

1 Introduction
Taxonomic studies of sharks and rays have led to an upsurge in new species descriptions within the last decade ([1,2]; and references therein). In particular, it appears that the number of species in the Coral Triangle region has been considerably under-estimated until recently [1]. Meanwhile, grave concern has been expressed over the risk of extinction in shallow-water species from a number of shark and ray families including Dasyatidae or stingrays. Overfishing of stingrays is particularly severe in the Coral Triangle region [3–5] and management is urgently needed. Species are the fundamental units in many studies of biogeography, community ecology and conservation ecology. Both conservation and fisheries management require that species be clearly identified and populations be delineated [6].
This paper focuses on the blue-spotted maskray, previously Neotrygon kuhlii (Müller and Henle, 1841) [7], a stingray species that inhabits Indo-West Pacific coral reefs, lagoons and slopes [8]. The blue-spotted maskray is heavily exploited in Southeast Asia, but its catch rate and mortality rates are poorly known and its population trends are unknown [9,10]. Authors have distinguished the “Java” (Java Sea) form of blue-spotted maskray from the “Bali” (Kedonganan) form, based on differences in size at birth and male size at maturity and treated them as different species [9]. Molecular population genetics offer the concepts and the practical tools for delineating populations, diagnosing closely related species, and detecting cryptic species. Cryptic species are defined as lineages with a substantial amount of genetic distinctiveness and no detectable morphological differences [11–13]. Ward et al. ([14]: 60–62) have noted that at the CO1 locus, “the D. kuhlii group showed considerable within species diversity … with four subgroups… One was the sole specimen from Australia (Queensland), one from the six specimens taken from Kedonganan fish market on Bali (Indonesia), one from the five specimens from Muara Angke fish market at Jakarta, Java (Indonesia) and one from the three specimens from the Penghu Islands (Taiwan). Average distances among and within these four groups were 2.80% and 0.18% respectively”. Subsequent genetic studies in the genus Neotrygon have focused on its phylogeny [15–17], and on the population genetic structure and phylogeography of the blue-spotted maskray [18,19]. Additional mitochondrial clades have been uncovered within the blue-spotted maskray [15,17,19]. These clades have a parapatric-like distribution [19]. Meanwhile, molecular markers have advanced the systematics of species in the genus Neotrygon: cryptic species have been uncovered within N. ningalooensis and N. picta [17], and the New Caledonian maskray N. trigonoides (Castelnau, 1873) [20] has been resurrected [16,21]. The hypothesis that the blue-spotted maskray may itself consist of a complex of multiple species has been raised repeatedly [14,15,17,22] and was also discussed by us [19,23]. We emphasized that the parapatric-like population structure uncovered in the Coral Triangle region “points to incipient speciation, where some degree of reproductive isolation has been achieved but ecological compatibility has not yet been reached”. Recently, Last et al. [24] described three new species (N. australiae, N. caeruleopunctata, N. orientale) previously under N. kuhlii and resurrected a fourth one, N. varidens (Garman 1885) [25]. Diagnostic morphological differences between the species were proposed but no in-depth assessment of inter-specific against infra-specific differences was included [24].
In the present paper, we compile all CO1 and cytochrome b gene sequences published thus far for the blue-spotted maskray and we add new sequences from samples collected in the western Indian Ocean and throughout the Indo-Malay archipelago, to construct a robust mitochondrial phylogeny and establish an updated distribution of the clades previously uncovered in the Coral Triangle region [14,15,17,19]. We assess whether the different clades, including those recently resurrected or erected as new species [24] correspond to evolutionary significant units that deserve the status of separate species.
2 Materials and methods
2.1 Ethics in sampling and information sharing
All specimens examined for the present study were independently captured by commercial fishers prior to being sub-sampled for DNA. Some of the sampling localities in our survey (Fig. 1) were in countries that have adopted a principle of state sovereignty over biological resources, including genetic resources with no commercial use [26]. This includes the Philippines, where sampling took place under a collaborative research agreement between the National Taiwan University, Taipei and the University of San Carlos, Cebu; and Indonesia, within the framework of a memorandum of understanding between LIPI, Jakarta and IRD, Marseille. Some of the samples were from West Papua, a former Dutch colony currently administered by Indonesia [27,28] that has no control over its own natural resources [29]. For equitable sharing of results and knowledge, the data produced in the course of our study will be made accessible from the following open-access repositories: Hal-IRD (http://www.hal.ird.fr/; hal@ird.fr), GenBank (http://www.ncbi.nlm.nih.gov) [30], and DIPnet (http://www.indopacificnetwork.wikispaces.com/).

