


 CC-BY 4.0
CC-BY 4.0
La version française de l’article est disponible à la suite de la version anglaise
1. Translational research from medical to plant research
Translational research is a concept developed in the field of medical research in the 80’s consisting in translating (moving) basic science discoveries more quickly and efficiently into practice, and summarize as “from bench to bed” [1]. In this context, translational research in medicine consists in favoring exchanges between laboratories and hospitals for turning appropriate biological discoveries into drugs, medical or therapeutic applications that can be used in the treatment of patients. Translational research aims also at exploiting preclinical research in animal models (predominantly murine species) in human health [2], questioning about the adequate model species to be used for each investigated human pathology and disease [3] to conduct translational research. This concept has been transposed to plant research and more precisely into crop breeding, consisting in converting basic knowledge from models (also referenced to as pivot species) into applications in crops to sustainably support food security and agriculture, and summarize as “from pot to plot” [4]. In this context, translational research consists in developing and exploiting methods and tools to make findings obtained in one species on a particular trait or phenotype exploitable to all species for which the considered phenotype or trait is of interest in breeding. Translational research in crops then raise the question on how to optimize the use of genomic and genetic knowledge from model species to improve crop breeding?
Since the beginning of the current century, with the first release of the Human (finally completed in 2022 [5]) and Arabidopsis thaliana [6] genomes, nowadays more than 100 plant (mainly crops) genomes of high quality (i.e. fully assembled at the chromosome level) are now available in the public domain (Figure 1). The constant improvement of sequencing technologies and reduction of costs make possible now the access to the genome of any living (as well as extinct [7]) organisms. Taking advantage of such resources, we illustrate in the Figure 2 the theoretical process of conducting translational research from models (such as Arabidopsis) to crops as well as from crops to crops with (1) the identification (from the literature) of genes driving key traits or phenotypes in one model species or a particular crop species (then referenced as pivot), (2) the identification of the best conserved genes in species for which these traits or phenotypes are of interest in breeding, (3) the functional validation of the conserved genes to show that they determine the same traits or phenotypes in the considered crops as it is known in the model or pivot species, (4) the identification of the genotypes carrying the best alleles for breeding for the conserved gene driving the trait or phenotype of interest, and (5) the field evaluation of such genotypes to quantify phenotype and trait improvement.
Sequenced plant genomes. The figure illustrates high-quality plant genome sequences (assembled at the chromosome level) according to the year of public release (x-axis) and size in megabase (y-axis).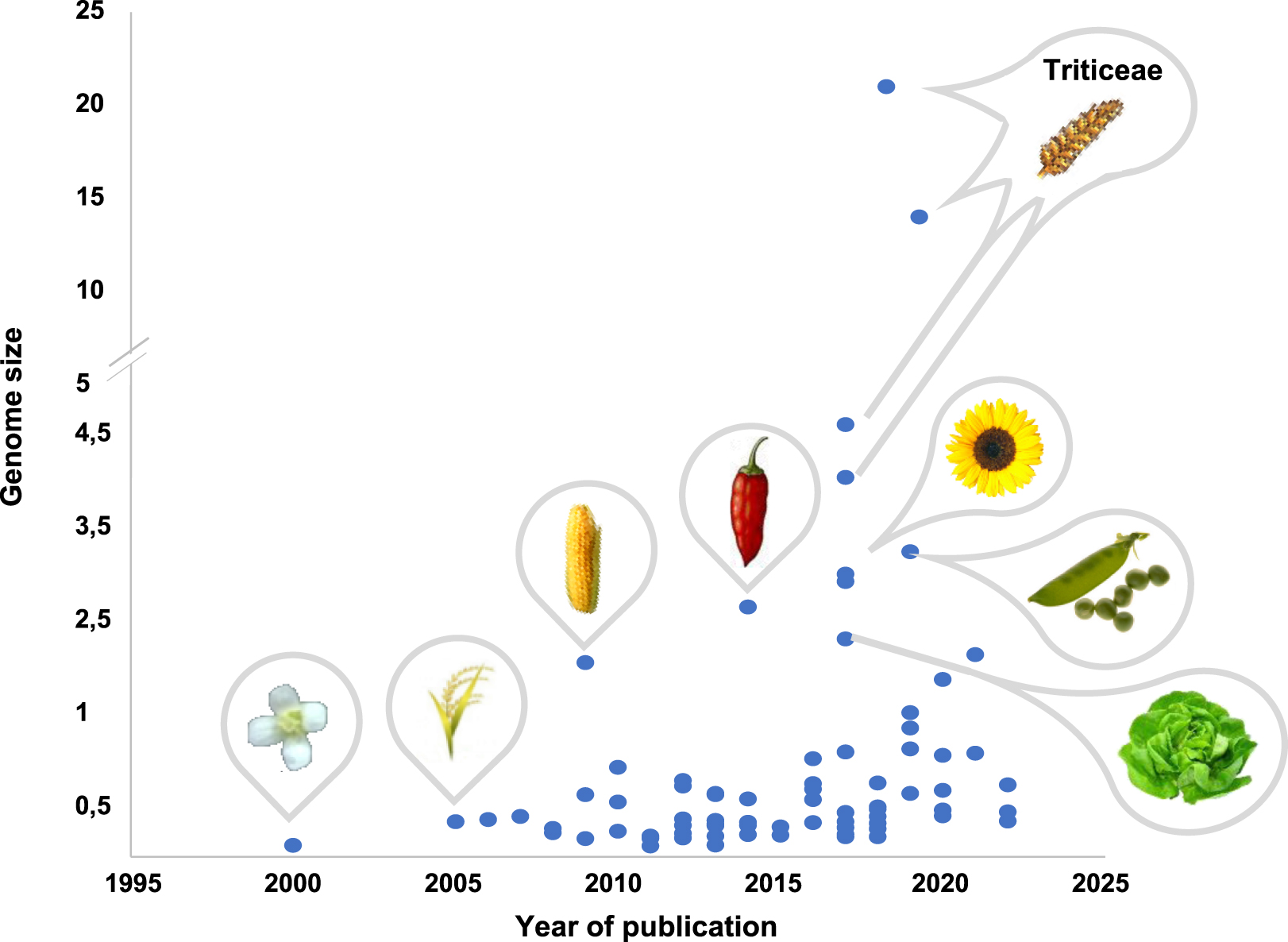
Process of translational research in crops. Top—Illustration of the method in reconstructing ancestral genomes in silico from the identification of genes (colored rectangles) completely conserved-duplicated (in red), partially conserved-duplicated (in blue) or specific from one species (in black) inherited from speciation and duplication events in the course of plant evolution (adapted from Pont et al. [8]). Bottom—Illustration of the process in conducting translational research in crops through (1) the identification of genes driving key traits or phenotypes in model or pivot species, (2) the identification of the best conserved genes between species, (3) the functional validation of the conserved genes, (4) the identification of genotypes carrying the best alleles, (5) the field evaluation of the genotypes in breeding schemes.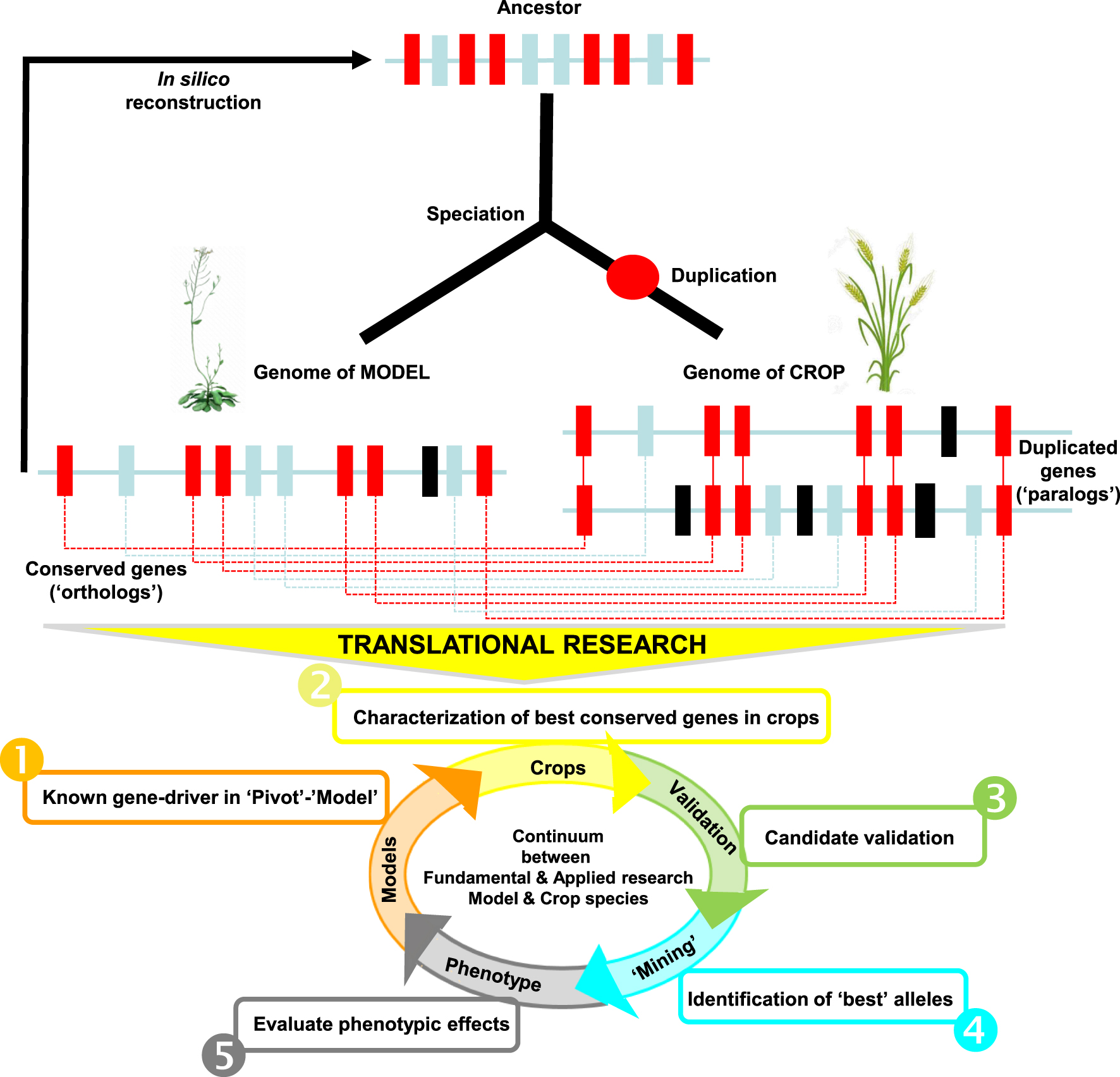
