

1 Introduction
A major function encoded within plant genomes is devoted to defense against pests and pathogens. In this context, genes coding for nucleotide binding site (NBS) domain proteins represent a major plant disease-resistance family, associated with effector-triggered immunity (ETI) [1]. The NBS domain (∼170 amino acids) is part of the larger ∼300 amino acid NB-ARC domain and contains strictly ordered motifs [2]. The NBS region binds and hydrolyzes ATP and GTP and primarily works as a signal transduction switch, following pathogen recognition. In most NBS-type proteins, the C-terminal domain is known as the leucine-rich-repeat (LRR) domain and consists of multiple copies of an imperfect leucine-rich-repeat sequence [3]. This domain, ranging between 22 and 25 residues, is often implicated in protein-protein interaction and, more precisely, binding to pathogen-derived molecules [4]. NBS-LRR genes are currently thought to encode cytoplasmic receptors, capable of detecting the presence of pathogen-borne avirulence proteins in the host's cytoplasm. NBS-containing proteins are known to recognize a wide variety of taxonomically unrelated pathogens, including viruses, bacteria, fungi, nematodes, and insects [5]. The activation of these genes results in a hypersensitive response (HR), a localized form of host-programmed cell death [6].
Studies of the NBS gene family have been performed in many monocot and dicot plant species, such as Arabidopsis [7], rice [8], Medicago truncatula [9], Poplar [10], grape [11], sorghum [12], Lotus japonica [13], Brassica rapa [14], Triticum aestivum [15], Brachypodium distachyon [16] and Hordeum vulgare (Y. Habachi-Houimli, Y. Khalfallah, H. Makni, M. Makni, D. Bouktila, unpublished data). All results have shown that the size of this gene family differs in each species. In general, approximately 0.2–1.76% of genes predicted in plant genomes encode NBS-type proteins [17]. Plant NBS-LRR proteins (NB-LRR or NB-ARC-LRR proteins) are typically categorized into the TIR or non-TIR class, based on the identity of the sequence that precedes the NB domain [2]. The TIR class of plant NBS-LRR proteins contains an amino-terminal domain with homology to the Toll and interleukin 1 receptors. The non-TIR class is less well-defined, but most NBS-LRR proteins of this class contain α-helical coiled-coil-like sequences in their amino-terminal domain [18]. While both classes are present in dicots, studies in monocots have reported only non-TIR-NBS-LRRs [3]. To explain this fact, two hypotheses have been made: either TIR-NBS-LRRs arose later after the monocotyledon/dicotyledon separation [19] or have been lost from monocots [18]. Currently, the latter hypothesis is more believed, because in rice, it was possible to identify several genes encoding proteins containing the TIR domain, although they do not seem to be related to NBS-LRR proteins [3]. Consequently, NBS-type proteins in cereals may adopt various domain architectures, such as NBS, CC-NBS, NBS-LRR and CC-NBS-LRR. Comparative analysis of NBS genes in different species of Poaceae such as rice, maize, sorghum, Brachypodium, Triticum aestivum, Triticum urartu, Aegilops tauschii and Hordeum vulgare showed considerable copy number variation ranging from 109 in maize [20] to 1,219 in Aegilops tauschii [21]. In rice, 21 of the 22 cloned functional resistance (R) genes against rice blast and one of the seven against rice bacterial blight encode NBS-LRR domains [22–24]. In wheat, eight cloned functional resistance genes encode NBS-LRR domains [25–33]. In barley, three functional resistance genes, encoding NBS-LRR domains, and conferring resistance against the powdery mildew caused by Blumeria graminis, were cloned [34–36]. In maize, one of two cloned functional resistance genes against rust encodes an NBS-LRR domain [37]. Further, the expression profiles of some NBS-LRR-encoding R genes have been shown to be affected by factors other than pathogen infection, such as tissue type, developmental stage, or environmental conditions [37]. Such a regulation of R genes may contribute to the optimization of defense responses in terms of reducing fitness costs of defense signaling. For this reason, it would be very important to study the influence of microRNAs on R genes.