Sampling sites of blue-spotted maskrays, Neotrygon spp. previously under Neotrygon kuhlii, including new samples from present study, samples from the literature, and additional haplotypes from BOLD, Cryobank and GenBank (details in Supplementary Table S1). Individuals were identified to species according to present revision, from either their COI or cytochrome b gene sequence. Roman numbers I–VIII designate the haplogroups or clades reported previously [19,23], four of which have since been described or redescribed [24] (Table 1). Surface of symbol (circle or triangle) proportional to sample size. A. I1, I2 and I3: clade I from, respectively, Tanzania (N = 7), Tamil Nadu (N = 7) and Vizakhapatnam (N = 1). V1 and V7: N. australiae from, respectively, Ningaloo Reef (N = 2) and Gulf of Carpentaria (N = 5). G: Guadalcanal maskray (N. kuhlii according to [24]), Honiara, Solomon archipelago (N = 1); open triangles: N. trigonoides; solid triangle: Vanikoro, type locality of N. kuhlii; inset: see Fig. 1B. B. Enlarged map of the Indo-Malay archipelago. II1, II2, II3 and II4: clade II from, respectively, Banda Aceh region (N = 11), Meulaboh (N = 3), Sibolga (N = 10) and Padang (N = 10). III1 and III2: clade III from, respectively, Andaman Sea coast of Thailand (N = 5) and Kuala Lama region (N = 25). N. varidens: IV1 from Kuala Selangor (N = 1), IV3 from Haiphong (N = 2), IV5 from Karimata Strait (N = 1/8), IV7 from Beibu Gulf (N = 1), IV8 from Sarawak (N = 1/4), IV13 from Penghu (N = 4), IV14 from Taiwan (N = 8). N. orientale: IV2 from Riau archipelago (N = 4), IV4 from the western Java Sea (N = 40), IV5 from Karimata Strait (N = 7/8), IV6 from Batang (N = 7), IV8 from Sarawak (N = 3/4), IV9 from Bali Strait (N = 7), IV10 from the western Sulawesi Sea (N = 11), IV11 from Sandakan (N = 2), IV12 from Makassar (N = 7), IV15 from Bone Bay (N = 2), IV16 from Tomini Bay (N = 3), IV17 from Cagayan (N = 12), IV18 from Kendari (N = 7), IV19 from Lapu-Lapu (N = 7), IV20 from Bitung (N = 12), IV21 from Tanjung Luar (N = 2). VI1, VI2, VI3 and VI4: N. caeruleopunctata from, respectively, Pelabuhan Ratu (N = 8), Sadang (N = 4), Bali Strait (N = 3) and southern Bali Island (N = 14). N. australiae: V2 from Tanjung Luar (N = 5), V3 from Labuan Bajo (N = 8), V4 from off Rote Island (N = 3), V5 from Tanjung Sulamo (N = 1), V6 from Kupang (N = 3), V8 from Torres Strait (N = 1), V9 from the northern Great Barrier Reef (N = 1). VII1, VII2: clade VII from, respectively, Ambon (N = 6) and Kei Islands (N = 20). VIII1, VIII2: clade VIII from, respectively, Pulau Numfor (N = 2) and Biak (N = 18). R: Ryukyu maskray, Ishigaki-shima, Ryukyu archipelago (N = 1).
2.2 Sampling
A total of 362 individuals, including 341 blue-spotted maskray and 21 New Caledonian maskray N. trigonoides, were utilized in the present study. This total includes 203 individuals whose CO1 or cytochrome b gene sequences were compiled from either the literature [14,16,17,19,31–38], the BOLD barcode database, the Cryobank DNA-barcode database (http://cryobank.sinica.edu.tw/chi/), or the GenBank nucleotide-sequence database; and 159 new individuals sampled from local fish-landing places throughout the Indo-Malay archipelago, in eastern Africa, and in New Caledonia. All individuals included in the present survey are listed in Supplementary Table S1, along with sampling details, GenBank accession numbers, voucher specimen details, and references. An overview of the density of blue-spotted maskray samples across the Indo-West Pacific, with a focus on the Coral Triangle region is given in Fig. 1.
