molecular analyses has provided new insights into the evolution of bears, by suggesting that during glacial periods of the Pleistocene, brown bear populations isolated in a few northern refugia were introgressed by female polar bears. Most previous genomic studies have focused on polar bears rather than brown bears. For instance, Liu et al. [20] analysed the complete nuclear genome of 79 polar bears, but that of only 10 brown bears. Future research should pay greater attention to brown bear populations. In particular, it could be very interesting to sequence the complete nuclear genome of several geographically isolated populations of southern Europe (e.g., Spain, France, Italy, and Romania) and Asia (e.g., Iran, Pakistan, Tibet, Gobi Desert, and Japanese Islands), as well as the complete mitochondrial genome of the extinct subspecies of North Africa (U. arctos crowtheri). These data would be very useful to estimate more precisely the age of the MRCA of polar and brown bears, to better understand the demography history of brown bears during the Pleistocene, including their recurrent introgressive hybridization by female polar bears, and to study potential gene flow of alleles involved in cold adaptations.
Acknowledgments
I acknowledge Andy Jennings and two anonymous reviewers for their helpful comments on the manuscript.