2. Comparative genomics in Angiosperms
Following the access to plant genomes since the year 2000, the constant development of methods and strategies offer the opportunity to identify conserved genes to conduct translational research from models to crops and crops to crops. To that regards, several public tools are available to query comparative genomics data between plant genomes such as PLAZA [9], Gramene [10], Ensembl [11], CoGe [12], Genomicus [13]. Flowering plants, or angiosperms, emerged some 120 to 250 million years ago [14] to rapidly diversify into 350,000 species alive today divided into two main groups, the monocots and eudicots [15]. Angiosperm evolution is characterized by recurrent whole genome duplication (WGD), also referenced as polyploidy events [16] that makes difficult the accurate identification of conserved genes between plant genomes from publicly available web tools (Figure 1).
As example and based on the known pattern of polyploidization events in flowering plants, genome comparison of rice (a crop from the monocot family) and Arabidopsis (a model from the eudicot family) involves the identification and investigation of similarities and differences between eight genomic regions in rice (that experienced 𝜏, 𝜎 and 𝜌 polyploidization events since the angiosperm ancestor) and twelve in Arabidopsis (that experienced 𝛼, 𝛽 and 𝛾 polyploidization events since the angiosperm ancestor), Figure 3. Conducting efficient translational research between Arabidopsis (model) and rice (crop) then necessitates for each gene of interest identified in Arabidopsis to deliver the complete catalogue of eight possible conserved gene in rice in order to select the one that may have retained the function of interest identified in Arabidopsis. Available tools and methods often fail to identify all the previous expected conserved genes between modern genomes inherited by known evolutionary events since the angiosperm ancestor, then resulting in an underestimation of gene conservation between species and, finally, reducing the efficiency of translational research in crops.
Evolution of modern flowering plants from inferred ancestral karyotypes. Illustration of the phylogenetic tree of angiosperm crop and model species, with polyploidization events indicated with coloured dots (duplications in red and triplications in blue), using a colour code highlighting the ancestral protochromosomes (left) and their evolution into modern genomes (right) for major botanical families (grasses, Solanaceae, Cucurbitaceae, Brassicaceae, Rosaceae and legumes), adapted from Pont et al. [7].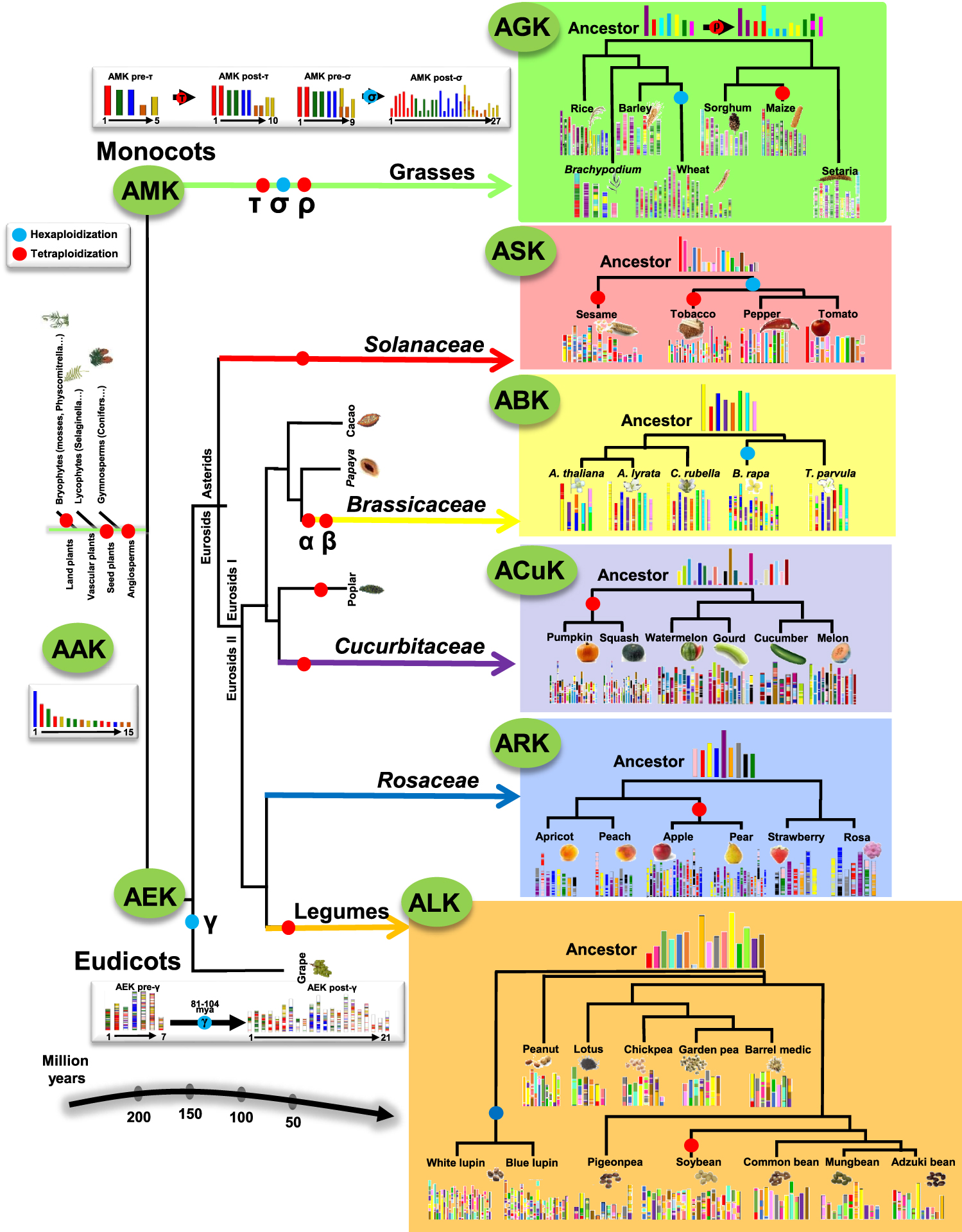
3. Translational research from inferred ancestral genomes
All angiosperm species experienced numerous whole genome duplication (or polyploidization) events that created different degrees of gene conservation between species so that such evolutionary events need to be precisely taken into account to preform translation research in transferring efficiently information about gene structure and function from one species to another. This is the goal of paleogenomics, which consists of reconstructing ancestral genomes of extinct founders, by comparing the genomes of modern species. From an ancestral (possibly extinct) genome that evolved into different extant species through speciation and distinct chromosome shuffling events (fusion, fissions, inversions and translocations), each of the ancestral chromosomes will derive a subset of modern chromosomal regions sharing conserved genes. Following this evolutionary evidence when reconstructing ancestral karyotypes in silico, comparative genomics of modern genomes should produce genomic fragments showing independent (non-shared) syntenic blocks, referred to as conserved ancestral regions (CARs), which are considered as ancestral chromosomes in the inferred ancestral karyotype [7]. The genomes of modern monocot and eudicot crops can then be connected (or “bridged”) via the inferred ancestral genomes delivering the complete repertoire of conserved genes, and putatively functions and traits, between for example the model species Arabidopsis and major cereals such as wheat, maize and rice (Figure 2).