2.3 Molecular analyses
A piece of skin or flesh ∼0.05 to ∼1 cm3 was removed, using surgical scissors, from the pelvic fin, or the pectoral fin, or the tail and was preserved in 95% ethanol at ambient temperature. DNA extraction was done using either the Viogene (Taiwan) tissue genomic DNA extraction protocol, or the DNEasy DNA extraction kit (Qiagen GmbH, Hilden, Germany). DNA was stored in 1X, pH 8.0 TE buffer (AppliChem, Darmstadt, Germany). Polymerase chain reaction (PCR) amplification of a fragment of the CO1 gene was done according to [19]. For the PCR-amplification of the cytochrome b gene, we used primers L14735 and CB7 [39,40]. The reaction volume was 15 μL and the reaction mixture contained 0.2 mM dNTPs, 1.5 μL 10× PCR buffer (Bioman, Taipei), 0.5 μM of each forward and reverse primers, 0.2 U Taq DNA polymerase (Bioman), and 1.0 μL template DNA. The PCR parameters were: initial denaturation at 94 °C for 4 min, followed by 35 cycles of denaturation (94 °C for 45 s), annealing (48 °C for 1 min), and extension (72 °C for 1 min), and a final extension step at 72 °C for 10 min. Intron 5 of the growth hormone gene, Gh5, was PCR-amplified using exon-anchored primers Gh5F (5′- A G G C C A A T C A G G A C G G A G C -3′) and Gh6R (5′-T G C C A C T G T C A G A T A A G T C T C C -3′) [41] setting the annealing temperature to 64.5 °C and the number of cycles to 35.
The PCR products were sequenced directly using the same primers as those used for the PCR. Sequences were analysed in an automated ABI Prism 377 sequencer (Applied Biosystems, Foster City, CA) at the sequencing facility of the Taiwan Normal University (Taipei).
2.4 Genetic data analysis
Nucleotide sequences, including those obtained in the present study, and homologous sequences retrieved from the BOLD, Cryobank, and GenBank databases (see Supplementary Table S1) were aligned using BioEdit [42]. Four datasets were analysed: (1) the CO1 gene sequence dataset, consisting of 330 blue-spotted maskray and N. trigonoides sequences aligned over a maximum length of 722 base pairs (bp); (2) the cytochrome b gene sequence dataset, consisting of 165 blue-spotted maskray and N. trigonoides sequences aligned over a maximum length of 1142 bp; (3) the concatenated CO1 + cytochrome b gene sequence dataset comprising 127 blue-spotted maskray and 6 N. trigonoides sequences; (4) the Gh5 intron sequence dataset, consisting of 18 blue-spotted maskray and N. trigonoides sequences aligned over a maximum length of 359 bp.
The phylogeny of concatenated CO1 + cytochrome b gene haplotypes was inferred by using the maximum-likelihood (ML) method under Mega6 [43]. The most likely nucleotide-substitution model, which was determined according to the Bayesian information criterion, was the Tamura–Nei model [44] where a discrete Gamma distribution (Γ = 0.76) was used to model evolutionary rate differences among nucleotide sites and invariable sites were allowed. The ML tree was rooted by choosing New Caledonian maskray N. trigonoides as outgroup [16]. The robustness of nodes in the tree was tested by bootstrap resampling.
The CO1 and cytochrome b gene sequence datasets (Supplementary Table S1) were used separately to assign individuals that were sequenced at only one of either locus to a clade uncovered from the phylogenetic reconstruction described above. For this, an ML tree was constructed using each of the two datasets under Mega6 and the assignment of a haplotype to a clade was determined visually from its relative position on the tree.
2.5 Delineation of cryptic species
The Automatic Barcode Gap Discovery (ABGD) algorithm proposed by Puillandre et al. [45] was used to identify gaps in the distribution of pairwise nucleotide distances. The ABGD algorithm detects the largest significant gap in the distribution of pairwise nucleotide distances and uses it for partitioning the dataset. Gap detection is then recursively applied to previously obtained groups to get finer partitions until there is no further partitioning [45]. The ABGD analysis was run on a matrix of 127 CO1 + cytochrome b gene sequences, trimmed to a core length of 1415 bp. Kimura's [46] two-parameter substitution model was selected and the minimum barcoding gap width was set to 1%.
Branch lengths between species are determined by speciation and extinction rates whereas branch lengths within a species reflect coalescent processes at the level of populations [47]. This results in the distinction of species when mitochondrial clades are substantially deeper than the coalescent haplogroups at the extremities of the tree. We ensured that each of the previously identified deep mitochondrial clades was genetically distant from its nearest neighbour by several times the mean intra-clade distance. For this, pairwise intra-clade and between-clades genetic distances were estimated on the concatenated CO1 + cytochrome b gene sequence matrix using Mega6.
