

1 Introduction
Autism spectrum disorders (ASD) are a group of neuropsychiatric disorders characterized by problems in social communication, as well as the presence of restricted interests, stereotyped and repetitive behaviours [1,2]. Epidemiological studies estimate that more than 1% of the population could receive a diagnosis of ASD [3]. Individuals with ASD can also suffer from other psychiatric and medical conditions including intellectual disability (ID), epilepsy, motor control difficulties, attention-deficit hyperactivity disorder (ADHD), tics, anxiety, sleep disorders, depression or gastrointestinal problems [4]. The term ESSENCE for ‘Early Symptomatic Syndromes Eliciting Neurodevelopmental Clinical Examinations’ was coined by Christopher Gillberg to take into account this clinical heterogeneity and syndrome overlap [4]. There are 4–8 times more males than females in ASD, but the sex ratio is more balanced in patients with ID and/or dysmorphic features. Autism can be studied as a category (affected vs. unaffected) or as a quantitative trait using auto- or hetero-questionnaires such as the social responsiveness scale (SRS) or the autism quotient (AQ). Using these tools, autistic traits seem to be normally distributed in clinical cases as well as in the general population.
The causes of autism remain largely unknown, but twin studies have constantly shown a high genetic contribution to ASD. Molecular genetic studies have identified more than 100 ASD-risk genes carrying rare and penetrant deleterious mutations in approximately 10–25% of the patients [5]. In addition, quantitative genetic studies have shown that common genetic variants could capture almost all the heritability of ASD [5]. The genetic landscape of ASD is therefore shaped by a complex interplay between common and rare variants and is most likely different from one individual to another [6]. Remarkably, the susceptibility genes seem to converge in a limited number of biological pathways including chromatin remodelling, protein translation, actin dynamics, and synaptic functions [6].
In this chapter, I will detail the advances of the genetics of ASD in the last four decades starting from twin studies, following with molecular genetics and neurobiological studies (Fig. 1) and end with propositions to foster research in this field.

The history of the genetics of autism from 1975 to 2015. The increase in the number of genes associated with ASD (SFARI, March 2015) is represented together with the prevalence of ASD (data taken from the Center for Disease Control and Prevention), the different versions of the Diagnostic Statistical Manual (from DSM II to DSM-5.0), and the advance in genetics technology.
2 Twin and family studies in ASD
Based on more than 13 twin studies, published between 1977 and 2015, researchers have estimated the genetic and environmental contribution to ASD (Fig. 2). In 1977, the first twin study of autism by Folstein and Rutter reported a cohort of 11 monozygotic (MZ) twins and 10 dizygotic (DZ) twins. This study showed that MZ twins were more concordant for autism in 36% (4/11) compared with 0% (0/10) for DZ twins. When a “broader autism phenotype” was used, the concordance increased to 92% for MZ twins and to 10% for DZ twins. Since this first small-scale study, twin studies have constantly reported higher concordance for ASD in MZ compared with DZ twins. Between 2005 and 2009, three twin studies with relatively large group of twins (up to more than 3000 twin pairs) have reported high concordances for ASD in MZ twins (77–95%) compared with DZ twins (31%). Interestingly, a significant proportion of the genetic contribution to ASD is shared with other neurodevelopmental disorders such as ADHD (> 50%) and learning disability (> 40%) [7]. In summary, when all twin studies are taken into account, concordance for ASD is roughly 45% for MZ twins and 16% for DZ twins.