High resolution plant paleogenomics data are now available to conduct translational research between angiosperm species using several inferred ancestors (see Figure 3): AAK (Ancestral Angiosperm Karyotype of 15 chromosomes carrying 10,000 to 20,000 genes conserved between modern angiosperms [17]), AGK (for Ancestral Grass Karyotype with 12 protochromosomes and 14,241 protogenes [18, 19]), AEK (for Ancestral Eudicot Karyotype with 21 chromosomes and 9022 protogenes [20]), ARK (for Ancestral Rosaceae Karyotype with 9 protochromosomes and 8861 protogenes [21]), ABK (for Ancestral Brassicaceae Karyotype with 8 protochromosomes and 20,037 protogenes [22]), ACuK (for Ancestral Cucurbitaceae Karyotype with 22 protochromosomes and 17,969 protogenes [23]), ALK (for Ancestral Legume Karyotype with 16 protochromosomes and 13,181 protogenes [24, 25]) and ASK (for Ancestral Solanaceae Karyotype with 17 protochromosomes and 17,879 protogenes). Such inferred ancestral genomes provide the robust and complete catalog of conserved genes between plant genomes taking into account evolutionary events such as polyploidizations of modern species of interest as illustrated in the Figure 3 and made publicly available at https://urgi.versailles.inrae.fr/synteny. The inferred ancestral genomes can be considered as a very useful resource for translational genomics research comparing sequenced flowering plant genomes, particularly model and crop species. For example, ALK delivers a catalog of 13,181 conserved genes between legumes offering the opportunity, for each gene of interest identified in Medicago Truncatula (model) to get access to the complete set of conserved genes in pea or lentil for example. In the same way, AGK delivers a catalog of 14,241 conserved genes to conduct translational research from Brachypodium (model) or rice (crop) to wheat (crop).
4. Translational research of traits and genes in crops
Paleogenomics data makes possible to efficiently transfer the knowledge acquired in a species (considered as a model or pivot) from any gene (driving a particular function or phenotype) to all other plant species for which the driven trait is of interest. For example, such approach has been successfully applied in cucurbits on fruit size and shape [26]. Consensus Quantitative Trait Loci (QTL) for cucumber, melon, and watermelon were associated with genes cloned in Arabidopsis, tomato, and rice that encode proteins containing the characteristic CNR (cell number regulator), CSR (cell size regulator), CYP78A (cytochrome P450), SUN, OVATE, TRM (TONNEAU1 Recruiting Motif), YABBY, and WOX domains that can be considered as best candidate driving the investigated traits. Applied to cereals, capitalizing on the knowledge gained on gene functions in rice or Brachypodium, FZP gene for FriZzy Panicle from the APETALA (AP2)/Ethylene Response Factor (ERF) family has been shown to be conserved [27] and driving surnumerary spikelets (a yield component) in the spike of several species [28, 29]. In the same vein, Trehalose 6-phosphate (T6P) has also been shown to be implicated in the improvement of yield in different environments (such as grain number and grain filling traits and abiotic stress resilience) for several crops [30]. As a case example of the paleogenomics-based translational research approach, the Figure 4a illustrates the PEPC genes known to be involved in the major C3/C4 trait, with C4 photosynthesis resulting in a greater photosynthetic capacity than C3 species. Starting from the Arabidopsis candidate gene (AT1G53310, [31]), the reconstructed Ancestral Angiosperm Karyotype (AAK) automatically delivers 8 to 3 gene-to-genes relationships expected from the known evolutionary history of between monocots and eudicots (Figure 2), all these chromosomal regions being derived from the same ancestral chromosome. A broader view of the conservation between 12 angiosperm modern species of one of the PEPC gene copies revealed an interesting evolutionary pattern, in which PEPC was conserved only in C3 species and was entirely absent from C4 plants (maize and sorghum). This example highlights the power of information delivered from the reconstructed ancestor for the extraction of candidate genes (i.e. from the Arabidopsis eudicot model to grass crops representative of monocots, such as rice) for traits of interest, for subsequent validation of their role in driving C3 to C4 transition. Translation research of key genes driving agronomically important traits can also benefit from knowledge gained outside the plant kingdom. The DGAT1 gene identified to be involved in milk and meat production traits in cattle, buffalo, goat and sheep [32] has been proven to also be a key determinant of oil content and composition in maize [33]. This result opens novel possibilities to perform translational research between plants and animals in the future.