MicroRNAs (miRNAs or miRs) are a class of endogenous single-stranded non-coding small RNAs of 18–25 nucleotides, that are processed from stem-loop structures of long primary transcripts, by a Dicer-Like 1 (DCL1) enzyme, leading to miRNA precursors that are cleaved into miRNA/miRNA* duplexes [38]. A strand, termed mature miRNA, is retained within the RNA-induced silencing complex (RISC), whose core component is the protein ARGONAUTE1 (AGO1). This strand (mature miRNA) regulates gene expression, while the near complement reverse strand (miRNA*) is released and usually degraded by exonucleases [39], but sometimes functions as a mature miRNA [40]. MicroRNAs suppress gene expression either by directing cleavage of their highly complementary target transcripts [41] or by translational repression. Recent studies have indicated that translational repression might be more prominent in plants than first thought [41,42].
The first plant miRNA was identified in Arabidopsis thaliana [43], followed by several miRNAs in other plant species, such as rice [44], tobacco [45], maize [46], sorghum [47], cotton [48], wheat [49], and barley [50]. Among fundamental functions regulated by miRNAs in plants, we can cite organogenesis, meristem development, leaf and flower morphogenesis, signal transduction, response to environmental stresses [51,52]. At the time of writing this article, 8496 mature miRNAs from 73 plant species have been discovered and deposited in the public miRNA database, miRBase (Release 21.0, http://www.mirbase.org/), among which 713 in rice, 525 in B. distachyon, 321 in maize, 241 in sorghum, 119 in wheat, and 71 in barley. Understanding the specific roles of miRNAs in response to predefined stresses will provide new insights into regulating their responses to environmental stresses, especially through miRNA-based genetic modification technology where artificial miRNAs (amiRNAs) can be utilized for improving several desirable plant traits [53]. Based on this, our goal in this study was to perform a comprehensive characterization of a large set of miRNAs in Poaceae, and attempt to characterize their impact on NBS-LRR mRNAs.
2 Methods
2.1 Data mining of NBS genes in Poaceae
Proteomes of S. bicolor, O. sativa and B. distachyon were downloaded from Phytozome v9.1 database (http://www.phytozome.net/sorghum.php), Rice Genome Annotation Project (RGAP 7.0, http://rice.plantbiology.msu.edu/) and Munich Information Center for Protein Sequences (MIPS, ftp://ftpmips.helmholtz-muenchen.de/plants/brachypodium/genes/v1.2/), respectively. From these three species, all proteins predicted to contain an NB-ARC domain (Pfam PF00931) were retrieved using the seqret tool from the European Molecular Biology Open Software Suite (EMBOSS version 6.5.0.0, ftp://emboss.open-bio.org/pub/EMBOSS/) and placed in separate files. MEME software [54] was used with restrictive parameters (maximum number of patterns = 4), to check the presence of core NBS domains with a conserved length (∼170 aa) and distribution of the four major motifs. Sequences that contained no or only a partial core NBS domain were excluded. This procedure allowed the identification of 139, 269, and 119 protein sequences from S. bicolor, O. sativa and B. distachyon, respectively. The coding sequences (CDS) corresponding to these proteins were retrieved from each of the repositories mentioned above. For H. vulgare and T. aestivum, 318 and 580 NBS-type gene models previously predicted from the whole genome shotgun sequence assembly of barley cv. Bowman and the LCG assembly of T. aestivum, respectively [15] were used in this study. From both sets, we have selected 257 barley and 286 wheat NBS gene models, following examination of the conservation of core NBS with MEME.