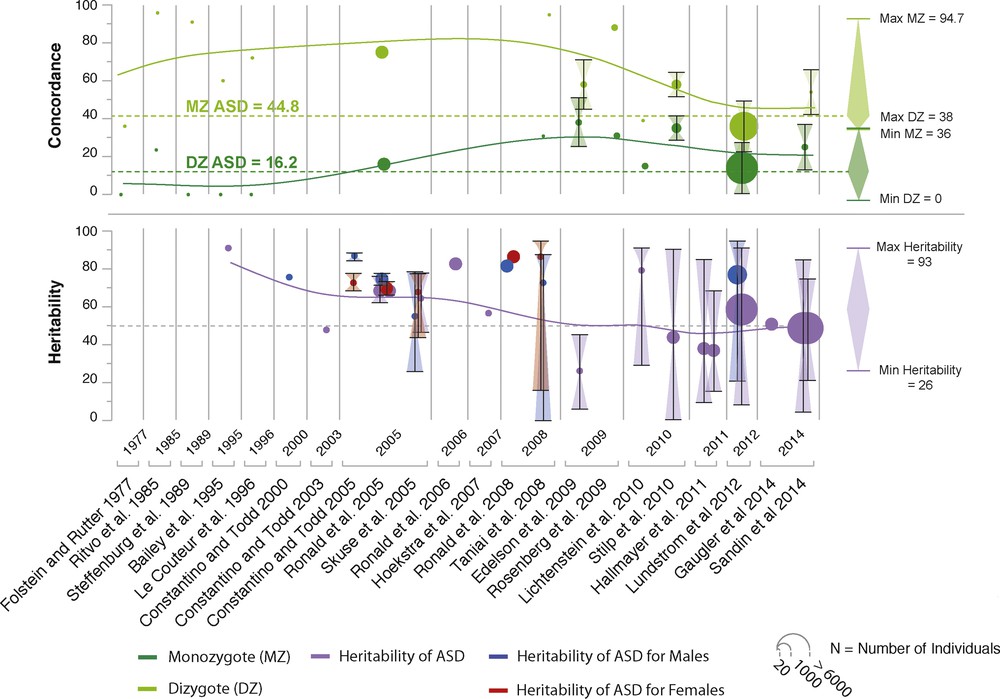
The main twins studies in ASD. A total of 13 twin studies and 17 heritability studies are depicted. Means of concordance and heritability weighted by sample size are presented on the right-hand side of the figure.
Family studies also revealed that the recurrence of having a child with ASD increases with the proportion of the genome that the individual shares with one affected sibling or parent. In a population-based sample of 14,516 children diagnosed with ASD [8], the relative risk for ASD (compared to the general population) was estimated to be 153.0 (95% confidence interval (CI): 56.7–412.8) for MZ twins, 8.2 (3.7–18.1) for DZ twins, 10.3 (9.4–11.3) for full siblings, 3.3 (95% CI: 2.6–4.2) for maternal half siblings, 2.9 (95% CI: 2.2–3.7) for paternal half siblings, and 2.0 (95% CI: 1.8–2.2) for cousins. The heritability was estimated to be 50% (95% CI: 45–56), and the non-shared environmental influence was also 50% (95% CI: 44–55). Surprisingly, only the additive genetic component and the non-shared environment seemed to account for the risk to develop ASD [8].
In summary, epidemiological studies have provided crucial information on the heritability of ASD. However, they do not inform us on the genes involved or on the number and frequency of their variants. In the last 15 years, candidate genes and whole-genome analyses have been performed to address these questions.
3 From chromosomal rearrangements to copy-number variants in ASD
The first genetic studies that associated genetic variants with autism used observations from cytogenetic studies. However, because of the low resolution of the karyotypes, it was almost impossible to associate a specific gene with ASD using this approach. The prevalence of large chromosomal abnormalities is estimated to be less than 2%. Thanks to progress in molecular technologies, the resolution in the detection of genomic imbalances has dramatically increased. Nowadays, the copy-number variants (CNVs)—that are small gain or loss of genomic DNA—are robustly detected. De novo CNVs are present in 4–7% of the patients with ASD compared with 1–2% in unaffected siblings and controls [9]. Beyond ASD, large CNVs (> 400 kb) affecting genes are present in 15% of patients with developmental delay (DD) or ID. Most of the CNVs are private to each individual, but some are recurrently observed in independent patients. The studies have also indicated that de novo CNVs identified in patients with ASD are most likely altering genes associated with synaptic functions [9].
In summary, large chromosomal rearrangements and CNVs increase the risk of having ASD in 5–10% of the individuals. In order to go further in the identification of the ASD-risk genes, candidate genes and whole exome/genome sequencing studies were performed.
4 From candidate genes to whole exome/genome sequencing studies in ASD
The first approach to associate a gene with ASD was to select specific candidate genes based on data coming from functional or genetic studies or a combination of both. This approach was successful in identifying several synaptic genes associated with ASD such as NLGN3, NLGN4X and SHANK3 [10,11]. Thanks to the advance in next-generation sequencing (NGS), we can now interrogate all genes of the genome in an unbiased manner using whole exome/genome sequencing (WES, WGS). These approaches allow the identification of virtually all the mutations within genes carried by an individual.