Translational research of genes and traits. (a) Simplified diagram of the carbon fixation pathways operating in C3 and C4 plants, adapted from (http://www.intechopen.com/books/abiotic-stress-in-plants-mechanisms-and-adaptations/c4-plants-adaptation-to-high-levels-of-co2-and-to-drought-environments). Abbreviations: C3, three-carbon organic acids; C4, four-carbon organic acids; C5, ribulose-1,5-bisphosphate; PCR, photosynthetic carbon reduction cycle; PEPC, phosphoenolpyruvate carboxylase; Rubisco, ribulose-1,5-bisphosphate carboxylase/oxygenase. PEPC (ATG1G53310 candidate from Arabidopsis thaliana) gene conservation between species is illustrated with genes as grey boxes connected by grey lines between 12 modern angiosperm genomes, with deletion events indicated by purple crosses. (b) Expansion and contraction in oak (y-axis) of gene families in their number of genes, relative to 15 other eudicot species (x-axis), with gene families expanded during oak evolution labelled from 1 to 9 (adapted from Plomion et al. [34]).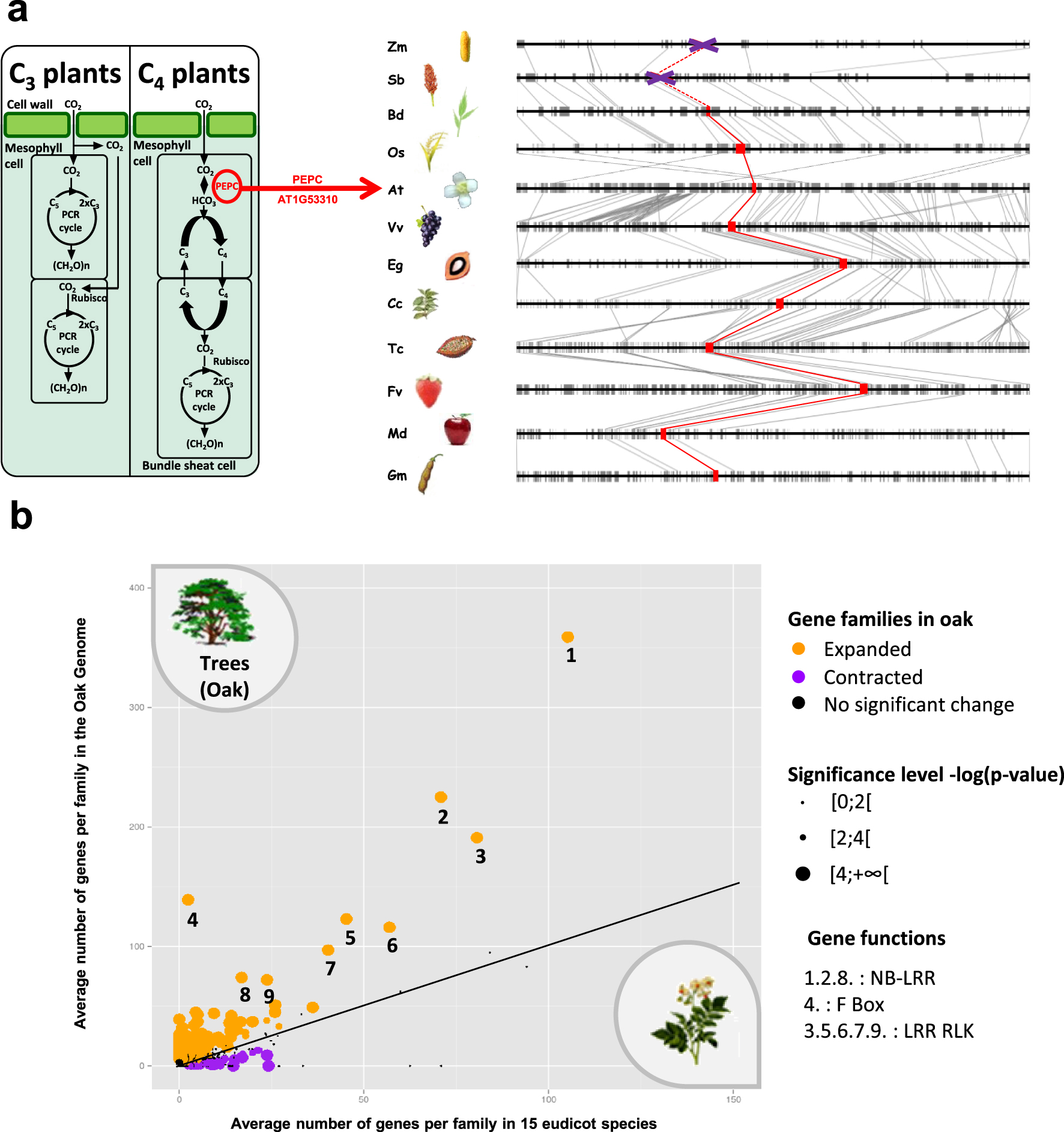
Which genes and associated functions controlling key traits of interest in Agriculture are best targets of translational research between plant species? Paleogenomics data makes possible to identify genes driving traits gained during the evolution in comparing the genomes of a group of species (genotypes or even varieties) that acquired this trait-phenotype to a group of species that did not gained this trait in evolution (literally, “Evo-Devo” for Evolutionary Developmental Biology). This translational research approach on traits can also provide a better understanding of the molecular bases of traits of major interest in Agriculture, such as seasonality (comparing annual versus perennial species), photosynthesis (comparing C3 versus C4 species) as well as grain and fruit development and quality traits. Plomion et al. [34] conducted such Evo-Devo approach in comparing the genome of nine trees and seven herbaceous species identifying nine gene families of conserved genes (i.e. orthogroups) that undergone expansion during trees (particularly in oak) evolution enriched in Gene Ontology (GO) terms relating to biotic interactions, particularly corresponding to disease resistance (R) genes (including NB-LRR for nucleotide-binding site leucine-rich repeat and RLK for receptor-like kinase), as illustrated in the Figure 4b. The expansion of disease-resistance related gene families during tree evolution is a genomic characteristics potentially underpinning longevity of long lifespan species. The identification such key gene functions driving particular life history traits of interest in Agriculture opens novel avenue in investigating specifically such gene functions in breeding programs.
5. Translational research for breeding improvement
Once genes of interest have been identified as conserved in several species of agronomic interest, as illustrated in the case example described previously, how to exploit this information into breeding schemes? This requires constant development in technologies aiming for functional validation of genes, automated phenotyping and large-scale genetic diversity screening, among others. The functional validation of the conserved genes is necessary to show that they drive the same traits or phenotypes in the different species (crops), as reported in the model or pivot species they have being initially investigated. To that regards, genome editing, efficient transformation protocols and mutant populations are necessary for a wide range of crop species to conduct translational research up to the field [35]. Beside the identification of the most promising conserved genes between species, exploitation in breeding relies on the identification of the best alleles (and accessions or progenitors carrying them) to be introduced in current (pre)breeding schemes. To that regards, reduction in sequencing cost is still necessary to be able to systematically sequence a wide range of genotypes covering the world-wide diversity of a given species, to unveil core-genomes (genes conserved between accession of a species) and more interestingly dispensable genomes (genes specific of accession of a species), all defining the concept of pan-genome, showing that the gene content from individuals of the same species varies considerably [36].
How to identify alleles of interest for breeding? In-depth resequencing of genotypes of a species delivers a catalog of polymorphisms that, associated to phenotypic data, allow to identify genomic regions and associated genes potentially driving the investigated trait (so called QTL and GWAS approaches, respectively for Quantitative Trait loci and Genome Wide Association Study). Another approach consists in identifying polymorphisms that have been selected during domestication of selection. Following this method, for example in wheat, the resequencing of the genomes (more precisely the exomes) of almost 500 genotypes selected from across the geographical range of the wheat species complex cultivated nowadays unveiled 5089 genomic regions and polymorphism that has been selected by breeders during the last two centuries of wheat improvement from which their impact on phenotypes or biological processes are still unknown, opening novel avenues in using this underexploited information in wheat breeding [8].
To accurately investigate the phenotypic gain obtained from the gene and associated allele resulting from a translational research approach from model or pivot species, requires to accurately and relevantly evaluate in the field the phenotypic gain in a high-throughput way. In complement to standardized conditions (in greenhouse or growth chamber), providing the maximal reproducibility in experimental conditions, dedicated field phenotyping platforms exist to adequately reflect a combination of fluctuating environmental conditions or stresses [37].
6. Conclusion
The efficient transfer of genomic information (such as gene sequence, annotation and functions) from one species (considered as model or pivot) to a less studied taxon and, ultimately, to crops (legume, fruit, and cereal species), requires the robust identification of genes that are functionally conserved between species, referenced as “phenologs” (i.e. gene encoding the same phenotype/trait between species). One of the most precise method for identifying putative phenologs between species involves the reconstruction of ancestral genomes and the identification of precisely conserved and lineage-specific genes. Providing ancestors of major botanical families as well as at the basis of the angiosperm delivering the entire set of conserved relationships between monocots and eudicots, is one of the tools currently available to the scientific community for the efficient transfer of genes and, more generally, genomic contexts/loci between angiosperms, from model to crops. Despite the development of translational research in plants from models or pivots to crops, this approach may fail for traits (1) that have a recent evolutionary history and not conserved between species (for example the tolerance to specific diseases) or for trait that are highly polygenic and driven by a complex interplay between several genes (for example the yield). However, the proposed paleogenomics-based translational research approach is a promising tool delivered for biotechnological and breeding applications, to develop high yielding stress-tolerant varieties in wide range of agronomically important plant species.