2.2 Data mining of microRNAs in Poaceae
All non-redundant mature miRNA sequences of H. vulgare, T. aestivum, S. bicolor, O. sativa, and B. distachyon were downloaded, in Fasta format, from miRBase (http://www.mirbase.org/ftp.shtml), accessed in August 15, 2015. These miRNAs included 71 Hvu-miRs, 119 Tae-miRs, 525 Bdi-miRs, 713 Osa-miRs and 241 Sbi-miRs. Analysis of Poaceae miRNAs according to their expression level was possible for only four of the total five studied species, due to the absence of tissue data for B. distachyon in miRBase (Table 1).
Statistical data of miRNAs from four Poaceae genomes, downloaded from miRBase (www.mirbase.org), according to their expression tissues and developmental stages.
| Experiment ID (www.mirbase.org) | Tissues and developmental stages | Raw number (including redundancies) | Number of miRNA species after exclusion of redundancies within experiments | Number of miRNA species after exclusion of redundancies within and among experiments | |
| Hordeum vulgare | ER0000000318 | 7-day leaf | 36 | 36 | 71 (Hvu-miRs) |
| ER0000000319 | Inflorescence | 42 | 42 | ||
| ER0000000320 | 7-day leaf inoculated with the fungus Blumeria, agent of barley powdery mildew | 39 | 39 | ||
| Oryza sativa | ER0000000276 | Callus: developmental stage: differentiated | 485 | 485 | 713 (Osa-miRs) |
| From ER0000000418 to ER0000000434 | Seedling | 6853 | 499 | ||
| From ER0000000435 to ER0000000448 | Root | 5448 | 490 | ||
| From ER0000000449 to ER0000000460 | Shoot | 5003 | 488 | ||
| From ER0000000461 to ER0000000479 | Panicle | 8561 | 560 | ||
| Triticum aestivum | ER0000000366 | 7-day leaf | 105 | 105 | 119 (Tae-miRs) |
| ER0000000367 | Spikelet inoculated in water | 110 | 110 | ||
| ER0000000368 | Spikelet inoculated with the fungus Fusarium | 105 | 105 | ||
| Sorghum bicolor | ER0000000351 | 3-week-old seedling (leaf) | 177 | 177 | 241 (Sbi-miRs) |
| ER0000000352 | 9-week-old plants (flower) | 159 | 159 | ||
| ER0000000353 | Panicle | 164 | 164 |
2.3 Identification and characterization of miRNA targets among Poaceae NBS genes
The identification of miRNA targets based on sequence complementarity is an essential step to further understand the regulatory function of miRNAs. Our final set of 1070 NBS-encoding sequences identified through data mining procedure (286 from T. aestivum, 257 from H. vulgare, 139 from S. bicolor, 269 from O. sativa and 119 from B. distachyon) was used, as coding sequences, in combination with 1669 mature miRNAs (71 Hvu-miRs, 119 Tae-miRs, 525 Bdi-miRs, 713 Osa-miRs and 241 Sbi-miRs), using the psRNATarget program [55] with default parameters, in order to predict all potential miRNA-NBS transcript pairs.
NBS transcripts, from the five studied Poaceae species, predicted as putative targets for miRNA regulation, underwent gene ontology (GO)-based functional characterization that was performed using Blast2GO software [56]. This operation was conducted at the protein level by searching for sequences similar to each gene model by BlastP, against the NCBI “nr” database. Based on the BlastP results, the Blast2GO extracted GO terms for each candidate R protein.
3 Results and discussion
3.1 Low conservation of Poaceae miRNAs pleads in favor of their recent origin
One thousand six hundred and sixty-nine (1669) non-redundant miRNAs, belonging to the five studied Poaceae species, were retrieved from miRBase. These miRNAs were distributed over a total of 625 MIR families. Fig. 1A illustrates the number of MIR families found in each grass species considered separately. Fig. 1B illustrates the relative size of the major MIR families (including at least 10 mature miRNA species). Only 54 MIR families were conserved across two or more plant species, while 571 were species-specific (Fig. 1C), with more than half of them (52%) expressed in rice (Fig. 1D).