To date, more than 18 of such WES/WGS studies have been performed, comprising altogether more than 4000 families. Based on these studies, 3.6–8.8% of the patients were shown to carry a de novo causative mutation with a twofold increase of deleterious mutations in the patients compared with their unaffected siblings. In a meta-analysis, using more than 2500 families, Iossifov et al. [12] found that de novo likely gene disrupting mutations (frameshift, nonsense and splice site) were more frequent in patients with ASD compared with unaffected siblings (P = 5 × 10−7). The carriers of these de novo mutations were more likely diagnosed with a low non-verbal IQ. Interestingly, the mutations were significantly enriched in genes involved in chromatin modelling factors (P = 4 × 10−6) and in genes regulated by the fragile X syndrome protein (FMRP) complex (P = 4 × 10−7).
Only few studies have analysed the contribution of recessive inherited mutations in ASD. It was estimated that likely gene disrupting mutations (present on both paternal and maternal chromosomes) could account for 3% of the patients with ASD. For the X chromosome, there was a significant 1.5-fold increase in complete loss of function mutations in affected males compared to unaffected males, who could account for 2% of males with ASD [13].
5 The common variants in ASD
In the general population, one individual carries on average 3 million genetic variants in comparison to the reference human genome sequence. The vast majority of the variants (> 95%) are the so-called common variants shared with more than 5% of the human population. Using quantitative genetics, it was estimated that common variants were contributing to a high proportion of the liability of ASD: 40% in simplex families and 60% in multiplex families. In 2014, the study of Gaugler et al. provided an estimation of the heritability (52.4%), which is almost exclusively due to common variation, leaving only 2.6% of the liability to the rare variants [14]. These results indicate that the contribution of common variants is important, but the causative SNPs still remain unknown since they are numerous (> 1000) and each associated with a low risk. To date, the largest genome wide association studies (GWAS) performed on < 5000 families with ASD were underpowered to identify a single SNP with genome wide significance. The recruitment of larger cohorts of patients with dimensional phenotypes is warranted to better ascertain the heritability of ASD and to identify the genetic variants, which explain most of the genetic variance.
6 The genetic architecture of ASD: an interplay between rare and common variants
Based on the results coming from epidemiological and molecular studies, it is now well accepted that genetic factors play an important role in the susceptibility to ASD. It was also shown that the genetic susceptibility to ASD could be different from one individual to another with a combination of rare deleterious variants and a myriad of low-risk alleles (also defined as the genetic background). For some individuals, a single de novo mutation will be enough to cause autism. For some individuals, the accumulation of many (> 1000) common risk alleles will additively increase the risk of autism.
The interplay between the rare or de novo variants and the background will also influence the phenotypic diversity observed in the patients carrying rare deleterious mutations. In some individuals, a genetic background will be able to buffer or compensate the impact of the rare genetic variations. In contrast, in some individuals, the buffering capacity of the genetic background will not be sufficient to compensate the impact of the deleterious mutations and they will develop ASD. In this model, ASD can be regarded as a collection of many genetic forms of autism, each with a different aetiology ranging from monogenic to polygenic models.
Fortunately, although the ASD-risk genes are numerous, they seem to converge in a limited number of biological pathways that are currently scrutinized by many researchers.
7 The biological pathways associated with ASD
Based on results from the genetics of ASD, cellular and animal models have been analysed in order to identify the main mechanisms leading to this complex syndrome. Remarkably, several studies showed that neuronal activity seems to regulate the function of many of the ASD-risk genes. This has led to the hypothesis that abnormal synaptic plasticity and failure of neuronal/synaptic homeostasis could play a key role in the susceptibility to ASD. Fig. 3 summarizes the main gens/pathways associated with ASD: chromatin remodelling, protein synthesis, protein degradation, and synaptic function (Fig. 3).