Conflicts of interest
The author has no conflict of interest to declare.
Version française
1. La recherche translationnelle, du domaine médical à la recherche végétale
La recherche translationnelle est un concept développé dans le domaine de la recherche médicale dans les années 80 consistant à traduire (transférer) les découvertes de la science fondamentale plus rapidement et plus efficacement en applications, et résumée « de la paillasse au lit du patient » [1]. Dans ce contexte, la recherche translationnelle en médecine consiste à favoriser les échanges entre laboratoires et hôpitaux pour transformer les découvertes en biologie en applications médicales ou thérapeutiques utilisables dans le traitement des patients. La recherche translationnelle vise également à exploiter la recherche préclinique sur des modèles animaux (principalement murins) sous forme d’innovations pour la santé humaine [2], en s’interrogeant sur l’espèce modèle adéquate à utiliser pour chaque pathologie et maladie humaine étudiée [3] en recherche translationnelle. Ce concept a été transposé à la recherche sur les plantes et plus précisément à la sélection des cultures végétales, consistant à convertir les connaissances fondamentales des espèces modèles (également appelés espèces pivots) en applications pour l’amélioration des espèces cultivées, afin de soutenir durablement la sécurité alimentaire et l’agriculture, ce qui peut se résumer par l’expression « du pot à la parcelle » [4]. Dans ce contexte, la recherche translationnelle consiste à mettre au point et à exploiter des méthodes et outils permettant de rendre les résultats obtenus chez une espèce sur un caractère ou un phénotype particulier exploitables chez toutes les espèces pour lesquelles le phénotype ou caractère considéré présente un intérêt en matière de sélection. La recherche translationnelle chez les espèces végétales cultivées à partir des espèces modèles soulève alors la question suivante : comment optimiser l’utilisation des connaissances génomiques et génétiques des espèces modèles pour améliorer la sélection des espèces cultivées ?
Depuis le début du siècle actuel, avec la première publication des génomes Humain (finalement achevée en 2022 [5]) et d’Arabidopsis thaliana [6], plus de 100 génomes de plantes (principalement des espèces cultivées) de haute qualité (c’est-à-dire entièrement assemblés à l’échelle de chromosomes) sont désormais disponibles dans le domaine public (Figure 1). L’amélioration constante des technologies de séquençage et la réduction des coûts rendent désormais possible l’accès au génome de n’importe quel organisme vivant (ou éteint [7]). En profitant de ces ressources, nous illustrons dans la Figure 2 le processus théorique de la recherche translationnelle des espèces modèles (comme Arabidopsis) aux espèces cultivées (comme les céréales) ainsi qu’entre espèces cultivées avec (1) l’identification (à partir de la littérature) des gènes contrôlant les traits ou phénotypes clés chez une espèce modèle ou une espèce cultivée particulière (alors référencée comme pivot), (2) l’identification des gènes les mieux conservés entre espèces pour lesquelles ces traits ou phénotypes sont d’intérêt pour la sélection, (3) la validation fonctionnelle des gènes conservés pour montrer qu’ils contrôlent les mêmes traits ou phénotypes chez les espèces modèle (ou pivot) et les espèces cultivées, (4) l’identification des génotypes portant les meilleurs allèles pour le gène conservé contrôlant le trait ou le phénotype d’intérêt, et (5) l’évaluation en conditions réelles au champ de ces génotypes pour quantifier l’amélioration du phénotype et du trait.
Génomes végétaux séquencés. La figure illustre les séquences de génomes végétaux de haute qualité (assemblés au niveau des chromosomes) en fonction de l’année de leur diffusion publique (axe des x) et de leur taille en mégabase (axe des y).
Processus de recherche translationnelle chez les espèces végétales cultivées. HAUT — Illustration de la méthode de reconstruction des génomes ancestraux in silico à partir de l’identification des gènes (rectangles colorés) complètement conservés et dupliqués (en rouge), partiellement conservés et dupliqués (en bleu) ou spécifiques d’une espèce (en noir) hérités d’événements de spéciation et de duplications au cours de l’évolution des plantes (adapté de Pont et al. [8]). BAS — Illustration du processus de recherche translationnelle chez les espèces végétales cultivées à travers (1) l’identification des gènes pilotant les traits ou phénotypes clés chez les espèces modèles ou pivots, (2) l’identification des meilleurs gènes conservés chez les espèces cultivées, (3) la validation fonctionnelle des gènes conservés, (4) l’identification des génotypes portant les meilleurs allèles, (5) l’évaluation au champ des génotypes dans les schémas de sélection.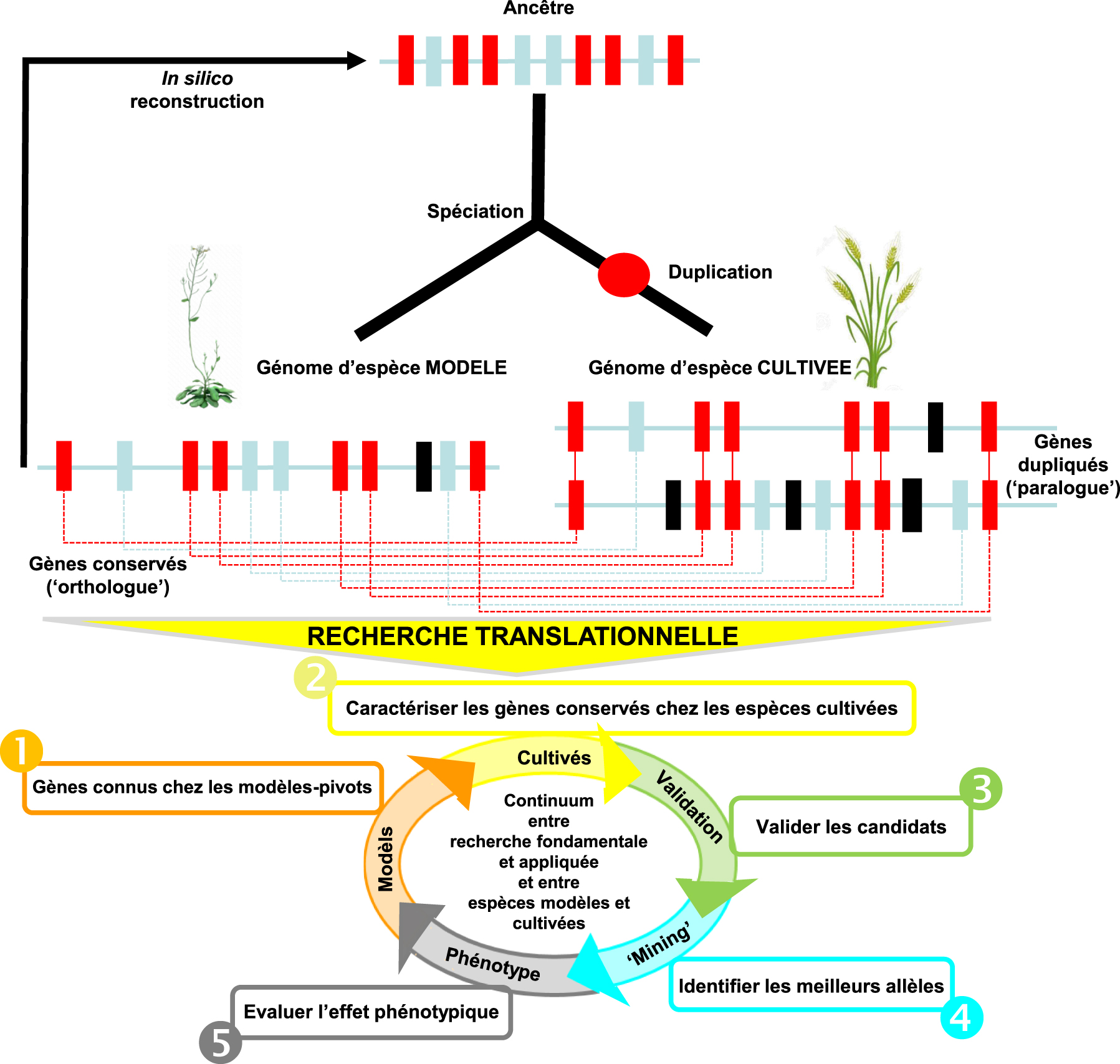
2. La génomique comparative chez les angiospermes
Suite à l’accès aux génomes des plantes depuis l’an 2000, le développement constant de méthodes et de stratégies de génomique comparative offre l’opportunité d’identifier des gènes conservés pour mener des recherches translationnelles des espèces modèles aux espèces cultivées. À cet égard, plusieurs outils publics sont disponibles pour interroger les données de génomique comparative entre les génomes végétaux, tels que PLAZA [9], Gramene [10], Ensembl [11], CoGe [12], Genomicus [13]. Les plantes à fleurs, ou angiospermes, sont apparues il y a environ 120 à 250 millions d’années [14] pour se diversifier rapidement en plus de 350 000 espèces vivantes aujourd’hui et divisées en deux groupes principaux, les monocotylédones et les eudicotylédones [15]. L’évolution des angiospermes est caractérisée par la duplication récurrente de l’ensemble du génome (WGD), également appelée événements de polyploïdie [16], ce qui rend difficile l’identification précise des gènes conservés entre les génomes végétaux à partir d’outils web publiquement accessibles (Figure 1).