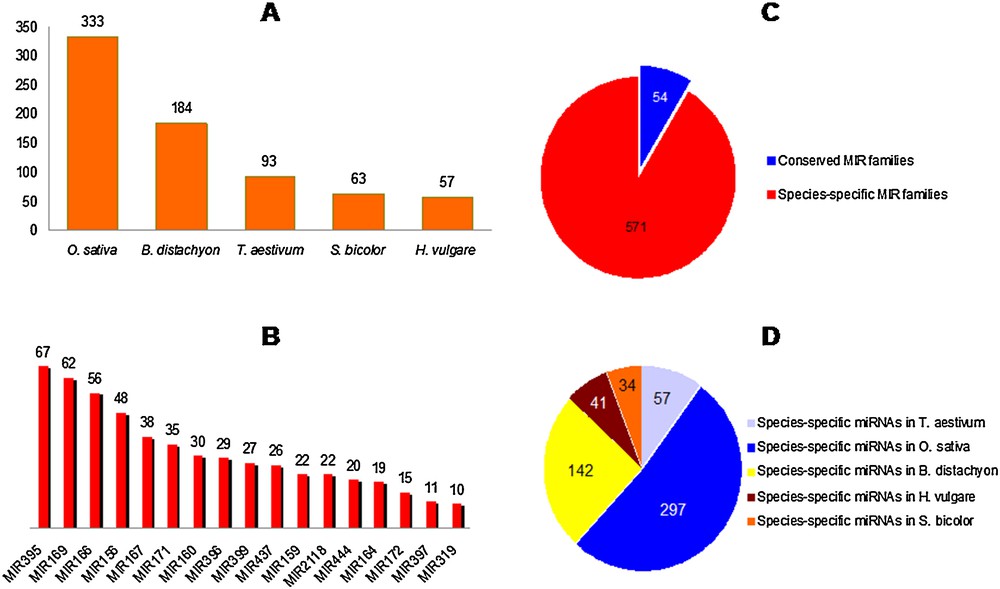
Number of MIR families detected in each studied species of Poaceae, their sizes; and their conservation among Poaceae. A. Number of MIR families found in each grass species considered separately. B. Relative size of major MIR families, including at least 10 mature miRNA species per family. C. Species-specific and conserved MIR families. D. Distribution of species-specific MIR families among Poaceae.
It has been already known that only a minority of miRNA gene families were conserved between plant families, while the majority are family or species-specific, as previously established by [57], suggesting that most miRNA genes arose relatively recently in evolutionary time. In our study, some of the miRNA families, conserved across the five Poaceae species (e.g., MIR156 and MIR319), have been reported to be of ancient origin, as they have been conserved from mosses to flowering plants [58].
3.2 Regulation of Poaceae NBS genes by miRNAs
3.2.1 Between Poaceae species cross-regulation
We have predicted 265 miRNAs to play the role of potential regulators of NBS genes in Poaceae. These miRNAs were distributed across the five studied Poaceae species, as follows: 116 Osa-miRs, 86 Bdi-miRs, 28 Tae-miRs, 26 Sbi-miRs and 9 Hvu-miRs (Additional file 1). It was revealed that miRNAs from a Poaceae species would potentially regulate another species’ NBS genes. For example, bdi-miR2118 regulates B. distachyon gene Bradi2g39517.1, but also gene Sb09g004410.1 of S. bicolor, H. vulgare CAJX010086000.1-1, T. aestivum CALP010039172.1-1, and O. sativa LOC_Os01g36640.1. This observation is consistent with previous predictions that several miRNAs are likely to target the NBS–LRR protein class genes in other plant species [59,60]. As a practical application in Poaceae, this finding will enhance pathogen resistance through the creation of transgenic crop plants that express miRNA regulators of R. genes from other plants.