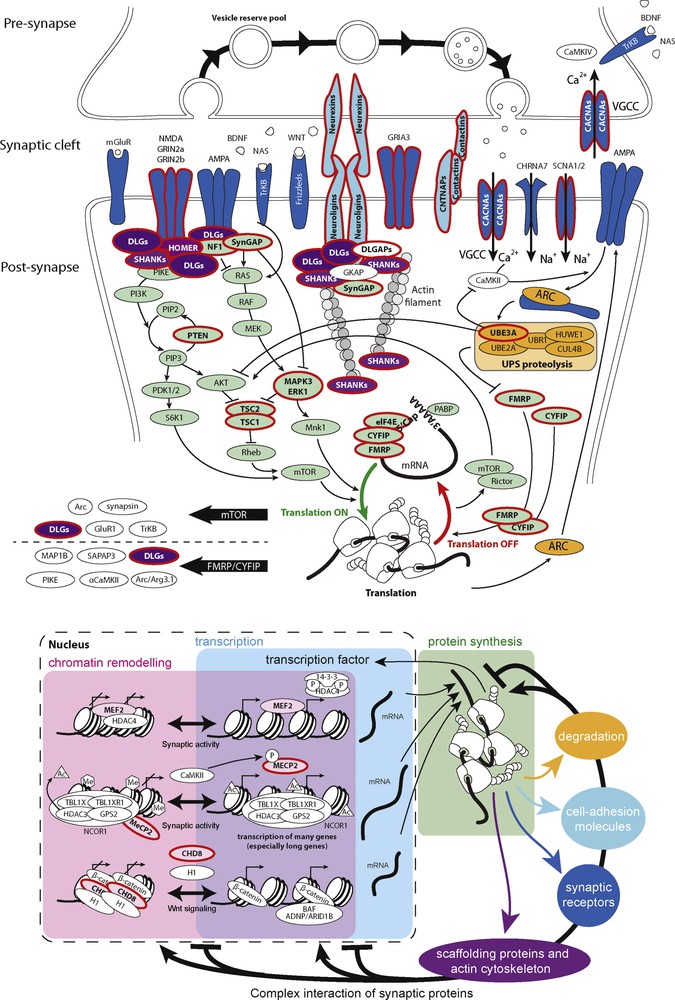
Examples of the biological pathways associated with ASD. The ASD-risk genes code for proteins involved in chromatin remodeling, transcription, protein synthesis, and degradation, cytoskeleton dynamics, and synaptic functions. Proteins associated with ASD are circled in red.
8 Perspectives: three propositions to improve research on autism
While tremendous progress has been made in the understanding of the causes of autism, I will detail three propositions that could foster the research in the field.
8.1 Proposition #1: fewer categories, more dimensions
The recent advances in genomics have demonstrated that an identical genetic variant may increase the risk for a wide range of diagnoses formerly thought of as distinct. These findings are contributing to a re-conceptualization of the current psychiatric nosology. The fifth edition of the Diagnostic and Statistical Manual (DSM-5) from the American Psychiatric Association now makes it easier to recognize overlap between different diagnostic categories, but in the main, the existing narrow and rigid categories tend to disconnect researchers from the real phenotypes. Recently, several initiatives such as the ESSENCE from Christopher Gillberg were undertaken to improve phenotype characterization using more dimensional approaches. The Research Domain Criteria (RDoC) has been launched by NIMH calling for the development, for research purposes, of new ways of classifying psychopathology based on dimensions of observable behaviour and neurobiological and genetic measures (http://www.nimh.nih.gov/research-priorities/rdoc/nimh-research-domain-criteria-rdoc.shtml). This effort is attempting to define basic dimensions of functioning related to known neural circuitry to be studied across multiple units of analysis, from genes to neural circuits to behaviours, cutting across disorders as traditionally defined.
In summary, it is most likely that progress in the comprehension of the risk factors for autism will come from dimensional and quantitative data that goes well beyond current psychiatric classification. One first step would be to gather the information that is currently separated by DSM-5 diagnostic categories and dispersed in different laboratories that may fail to communicate. To achieve this, there is a need of more data sharing (see below).
8.2 Proposition #2: more research on genetic and brain diversity in human populations and more data sharing
Based on current case-control design, there is a tendency for researchers to know better the genotypes and phenotypes of the patients than those of the controls. Indeed, in the vast majority of the genetic studies, controls are often not investigated at the phenotypic level, and in phenotypic studies, controls are very limited in their number and cultural and social economic status diversity. As a consequence, early onset developmental disorders are therefore considered as binary traits “affected” vs. “non-affected” without taking into account the genetic and phenotypic diversity of both “affected” and “non-affected” individuals.
Integrating diversity in our experimental design will require increasing the sample size of our study populations. Indeed, risk factors for early onset neurodevelopmental disorders are either rare with large effect or frequent, but with a small effect. In both situations, robust genotype–phenotype relationships are difficult to ascertain in small samples. One opportunity to increase sample size is to foster data sharing. Many brakes reduce efficient data sharing. Hence, there is a need:
- • to agree on an ethical informed consent for research subjects that will allow data sharing;
- • to agree on standardized measures;
- • to change the reward system regarding publications;
- • to set up a system to make data sharing easy and secure.
There is an emerging community of researchers involved in data sharing. Specifically in neuroscience, initiatives such as the Neuroscience Information Framework (NIF) or the International Neuroinformatics Coordinating Facility (INCF) were launched recently. NIF (http://www.neuinfo.org) is a dynamic inventory of Web-based neuroscience resources: data, materials, and tools accessible via any computer connected to the Internet. This should advance neuroscience research by enabling discovery and access to public research data and tools worldwide through an open source, networked environment. INCF (http://www.incf.org/) develops a collaborative neuroinformatics infrastructure and promotes the sharing of data and computing resources with the international research community.