To delimit potential cryptic species, we applied the general mixed Yule-coalescent (GMYC) algorithm [47] as implemented in the program Splits under R [48,49]. This algorithm uses an ultrametric tree to differentiate branching patterns consistent with coalescent vs. speciation processes, which provides a threshold time for the transition between populational and species-level processes. We selected the single-threshold option. The ultrametric tree was constructed using Beast v. 1.7 [50] based on the same matrix (127 individuals × 1415 bp) as that used for the ABGD analysis. Outgroup N. trigonoides was excluded from the analysis, which focused on the blue-spotted maskray exclusively. A non-calibrated relaxed lognormal clock, and a constant coalescent tree were assumed. The model of nucleotide substitution was TN93 + G + I (see Section 2.4). Ten million generations of Markov-Chain Monte Carlo analysis were run, of which we sampled a total of 10,000 genealogies. Length of burn-in was determined by visual inspection of traces in Tracer v. 1.5 [50]. A single analysis was run, for which convergence was reached rapidly. The TreeAnnotator v. 1.7. software [50] was used to produce the final tree, based on maximum clade credibility and mean node height.
3 Results
The complete set of partial CO1- and cytochrome b gene sequences, which comprises both those compiled from the literature and from nucleotide databases, and those produced in the present study are presented in Supplementary Table S2. New CO1 gene sequences have GenBank accession numbers KU497912-KU498038 and KU521523; new cytochrome b gene sequences have GenBank accession numbers KU497752-KU497911 (details in Supplementary Table S1).
The ML tree of concatenated (CO1 + cytochrome b) gene nucleotide sequences, rooted by N. trigonoides, showed 9 main statistically supported clades (Fig. 2). Here, we distinguished the sister-subclades IVa (N. orientale) vs. IVb (N. varidens) within previous Clade IV [19,23]. The 7 other clades were Clades I–III, VII, VIII identified previously [19,23], N. australiae (our former Clade V), and N. caeruleopunctata (our former Clade VI). Variation in clade nomenclature across studies [15,17,19,23,24] is summarized in Table 1. Pairwise nucleotide distances between clades ranged from 0.014 to 0.029 (average 0.022), that is, two to 29 times higher than within-clade average distances (0.001–0.007; average 0.004) (Table 2).
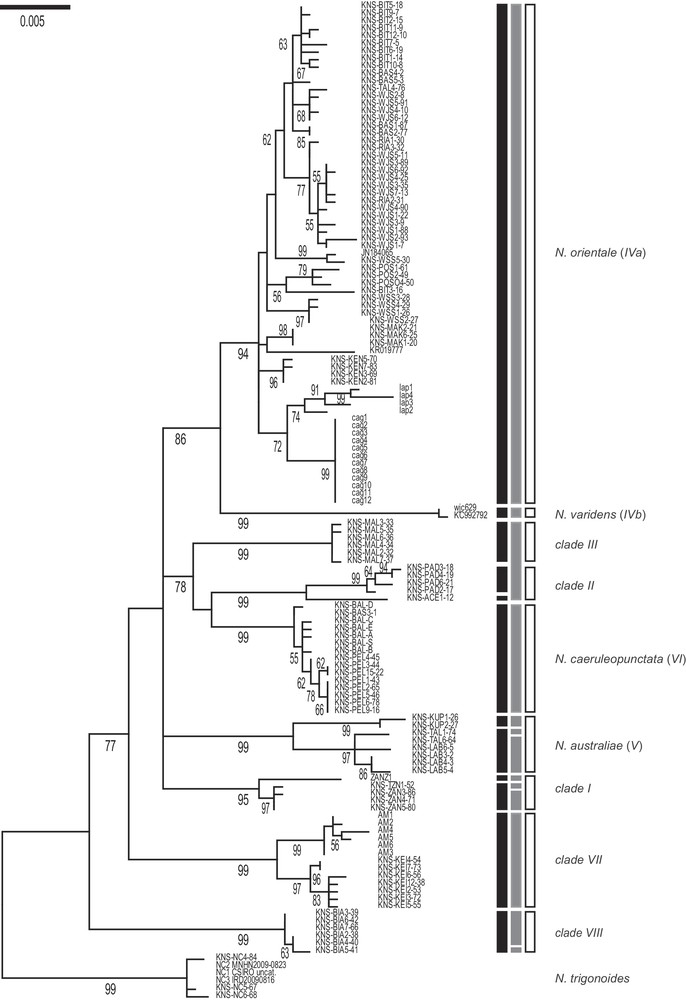
Maximum-likelihood phylogeny (Mega6 [43]; Tamura-Nei model with gamma-distributed rate differences among sites + invariant sites [44]; partial deletion) of blue-spotted maskrays Neotrygon spp. previously under N. kuhlii, based on nucleotide sequences of the concatenated CO1+ cytochrome b gene fragments. A total of 133 individual sequences (Supplementary Table S1), aligned over 1640 bp, was retained in the final dataset, after all positions with less than 95% site coverage had been eliminated. N. trigonoides was used as outgroup [16]. Roman numbers I–VIII designate the haplogroups or clades reported previously [19]. Numbers at a node are bootstrap scores (from 600 bootstrap resampling runs). Vertical black bars summarize the partition obtained according to Puillandre's gap detection analysis, ABGD. Vertical grey bars summarize the partition into species obtained from GMYC analysis (Splits; single-threshold option). Open bars: partition into species finally retained considering tree topology, nucleotide divergence between lineages, and results of ABGD and GMYC analyses.