3.2.2 Multi-tissue miRNA networks
From the 265 miRNA species shown to have a probable role in regulating disease resistance genes in Poaceae, only 179 could be recognized with their tissue expression levels (Fig. 2). Our results clearly indicate that most of these miRNAs seem to be simultaneously co-expressed in distinct tissues. The tissue range in which a miRNA is expressed places an obvious constraint on its physiological role(s), as well as on its possible mRNA target(s). In addition, we have found that all miRNAs (100%) of T. aestivum were expressed at least in leaves. In both H. vulgare and S. bicolor, more than 75% of miRNAs were expressed in leaves. The expression of a large number of miRNAs in the leaf reflects the complexity of regulatory activities required for pathogen resistance in this organ [61]. The majority of the miRNAs detected in O. sativa plants were expressed in multiple plant organs (i.e. panicle, roots, shoots, seedlings, and callus). These findings may be interpreted in two ways: either as reflecting the genetic expression of a given miRNA precursor gene in various plant organs, and/or as reflecting the systemic transmission of miRNAs as long-distance signals between the plant organs, in order to adapt to multiple environmental stimuli to confer long-lasting effects against stresses and maintain plant growth and development [62].
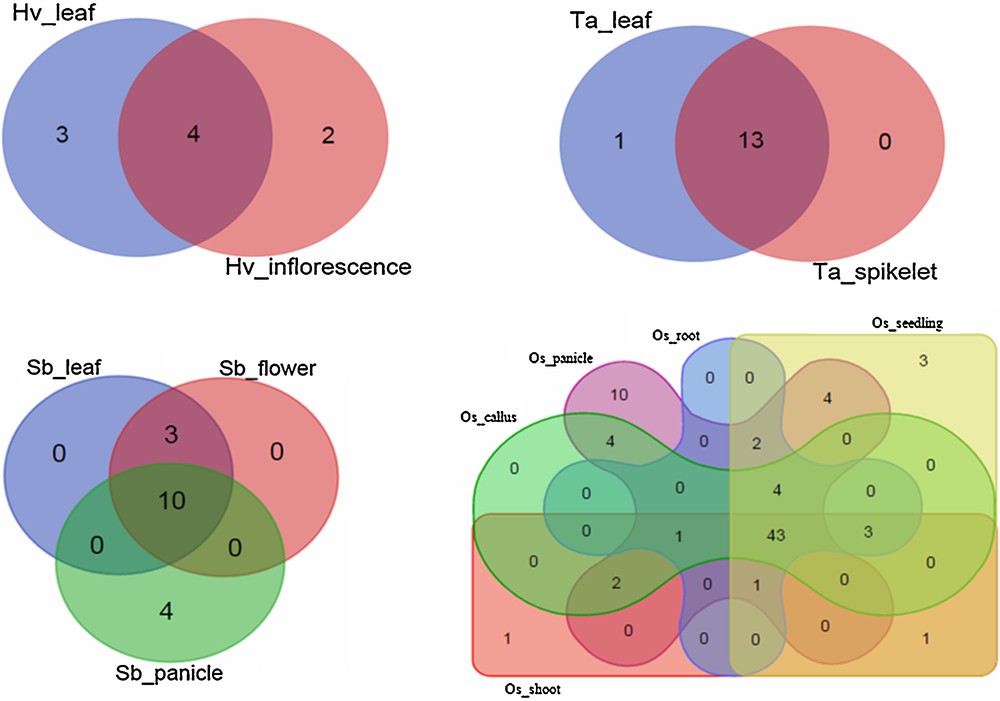
Venn diagrams illustrating tissue co-expression profiles among 179 miRNAs shown to have a potential role in regulating disease resistance genes in four Poaceae species. Hv, Ta, Sb, and Os refer to Hordeum vulgare, Triticum aestivum, Sorghum bicolor, and Oryza sativa, respectively. Terms following underscores refer to tissues in which miRNAs are expressed.