8.3 Proposition #3: patients and relatives as participants for research
Many aspects on the quality of life of the patients and their relatives are not adequately taken into account by researchers. For example, in ASDs, comorbidities such as gastrointestinal and sensory problems are under-explored. The movement “no research about me, without me” is calling for patients and their relatives to be more involved in research designs. For example, the UK National health Service (NHS) initiative INVOLVE (http://www.invo.org.uk) is a national advisory group that supports greater public involvement in NHS, public health and social care research. There is also the James Lind Alliance (http://www.lindalliance.org), the Patient-Centered Outcomes Research Institute (PCORI; http://www.pcori.org), and PatientsLikeMe (http://www.patientslikeme.org) initiatives for patients that want to monitor their own health and chronic illness. Another example is the initiative for Cancer research at Sage Bionetworks (http://www.sagebase.org). SAGE develops tools so that medical patients can keep their own data rather than storing that data in particular medical institutions. The aim is to offer predictive, personalized, preventive, participatory (P4) medicine.
9 Conclusion: towards a more inclusive world
In the last 40 years, very significant progresses were made in the genetics of ASD. We have now a better knowledge of the genetic architecture of this heterogeneous syndrome and some of the biological pathways are now investigated using different approaches such as cellular and animal models. There are however many aspects of the genetics of ASD that remain largely unknown.
The first challenge concerns the role of the common variants. These variants are most likely playing a key role in the susceptibility to ASD and in the severity of the symptoms. But, because the impact of each single SNP is very low, it is currently impossible to identify the risk alleles using conventional GWAS. In human quantitative traits such as height, neuroanatomical diversity or intelligent quotient, very large cohorts of many thousands of individuals are necessary to identify the main causative SNPs.
The second challenge concerns the stratification of the patients and the role of the ASD-risk genes during brain development/function. Based on our current knowledge, the genetic architecture of ASD seems to be different from one individual to another with possibly contrasting impact on when and where neuronal connectivity could be atypical compared to the general population. For example, in animal models, several mutations lead to higher connectivity whereas other mutations alter synaptic density. It is therefore crucial to increase our knowledge from a basic research perspective on the biological roles of the ASD-risk genes and their partners.
Finally, while we all agree that biological research is necessary to improve the quality of life of the patients and their families, progress should also be made for a better recognition and inclusion of people with neuropsychiatric conditions in our societies (no mind left behind). For patients, the burden of neurodevelopmental disorders makes daily activities difficult and lowers the odds of living an independent life. In addition to improved medical care, innovative initiatives towards a more inclusive world point toward other important advances. For example, Aspiritech (http://www.aspiritech.org), a non-profit organization based in Highland Park, Illinois, places people who have autism (mainly Asperger's syndrome) in software testing jobs. The Danish company Specialisterne has helped more than 170 individuals with autism obtain jobs since 2004. Its parent company, the Specialist People Foundation, aims to connect one million autistic people with meaningful work (http://www.specialistpeople.com). Laurent Mottron, a psychologist working in Montréal, has offered jobs for patients with ASD in his group and this new perspective has had a positive impact on his research on autism. As he said, “The hallmark of an enlightened society is its inclusion of non-dominant behaviours and phenotypes, such as homosexuality, ethnic differences and disabilities. Governments have spent time and money to accommodate people with visual and hearing impairments, helping them to navigate public places and find employment, for instance—we should take the same steps for autistics” [15]. As suggested by Gillberg, it might be better to abandon the belief that there is a single defining ASD brain dysfunction. Instead, we should understand the diversity of ASD (or autisms). Considering autism not as a single entity, but as a continuum of human diversity and tackling this heterogeneity using information coming from different fields of research (including direct information from the affected individuals and their family) should allow a better diagnosis, care and integration of individuals with autism [15].
Disclosure of interest
The author declares that he has no competing interest.
Acknowledgements
I thank Guillaume Huguet for drawing the figures. This work was funded by the Institut Pasteur, the Bettencourt-Schueller foundation, the “Centre national de la recherche scientifique”, Paris Diderot University, the “Agence nationale de la recherche” (SynDiv-ASD), the GenMed labex, the BioPsy labex, the Conny-Maeva Charitable Foundation, the Cognacq-Jay Foundation, the Orange Foundation, and the Fondamental Foundation.