Blue-spotted maskray, Neotrygon spp. (formerly N. kuhlii) and New Caledonian maskray, N. trigonoides. Lineage nomenclature utilized in present study, and its relationship to recent genetics-based studies. Homology between lineages was assessed by the phylogenetic placement of individuals sequenced at different loci and/or by the geographic origin of a sample. Genetic markers indicated in brackets.
| Present study | Naylor et al., 2012 [15] |
Arlyza et al., 2013 [19,23] |
Puckridge et al., 2013 [17] |
Last et al., 2016 [24] |
| (CO1, cytb) | (ND2) | (CO1) | (CO1 + 16S, RAG-1) | (CO1) |
| N. australiae | N. kuhlii 4 | N. kuhlii Clade V | Clade 5 | N. australiae |
| N. caeruleopunctata | – | N. kuhlii Clade VI | Clade 6 | N. caeruleopunctata |
| N. orientale | N. kuhlii 1 | N. kuhlii Clade IV | Clades 2, 3 | N. orientale |
| N. varidens | N. kuhlii 2 | N. kuhlii Clade IV | Clade 1 | N. varidens |
| Clade I | N. kuhlii 3 | N. kuhlii Haplogroup I | Clade 8 | – |
| Clade II | – | N. kuhlii Clade II | – | – |
| Clade III | – | N. kuhlii Clade III | Clade 7 | – |
| Clade VII | – | N. kuhlii Clade VII | – | – |
| Clade VIII | – | N. kuhlii Clade VIII | – | – |
| Guadalcanal maskray | – | – | – | N. kuhlii |
| Ryukyu maskray | – | N. kuhlii Clade IV | Clade 4 | – |
| N. trigonoides | – | N. trigonoides | Clade 9 | N. trigonoides |
Blue-spotted maskray, Neotrygon spp. Mean ± SD pairwise genetic distances (number of nucleotide substitutions per site), estimated from concatenated CO1 + cytochrome b gene fragment sequences (total N = 133) (Mega6 [43]; Tamura-Nei model [44]). N sample size.
| No. | Clade | N | Mean pairwise genetic distance | |||||||||
| Within clade | Between clades (net) | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||||
| 1 | N. australiae | 8 | 0.007 ± 0.001 |
|||||||||
| 2 | N. caeruleopunctata | 15 | 0.002 ± 0.001 |
0.020 ± 0.003 |
||||||||
| 3 | N. orientale | 67 | 0.007 ± 0.001 |
0.017 ± 0.003 |
0.014 ± 0.003 |
|||||||
| 4 | N. trigonoides | 6 | 0.001 ± 0.001 |
0.032 ± 0.005 |
0.032 ± 0.005 |
0.028 ± 0.004 |
||||||
| 5 | N. varidens | 2 | 0.001 ± 0.001 |
0.026 ± 0,004 |
0.025 ± 0.004 |
0.016 ± 0.003 |
0.037 ± 0.005 |
|||||
| 6 | Clade I | 5 | 0.003 ± 0.001 |
0.018 ± 0.003 |
0.016 ± 0.003 |
0.014 ± 0,003 |
0.028 ± 0.004 |
0.024 ± 0.004 |
||||
| 7 | Clade II | 5 | 0.006 ± 0.001 |
0.023 ± 0.004 |
0.015 ± 0.003 |
0.020 ± 0.003 |
0.033 ± 0.005 |
0.027 ± 0.004 |
0.020 ± 0.003 |
|||
| 8 | Clade III | 6 | 0.001 ± 0.000 |
0.022 ± 0.004 |
0.017 ± 0.003 |
0.017 ± 0.003 |
0.031 ± 0.005 |
0.026 ± 0.004 |
0.019 ± 0.003 |
0.018 ± 0.003 |
||
| 9 | Clade VII | 13 | 0.005 ± 0.001 |
0.022 ± 0.004 |
0.021 ± 0.003 |
0.019 ± 0.003 |
0.030 ± 0.005 |
0.027 ± 0.004 |
0.018 ± 0.003 |
0.027 ± 0.004 |
0.026 ± 0.004 |
|
| 10 | Clade VIII | 6 | 0.001 ± 0.000 |
0.026 ± 0.004 |
0.023 ± 0.004 |
0.024 ± 0.004 |
0.030 ± 0.005 |
0.030 ± 0.005 |
0.024 ± 0.004 |
0.029 ± 0.004 |
0.028 ± 0.004 |
0.027 ± 0.004 |
Through automatic gap determination using Puillandre's ABGD algorithm, 12 distinct mitochondrial lineages were found, separated from each other by a gap in genetic distance ≥ 1% (Fig. 2). The ABGD partitioning was consistent with the topology of the ML tree. In particular, Clades II and III were recognized as distinct. The distinction between Clades IVa (N. orientale) and IVb (N. varidens) was confirmed. Two subclades were identified within each Clade I (Indian-Ocean maskray), and N. australiae. All 9 main clades of the ML tree were confirmed by the ABGD partitioning (Fig. 2).