3.2.3 Implication of members of the MIR2118 family
In dicots, such as tomato, tobacco and Medicago, members of the MIR482/MIR2118 superfamily have been shown to regulate numerous NBS-LRR defense genes through targeting the conserved sequences encoding the P-loop motif of the NBS-LRR receptors [63]. The present study was conducted in Poaceae, where no MIR482 family members have been reported [63]. We have identified 21 miRNA species belonging to the MIR2118 family (Additional file 1). These miRNAs have been predicted to target eight NBS transcripts from S. bicolor, seven from B. distachyon, 10 from H. vulgare, 13 from T. aestivum, and 20 from O. sativa. Further analysis of the site, in the NBS genes of Poaceae, which is targeted by osa-miR2118, revealed the P-loop domain as a prime candidate, with a perfect match between the two sequences (Fig. 3). This result further indicates that MIR2118 regulates NBS-LRR protein-related fungal and bacterial disease resistance.

Regulation of Poaceae NBS genes, by MIR2118, recruits the P-loop domain as an interacting substratum. A. Alignment of 21 bdi-miR2118, osa-miR2118 and sbi-miR2118 species, identified in this study, with BioEdit [66]. B. Sequence complementarity, determined with psRNATarget program, between osa-miR2118p and the P-loop sequence of the wheat gene CALP010183577.1-1 [15].
3.2.4 A large host defense functional array subjected to regulation by miRNAs
Target analysis using psRNATarget revealed that 42% (454/1069) of the NBS genes from the five studied Poaceae species were probably regulated by 265 distinct miRNA species (Additional file 1). All the predicted miRNA targets from different species, identified in this study (454 NBS genes), were subjected to GO analysis, to gain a better understanding of their functions. This enabled their assignment to 16 functional groups (Fig. 4). The average similarity level for the 20 best hits ranged from 47.1% to 94.35%. The average e-value for the 20 best BlastP hits ranged from 6.2e−49 to 0.0e0 (data not shown). These data indicate that the annotation is highly reliable.
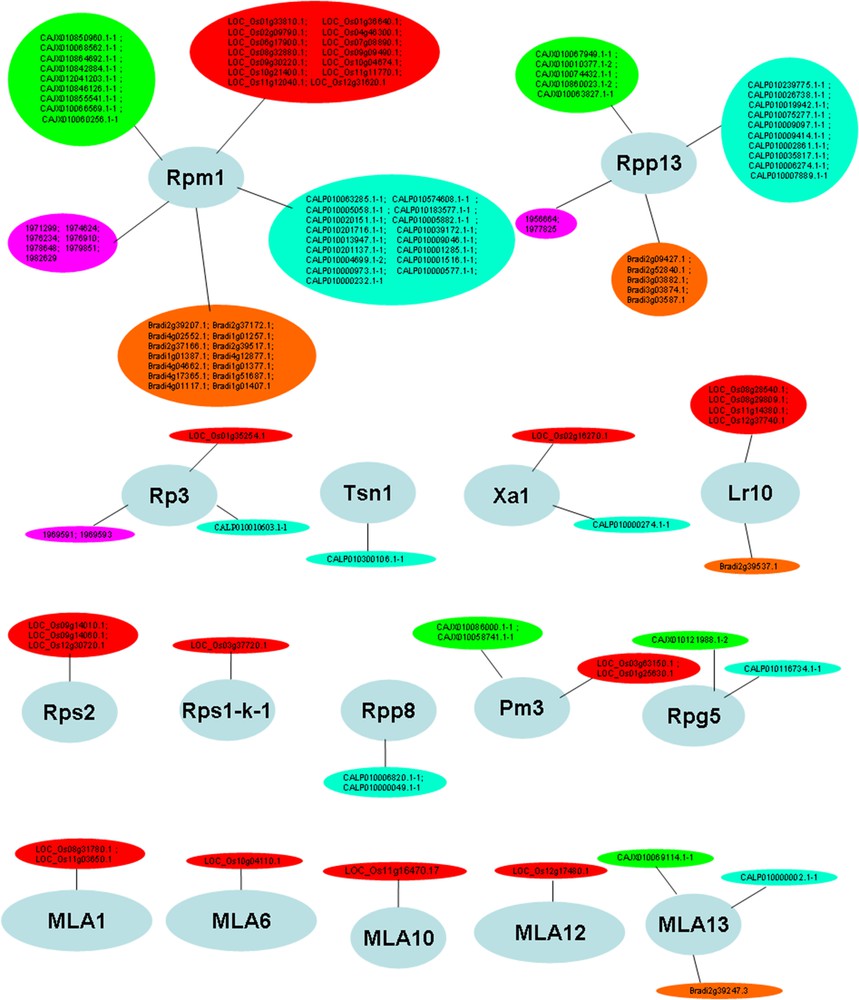
Functional categorization of major NBS genes potentially regulated by miRNAs, based on BlastP and gene ontology, using Blast2GO software [56]. Color-filled circles refer to NBS-LRR gene models of the five studied Poaceae species, as follows: red: Oryza sativa; green: Hordeum vulgare; cyan: Triticum aestivum; orange: Brachypodium distachyon and purple: Sorghum bicolor.