À titre d’exemple et sur la base du schéma connu des événements de polyploïdisation chez les plantes à fleurs, la comparaison des génomes du riz (espèce cultivée de la famille des monocotylédones) et d’Arabidopsis (modèle de la famille des eudicotylédones) implique l’identification des huit régions génomiques du riz (issues des événements de polyploïdisation 𝜏, 𝜎 et 𝜌) et douze régions génomiques d’Arabidopsis (issues des événements de polyploïdisation 𝛼, 𝛽 et 𝛾) héritées depuis l’ancêtre des angiospermes datant de 214 millions d’années (Figure 3). Pour mener une recherche translationnelle efficace entre Arabidopsis (espèce modèle) et le riz (espèce cultivée), il faut donc, pour chaque gène d’intérêt identifié chez Arabidopsis, fournir le catalogue complet des huit gènes potentiellement conservés chez le riz, et ceci afin de sélectionner celui qui peut avoir conservé la fonction d’intérêt identifiée chez Arabidopsis. Les outils et méthodes disponibles ne parviennent souvent pas à identifier tous les gènes conservés attendus entre les génomes modernes hérités d’événements évolutifs (notamment les polyploïdies successives) connus depuis l’ancêtre des angiospermes, ce qui entraîne une sous-estimation de la conservation des gènes entre les espèces et, ainsi, réduit l’efficacité de la recherche translationnelle.
Evolution des plantes à fleurs modernes à partir des génomes ancestraux reconstruits. Illustration de l’arbre phylogénétique des plantes cultivées et des espèces modèles d’angiospermes, les événements de polyploïdisation étant indiqués par des points de couleur (duplications en rouge et triplications en bleu), à l’aide d’un code couleur mettant en évidence les protochromosomes ancestraux (à gauche) et leur évolution vers les génomes modernes (à droite) pour les principales familles botaniques (graminées, Solanaceae, Cucurbitaceae, Brassicaceae, Rosaceae et légumineuses), adaptée de Pont et al. [7].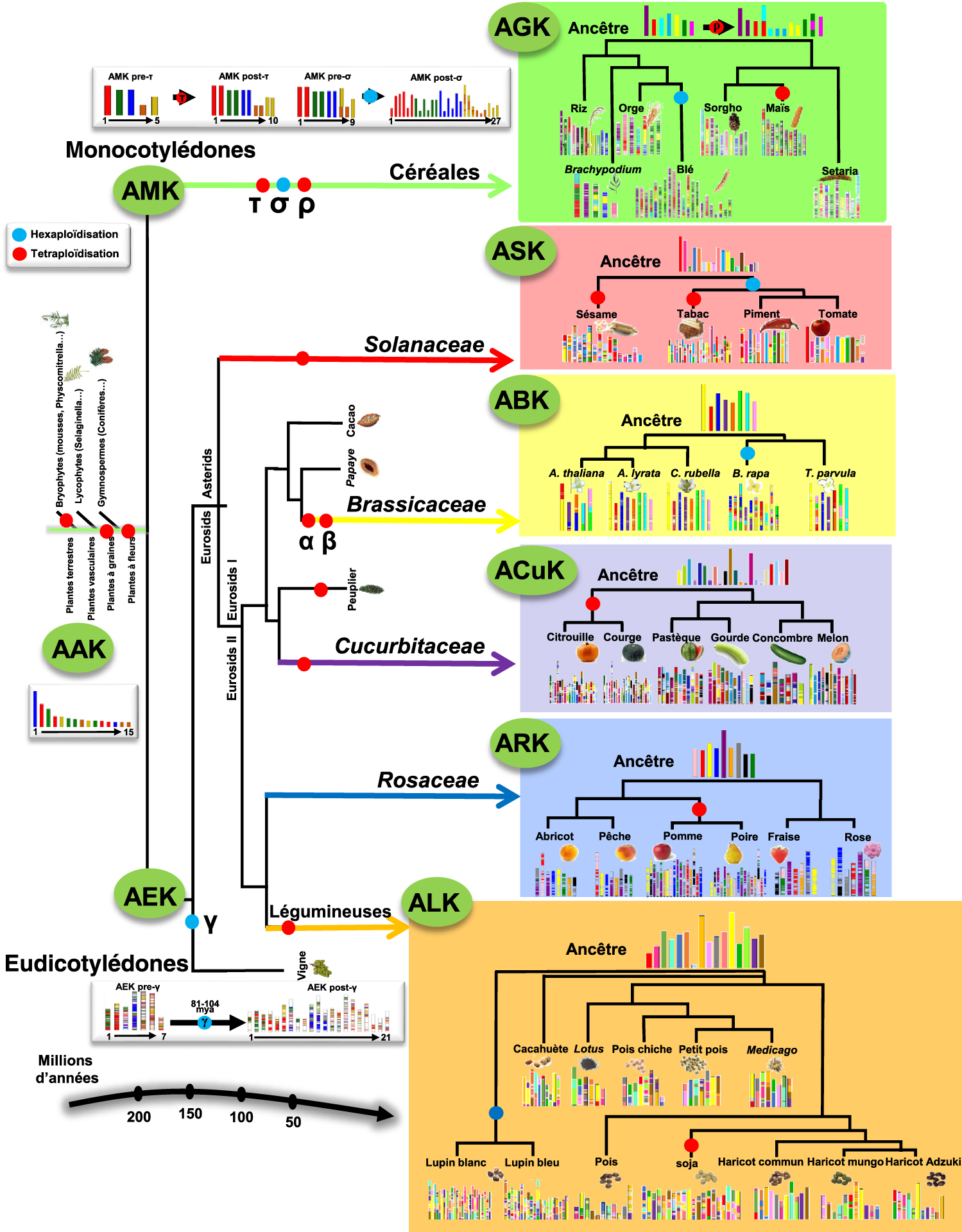
3. La recherche translationnelle à partir de génomes ancestraux reconstruits
Toutes les espèces d’angiospermes ont connu de nombreux événements de duplication du génome entier (ou de polyploïdisation) qui ont créé différents degrés de conservation des gènes entre les espèces, de sorte qu’un tel événement évolutif doit être précisément pris en compte pour transférer efficacement les informations sur la structure et la fonction des gènes d’une espèce à un autre. C’est le but de la paléogénomique qui consiste en la reconstruction des génomes ancestraux ou fondateurs disparus, par la comparaison des génomes d’espèces modernes. À partir d’un génome ancestral (possiblement éteint) qui a évolué en différentes espèces modernes par spéciation et par des événements distincts de réarrangements des chromosomes (fusion, fissions, inversions, duplications et translocations), chacun des chromosomes ancestraux dérive d’un sous-ensemble de régions chromosomiques modernes qui partagent des gènes conservés entre les espèces actuelles. Suite à ces évidences évolutives, lors de la reconstruction in silico des génomes (ou caryotypes) ancestraux, la comparaison des génomes modernes produit des fragments génomiques de chaque espèce qui partagent des groupes de gènes conservés (on parle de synténie), considérés comme régions ancestrales conservées (CAR), ou chromosomes ancestraux (appelés protochromosomes) des génomes ancestraux reconstruits [7]. Les génomes des espèces modernes de monocotylédones et d’eudicotylédones peuvent alors être connectés (ou « pontés ») via les génomes ancestraux reconstruits délivrant le répertoire complet des gènes conservés, et supposément des fonctions et des traits, entre par exemple l’espèce modèle Arabidopsis et les principales céréales telles que le blé, le maïs ou le riz (Figure 2). Des données de paléogénomique végétale à haute résolution sont maintenant disponibles pour mener des recherches translationnelles entre espèces d’angiospermes en utilisant plusieurs ancêtres inférés (Figure 3) : AAK (Ancestral Angiosperm Karyotype de 15 protochromosomes portant 10 000 à 20 000 gènes conservés entre les angiospermes modernes [17]), AGK (pour Ancestral Grass Karyotype à 12 protochromosomes et 14 241 protogènes [18, 19]), AEK (pour Ancestral Eudicot Karyotype à 21 chromosomes et 9022 protogènes [20]), ARK (pour Ancestral Rosaceae Karyotype à 9 protochromosomes et 8861 protogènes [21]), ABK (pour Ancestral Brassicaceae Karyotype à 8 protochromosomes et 20 037 protogènes [22]), ACuK (pour Ancestral Cucurbitaceae Karyotype à 22 protochromosomes et 17 969 protogènes [23]), ALK (pour Ancestral Legume Karyotype à 16 protochromosomes et 13 181 protogènes [24, 25]) et ASK (pour Ancestral Solanaceae Karyotype à 17 protochromosomes