Based on the ultrametric tree of blue-spotted maskray mitochondrial haplotypes (not shown), the likelihood of the null model (i.e., where all mitochondrial sequences belong to a single species) was 1141.9, significantly lower than the maximum likelihood of the GMYC model (1145.1); the likelihood ratio was 6.477 (P = 0.039). The estimated number of clusters within the blue-spotted maskray was 13 (95% confidence interval: 10–46), whose boundaries often coincided with the above 9 clades (Fig. 2). The distinction between clades IVa (N. orientale) and IVb (N. varidens) was thus confirmed, as was that of N. caeruleopunctata. The main exception was the lack of recognition of clades II and III as separate entities. The individuals collected in the Indian Ocean (our clade I [19]) formed a distinct series of three clusters, as did N. australiae, and a subcluster was found within clade VIII (Fig. 2).
The partition into species that was finally retained considered the tree topology, the nucleotide divergence between lineages, and the results of the ABGD and GMYC analyses. This partition was consistent with the current taxonomy of species, leaving unchallenged the four nominal species N. australiae, N. caeruleopunctata, N. orientale and N. varidens (Fig. 2). The inability of the GYMC algorithm to separate clade II from clade III may be caused by a lack of power, as the two clades were approximately equally distant from each other as they were each from N. caeruleopunctata (Table 2).
Once cryptic species of the blue-spotted maskray had been identified, it was possible to tentatively assign each individual in the total sample (Supplementary Table S1) to either of them, using the partial CO1- or cytochrome b gene sequence. The ML tree of CO1 gene sequences, rooted by N. trigonoides (Supplementary Fig. S1), showed the same 9 haplogroups as those highlighted in the phylogenetic tree of Fig. 2, plus a tenth lineage represented by the single CO1 gene haplotype from the Ryukyu Islands (GenBank AB485685; [31]). All haplogroups had acceptable to strong statistical support, except the haplogroup representing individuals from the Indian Ocean. All 330 individuals characterized by their CO1 gene sequence (except a single individual from India; GenBank HM467799) unambiguously clustered with either one of the 9 lineages of Fig. 2, or with N. trigonoides. The ML tree of cytochrome b gene sequences, rooted by N. trigonoides (Supplementary Fig. S2), similarly showed the same 9 haplogroups as those highlighted in Fig. 2. Eight out of the 9 haplogroups had strong statistical support, with bootstrap scores between 93 and 100%. All 165 individuals characterized by their cytochrome b gene sequence unambiguously clustered with either one of these 9 blue-spotted maskray lineages, or with N. trigonoides.
Using the sampling locations of the individuals (Supplementary Table S1), it was possible to delineate the geographic distribution of each clade (Fig. 1). The 9 clades of the blue-spotted maskray and the closely related N. trigonoides had parapatric distributions (Fig. 1). Zones of contact were identified between N. orientale and N. varidens in the southeastern part of the South China Sea, between N. caeruleopunctata and N. orientale in Bali Strait, and between N. australiae and N. orientale in the Lombok area (Fig. 1B). Similar zones of contact are expected between Clades II and III east of the northern tip of Aceh, between Clades III and N. varidens in the Malacca strait, between N. caeruleopunctata, N. orientale, and Clade II west of the southwestern tip of Java, and between N. australiae and N. caeruleopunctata east of southern Bali (Fig. 1B). Other contact zones may be possible between N. orientale and Clade VII, between N. australiae and Clade VII, and between Clade VII and Clade VIII (Fig. 1B).
Eighteen Neotrygon spp. individuals (out of 37 tested) were successfully sequenced at the Gh5 intron locus. The sequences have GenBank accession numbers KU497734–KU497751. The ML tree derived from the alignment of sequences, rooted by N. trigonoides, is presented in Fig. 3A.