As illustrated in Table 2, Rpm1, Rps2 and Xa1-like genes are regulated by 57 miRNA sequences. These genes mediate resistance against bacteria (Pseudomonas syringae, and Xanthomonas oryzae) in various Poaceae species. Besides, genes mediating resistance to a broad range of fungi (Rpp8, Rpp13, Rp3, Tsn1, Lr10, Rps1-k-1, Pm3, Rpg5 and MLA genes) were found to be potentially regulated by 61 distinct miRNA species (Table 2). Some miRNAs, including miR2118, miR7757, miR5071 and miR9664 are potential regulators of both fungi and bacteria-resistant genes (e.g., bdi77575p.1, osa2118 g, tae96643p, etc.). There are scarce data on the specific sequence-to-sequence interaction between miRNAs and NBS-LRRs in Poaceae, hence, the promise held by the present study in deciphering the regulatory networks developed during post-transcriptional regulation in diverse pathological contexts. MIR159, reported by Gupta et al. (2012) [64] as a potential regulator of defense responses in T. aestivum L. during Puccinia graminis infection was predicted, in our study, as a potential regulator of Rp3 conferring resistance to common rust caused by Puccinia sorghi, suggesting a multivalent role of this miRNA family during plant defense against rust (Puccinia) species. MIR166 targeting Poaceae transcripts similar to downy mildew genes, Rpp8 and Rpp13, was previously described as involved in miRNA-mediated gene expression regulation, during the pathological development of stem canker disease in poplar, Populus trichocarpa [65]. It is nonetheless true that the majority of miRNA families reported in the present study as possible regulators of NBS-LRR genes (Table 2) have not been previously associated with plant response to any specific biotic stresses. This fact is, most probably, due to a still poor understanding of the role played by these evolutionarily young, species-specific plant MIRNA genes [57]. A deeper understanding will be gained, in the future, through real-time analysis of gene expression patterns of most miRNAs reported in our study, during response to selected biotic stresses.
List of miRNA species predicted to regulate Poaceae NBS-type genes clustered by target Gene Ontology (GO) annotation.