et 17 879 protogènes). Ces génomes ancestraux reconstruits fournissent un catalogue robuste et complet de gènes conservés entre les génomes végétaux prenant en compte les événements évolutifs tels que les polyploïdisations comme illustré dans la Figure 3 et sont mis à la disposition de la communauté scientifique via https://urgi.versailles.inrae.fr/synteny. Les génomes ancestraux inférés peuvent être considérés comme une ressource très utile pour la recherche translationnelle comparant les génomes séquencés des plantes à fleurs, en particulier les espèces modèles et cultivées. Par exemple, ALK livre un catalogue de 13 181 gènes conservés entre légumineuses offrant la possibilité, pour chaque gène d’intérêt identifié chez Medicago Truncatula (modèle) d’avoir accès à l’ensemble des gènes conservés chez le pois ou la lentille (espèces cultivées) par exemple. De la même manière, AGK livre un catalogue de 14 241 gènes conservés pour mener des recherches translationnelles de Brachypodium (espèce modèle) ou du riz (espèce cultivée) vers le blé (espèce cultivée).
4. La recherche translationnelle de traits et de gènes chez les espèces cultivées
Les données de paléogénomique permettent de transférer efficacement les connaissances acquises chez une espèce (considérée comme un modèle ou un pivot) à partir de n’importe quel gène (pilotant une fonction ou un phénotype particulier) à toutes les autres espèces végétales pour lesquelles le caractère présente un intérêt. Par exemple, cette approche a été appliquée avec succès chez les cucurbitacées pour la taille et la forme des fruits [26]. Les locus de caractères quantitatifs (LCQ, QTL en anglais) consensus identifiés chez le concombre, le melon et la pastèque ont été associés chez Arabidopsis, la tomate et le riz à des gènes clonés qui codent pour des protéines contenant les domaines caractéristiques CNR (régulateur du nombre de cellules), CSR (régulateur de la taille des cellules), CYP78A (cytochrome P450), SUN, OVATE, TRM (motif de recrutement de TONNEAU1), YABBY et WOX qui peuvent être considérés comme les meilleurs candidats pour les caractères étudiés. Appliqué aux céréales, en capitalisant sur les connaissances acquises sur les fonctions des gènes chez le riz ou Brachypodium, le gène FZP pour FriZzy Panicle de la famille APETALA (AP2)/Ethylene Response Factor (ERF) s’est avéré être conservé [27] et produire des épillets surnuméraires (un composant du rendement) de l’épi de plusieurs espèces [28, 29]. Dans le même ordre d’idées, il a également été démontré que le tréhalose 6-phosphate (T6P) est impliqué dans l’amélioration du rendement (comme le nombre de grains, le remplissage des grains et la résistance aux stress abiotiques) dans différents environnements et ceci pour plusieurs espèces cultivées [30]. À titre d’exemple de l’approche de recherche translationnelle basée sur la paléogénomique, la Figure 4a illustre les gènes PEPC connus pour être impliqués dans le trait majeur « C3/C4 », les espèces dites « C4 » se traduisant par une capacité photosynthétique supérieure à celle des espèces « C3 ». En partant du gène candidat d’Arabidopsis (AT1G53310, [31]), le génome ancestral des angiospermes (AAK) reconstruit fournit automatiquement les 8 à 3 relations gènes-à-gènes attendues entre les monocotylédones et les eudicotylédones sur la base de l’histoire évolutive connue des angiospermes (Figure 2), toutes ces régions chromosomiques étant dérivées du même chromosome ancestral fondateur. Une vue plus large de la conservation entre 12 espèces modernes d’angiospermes de l’une des copies du gène PEPC impliqué dans le caractère « C3/C4 » a révélé un schéma évolutif intéressant, dans lequel PEPC ne semble conservé que chez les espèces « C3 » et potentiellement absent des plantes « C4 » (maïs et sorgho). Cet exemple met en évidence la puissance des informations fournies par les ancêtres reconstruits pour l’extraction de gènes candidats (issues du modèle Arabidopsis chez les eudicotylédones) chez les céréales (représentatives des monocotylédones, comme le riz), en vue de la validation ultérieure de leur rôle dans le contrôle de la transition entre plantes « C3 » et « C4 ». La recherche des gènes clés qui déterminent des caractéristiques agronomiques importantes peut également bénéficier des connaissances acquises en dehors du règne végétal. Le gène DGAT1, identifié comme étant impliqué dans la qualité de production de lait et de viande chez les bovins, les buffles, les chèvres et les moutons [32], s’est avéré être également un déterminant clé de la teneur et de la composition en huile chez le maïs [33]. Ce résultat ouvre de nouvelles possibilités de recherche translationnelle entre les plantes et les animaux à l’avenir.