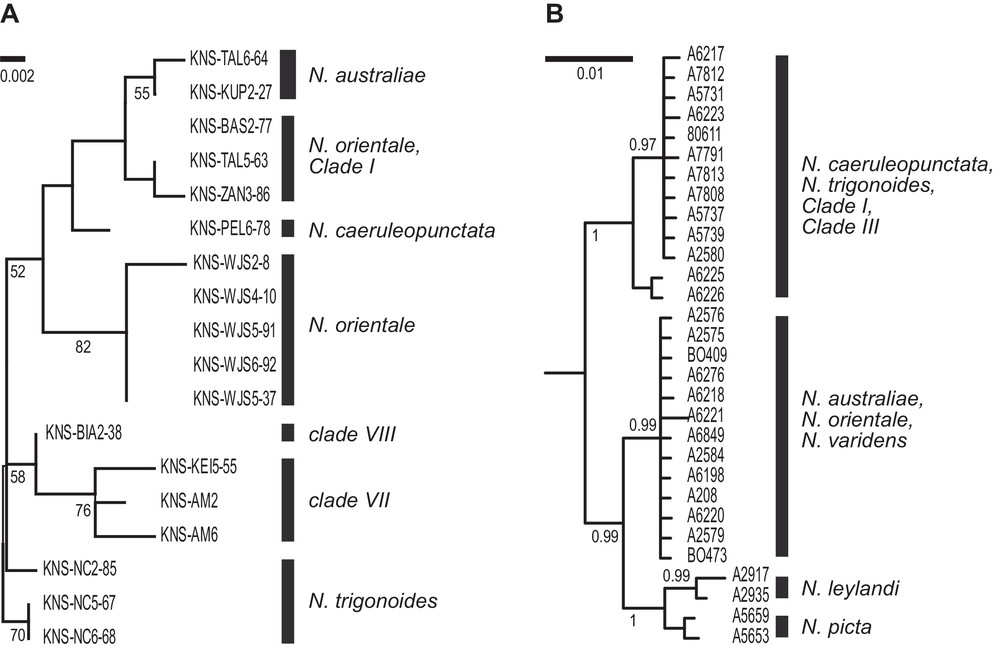
Nuclear phylogenies of the blue-spotted maskray, Neotrygon spp. A. Maximum-likelihood phylogeny (Mega6 [43]; Jukes-Cantor model [51]) with gamma-distributed rate differences among sites; partial deletion] of Gh5 intron sequences. A total of 18 individual sequences aligned over 356 bp were retained in the final dataset, after all positions with less than 95% site coverage had been eliminated. N. trigonoides was used as outgroup [16]. Numbers at a node are bootstrap scores (from 1000 bootstrap resampling runs). B. Summary of the Bayesian phylogeny based on partial RAG-1 sequences (redrawn from Fig. 2a of [17]; lineage names edited according to Table 1). Numbers at a node are posterior probabilities.
4 Discussion
The present results enforce Ward et al.’s [14] hypothesis that the blue-spotted maskray is a complex of cryptic species, for the reasons developed in the following.
4.1 Coalescence patterns
Nucleotide distances between major blue-spotted maskray clades were several times higher than distances within, confirming previous observations based on preliminary geographic sampling [14,15,17,19]. Denser geographic sampling (present study) did not lead to a dramatically altered ratio of genetic distance between clades to that within, compared to Ward et al.’s results [14]. The gaps in nucleotide distance remained consistent with the existence of 9 main lineages within the blue-spotted maskray. The GMYC algorithm determined that the coalescence patterns within a lineage vs. patterns between generally conformed to the expectations of infra-specific vs. inter-specific processes.
4.2 General concordance of mitochondrial and nuclear differences
In sharks and rays, short mitochondrial DNA sequences at the CO1 locus (or CO1 DNA barcodes) have proven their suitability to identify up to 99% species [14]. This high barcoding success might be related to the apparent absence of mitochondrial introgression in this vertebrate group [14], itself pointing to a likely low incidence of inter-specific hybridization. This may be an indirect consequence of complex pre-mating behaviour, which prevents hetero-specific pairs, or of mismatch in the morphologies of male and female genitalia, which hampers copulation. Complex pre-mating behaviour has been reported in sandtiger shark [52] and New Caledonian maskray [53]. Mismatch of male–female copulatory apparatuses has been hypothesized for whiprays of the Himantura uarnak (Forsskål, 1775) [54] species complex [35]. Other hypotheses for pre-mating isolation involve chemical or visual cues [17]. Because cases of mitochondrial introgression are virtually unknown in sharks and rays [6], one expects that patterns of inter-specific genetic differentiation at nuclear loci to be parallel to those at the mitochondrial locus.