| Pathogen | Targets GO cluster | miRNA species with a predicted regulatory role | |
| Bacteria | Pseudomonas syringae | Rpm1 | bdi2118a, bdi2118b, bdi5057, bdi5064, bdi51733p, bdi5174e5p.2, bdi5185l5p, bdi5198, bdi7723b5p, bdi7726a5p, bdi77575p.1, bdi77775p.2, bdi9497, hvu5049c, hvu6197, hvu6198, osa18543p, osa18545p, osa1876, osa1877, osa2118a, osa2118b, osa2118c osa2118d, osa2118e, osa2118f osa2118 g, osa2118 h, osa2118i, osa2118j, osa2118 K, osa2118l, osa2118 m, osa2118n, osa2118p, osa2118q, osa2118r, osa5071, osa5079a/b, osa5337b, osa5498, sbi5568c3p, sbi5568c5p, sbi6217a/b5p, sbi62313p, tae1122b3p, tae77575p, tae9657a3p, tae96643p, tae9773, tae9774, tae9778 |
| Xanthomonas oryzae pv. oryzae (causal agent of bacterial blight) | Xa1 | osa1871, osa2864.2 | |
| Pseudomonas syringae pv. tomato | Rps2 | bdi5163b3p, bdi77355p, bdi77443p, bdi77533p, bdi77575p.1, osa416, bdi77575p.2, bdi77633p, osa2118b, osa2118 g, osa2118n, osa5071, tae1120c5p, sbi62305p, tae77575p, tae96643p | |
| Fungi | Peronospora parasitica (causal agent of downy mildew) | Rpp8, Rpp13 | bdi166h5p, bdi2118b, bdi395, bdi5163a3p, bdi5169a/b, bdi5200a5p, bdi5200b5p, bdi5285p, bdi77575p.1, bdi77575p.2, bdi77813p, bdi9485, bdi9494, hvu6197, osa1439, osa18665p, osa2118f, osa2118j, osa2118 m, osa5071, osa5285p, osa5532, osa6248, sbi528, tae1121, tae395b, tae50493p, tae77575p, tae96605p, tae96643p, tae96643p |
| Puccinia sorghi (rust agent) | Rp3 | bdi159b5p.3, bdi397a, osa1850.3, osa395v, osa397a/b, sbi3975p, sbi6232a5p, tae96643p | |
| Tan spot and Stagonospora nodorum blotch | Tsn1 | bdi2118a, osa2118e, osa2118o, osa2118e, osa2118r | |
| Leaf rust | Lr10 | osa5071, osa51525p, tae77575p, tae96643p | |
| Phytophthora root, Phytophthora sojae | Rps1-k-1 | tae96615p, | |
| powdery mildew in wheat | Pm3 | bdi2118a, bdi77473p, osa1440b, osa2118b, osa2118 g, osa2118n, osa5832, | |
| Puccinia graminis (causal agent of wheat stem rust in barley) | Rpg5 | hvu6214, osa2926, osa51445p | |
| Powdery mildew (barley) | MLA genes | bdi5163b3p, bdi77355p, bdi77443p, bdi77533p, bdi77575p.1, bdi77575p.2, bdi77633p, osa416, osa5071, tae77575p, tae96643p |
4 Conclusion
Our study demonstrated that disease resistance NBS gene family, in Poaceae, is targeted by multiple, independent miRNA families. The great difference in miRNA family size and specificity between different species of Poaceae indicates that miRNA genes change their copy numbers and their sequence actively, probably to provide dosage effects or different regulatory patterns in target gene regulation. Crop improvement strategies based on small non-coding RNAs have an enormous potential to increase productivity as well as nutritional value. In this context, the present study especially puts the light on two aspects, namely:
- • the non-tissue-specificity of post-transcriptional silencing in plants, which is greatly mobile, so that it might be induced locally and then spreads throughout the plant;
- • between Poaceae species miRNA interactions that will, supposedly, be a new and exciting field of study.
Finally, much remains to be learned about the mechanisms and roles of miRNAs in both normal physiological and pathological contexts before we can fully understand exogenous miRNA-mediated cross-species communication.
Disclosure of interest
The authors declare that they have no competing interest.
Acknowledgements
This study was supported by the Tunisian Ministry of Higher Education and Scientific Research. We are most grateful to Abdennour Sébéi (CRRGC, Béja, Tunisia) and Mejda Chérif (INAT, Tunis, Tunisia) for their logistic help.