Nuclear markers have also been used to characterize blue-spotted maskray populations [17,18]. Significant allele-frequency differences at 2/4 intron loci were observed between adjacent populations from southern Java and Bali Strait [18], corroborating the distinction between previous Clades IVb and VI ([19,23]; present study), hence the recognition of, respectively, N. orientale and N. caeruleopunctata as separate species. Two major clades were observed at the nuclear locus RAG-1 [17] (Fig. 3B), that distinguished the lineage from the eastern Andaman Sea (our Clade III) and those from the adjacent South China Sea (N. varidens) and Java Sea (N. orientale). Similarly, the RAG-1 marker allowed the distinction between the lineage from southern Bali (N. caeruleopunctata; our previous Clade VI) and that of the adjacent Lombok Island (N. australiae; our previous Clade V) [17]. Additional information from nuclear marker Gh5 was produced in the present study. Nucleotide sequences at intron locus Gh5 confirmed the distinction between N. orientale on the one side, and N. australiae and N. caeruleopunctata on the other side, although some other Gh5 sequences of individuals determined as N. orientale according to their mitochondrial haplotypes appeared to cluster with N. australiae and with Clade I. This may either signal mitochondrial introgression, or reflect incomplete lineage sorting at the Gh5 locus. In addition, at this locus, samples from the Moluccas and West Papua (respectively, our lineages VII and VIII) were distinct from all the other samples, and also from each other.
4.3 Parapatric distribution
Repeated lowering of the sea level caused by global temperature oscillation [55] has led to the repeated fragmentation of the marine habitats of the Indo-Malay archipelago in the Pleistocene [56]. In shallow-water species like the blue-spotted maskray, this has caused repeated depletion and fragmentation of the populations, followed by repeated recolonisation of shallow-water habitats once the sea level rose again, and secondary contact between long-isolated populations. Population collapse reduces genetic diversity by the random fixation of a subset of alleles and subsequent population expansion favours new, nascent diversification. This succession of isolation episodes with cyclic population collapses, followed by rapid population expansions, is thought to have favoured speciation in the blue-spotted maskray [17,19].
Secondary contact after recolonisation of inundated shallow-water habitat may have been a factor reinforcing genetic isolation instead of promoting re-homogenisation. At the scale of an individual's lifetime, the blue-spotted maskray is a sedentary species. Long-term site fidelity has been inferred from tagging experiments in the closely-related N. trigonoides, with similar results for females and males [53]. Relatively poorly dispersing species often form well-delineated parapatric boundaries [57]. One of the proposed mechanisms is that of a narrow and stable hybrid zone, which acts as a geographic barrier to cross-dispersal [57]. While this hypothesis remains to be tested in the blue-spotted maskray species complex, the present study identified several of these parapatric boundaries, on which future genetic studies should focus.
5 Conclusion
Sound conservation and fisheries management requires knowledge on population genetic structure, so as to delineate demes, which are the basic units on which to conduct meaningful demographic analyses. Previous attempts at investigating the population genetic structure of blue-spotted maskray have found substantially higher levels of population differentiation than usual, leading to suspecting cryptic species. Here, molecular markers distinguished nine main separate lineages within the blue-spotted maskray previously under N. kuhlii. These lineages qualify as distinct species, based on levels of genetic divergence, coalescence patterns, concurrent differentiation at nuclear loci, and parapatric distribution, which points to reproductive isolation. We propose that repeated dramatic demographic lows and highs in the Pleistocene, combined with individual sedentarity and possibly homogamy, have driven speciation in the blue-spotted maskray.
Disclosure of interest
The authors declare that they have no competing interest.
Acknowledgments
We thank Jean-Dominique Durand (IRD, Ho-Chi Minh Ville), Nicolas Hubert (IRD, Cibinong), Samuel P. Iglesias (MNHN, 387 Concarneau) and Robert D. Ward (CSIRO, Hobart) for exchanging viewpoints, and five anonymous reviewers for insightful comments on previous versions of this paper. DeAnne Olsen Cravaritis (NCBI, Bethesda) kindly edited the files deposited in GenBank. A. Pavan Kumar (ICAR-CIFE, Mumbai) kindly provided details on specimen VIZNK-01. The fishers, researchers, and travellers who participated in the collection of samples are listed in Appendix A. The background map of the Indo-West Pacific was edited from images downloaded from Digital Vector Maps, San Diego (http://digital-vector-maps.com/). Funded by IRD-UMR 250, NTOU, and LIPI-P2O. The present report is a contribution of the IRD / LIPI project on the population genetics of Elasmobranchs in the Indonesian archipelago (PARI). Designed the study: PB, KNS. Contributed reagents or materials or analysis tools: ISA, PB, TBH, KNS. Did the experiments: ISA, KNS. Analyzed and interpreted the data: PB, TBH, KNS. Wrote the paper: PB.