La recherche translationnelle des gènes et des caractères. (a) Schéma simplifié des voies de fixation du carbone opérant dans les plantes « C3 » et « C4 », adapté de (http://www.intechopen.com/books/abiotic-stress-in-plants-mechanisms-and-adaptations/c4-plants-adaptation-to-high-levels-of-co2-and-to-drought-environments). Abréviations : C3, acides organiques à trois carbones ; C4, acides organiques à quatre carbones ; C5, ribulose-1,5-bisphosphate ; PCR, cycle de réduction du carbone photosynthétique ; PEPC, phosphoenolpyruvate carboxylase ; Rubisco, ribulose-1,5-bisphosphate carboxylase/oxygénase. La conservation du gène PEPC (candidat ATG1G53310 d’Arabidopsis thaliana) entre les espèces est illustrée par des gènes sous forme de boîtes grises reliées par des traits gris entre 12 génomes d’angiospermes modernes, les événements de délétion étant indiqués par des croix violettes. (b) Expansion et contraction chez le chêne (axe des ordonnées) de familles de gènes comparativement à 15 autres espèces d’eudicotylédones (axe des abscisses), les familles de gènes ayant subi une expansion dans leurs nombres de copies au cours de l’évolution du chêne sont étiquetées de 1 à 9 (adapté de Plomion et al. [34]).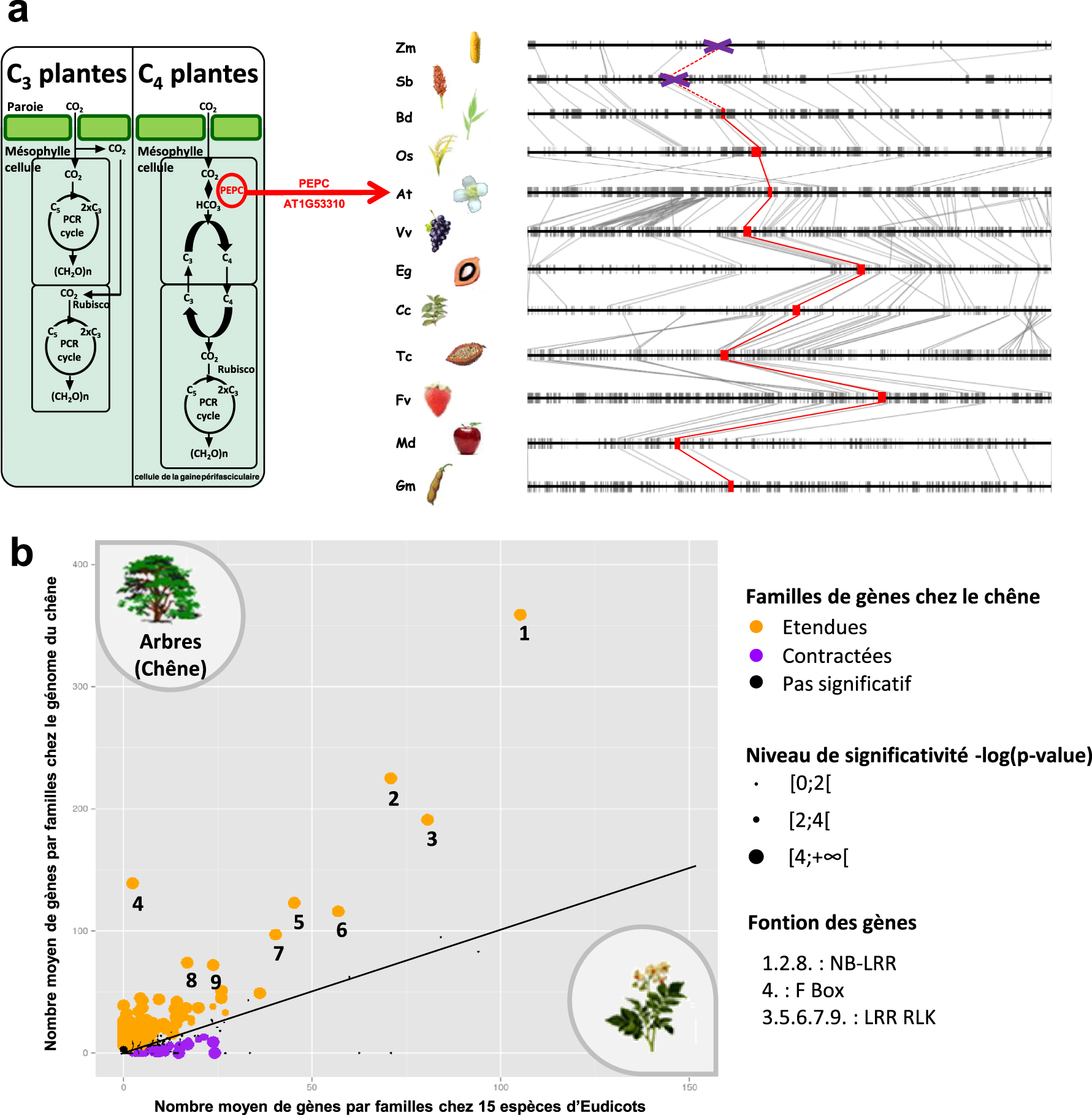
Quels sont les gènes et les fonctions associées qui contrôlent les principaux traits d’intérêt en agriculture et qui sont les meilleures cibles de la recherche translationnelle entre les espèces végétales ? Les données paléogénomiques permettent d’identifier les gènes qui contrôlent les traits acquis au cours de l’évolution en comparant les génomes d’un groupe d’espèces (de génotypes ou même de variétés) qui ont acquis ce trait ou phénotype à un groupe d’espèces qui n’a pas acquis ce trait au cours de l’évolution (littéralement, « Evo-Devo » pour Evolutionary Developmental Biology). Cette approche de recherche translationnelle sur les processus peut également permettre de mieux comprendre les bases moléculaires de traits d’intérêt majeur en agriculture, tels que la saisonnalité (en comparant les espèces annuelles et pérennes), la photosynthèse (en comparant les espèces « C3 » et « C4 ») ainsi que le développement et la qualité des grains et des fruits. Plomion et al. [34] ont mené une telle approche Evo-Devo, en comparant le génome de neuf arbres et de sept espèces herbacées, identifiant neuf familles de gènes conservés (c’est-à-dire des orthogroupes) qui ont subi une expansion au cours de l’évolution des arbres (en particulier du chêne), enrichis en termes d’ontologie de gènes (GO) relatifs aux interactions biotiques, correspondant particulièrement aux gènes de résistance (R) aux maladies (incluant les gènes NB-LRR pour nucleotide-binding site leucine-rich repeat et RLK pour receptor-like kinase), comme illustré dans la Figure 4b. L’expansion des familles de gènes liés à la résistance aux maladies au cours de l’évolution des arbres est une caractéristique génomique qui pourrait jouer un rôle majeur dans l’adaptation de ces espèces à longue durée de vie. L’identification de ces fonctions génétiques clés qui déterminent des traits particuliers du cycle de vie intéressant pour l’agriculture (ici l’arboriculture ou la sylviculture) ouvre une nouvelle voie pour l’étude spécifique de ces fonctions génétiques dans les programmes de sélection.
5. Recherche translationnelle pour l’amélioration de la sélection
Une fois que des gènes d’intérêt ont été identifiés comme conservés entre plusieurs espèces d’intérêt agronomique, comme l’illustrent les exemples décrits précédemment, comment exploiter ces informations dans les schémas de sélection ? Cela nécessite un développement constant des technologies visant à valider fonctionnellement ces gènes, le phénotypage automatisé et le criblage de la diversité génétique à grande échelle, entre autres. La validation fonctionnelle des gènes conservés est nécessaire pour montrer qu’ils sont à l’origine des mêmes traits ou phénotypes chez les différentes espèces cultivées, comme décrit dans l’espèce modèle ou pivot sur laquelle ils ont été initialement étudiés et décrits. À cet égard, l’édition du génome, des protocoles de transformation efficaces et des populations mutantes sont nécessaires pour un large éventail d’espèces cultivées afin de mener des recherches translationnelles jusqu’au champ [35]. Outre l’identification des gènes conservés les plus prometteurs entre les espèces, l’exploitation en sélection repose sur l’identification des meilleurs allèles (et des accessions ou géniteurs qui les portent) à introduire dans les programmes de (pré)sélection actuels. À cet égard, la réduction des coûts de séquençage est encore nécessaire pour pouvoir séquencer systématiquement une large gamme de génotypes couvrant la diversité mondiale d’une espèce donnée, pour dévoiler les core-génomes (les gènes conservés entre les accessions d’une espèce) et les génomes dispensables (gènes spécifiques de l’accession d’une espèce), définissant le concept de pan-génome qui montre que le contenu génétique des individus d’une même espèce varie considérablement [36].
Comment identifier les allèles d’intérêt pour la sélection ? Le reséquençage approfondi des génotypes d’une espèce fournit un catalogue de polymorphismes qui, associés aux données phénotypiques, permettent d’identifier les régions génomiques et les gènes associés qui sont potentiellement responsables du caractère étudié (approches dites QTL et GWAS, respectivement pour Quantitative Trait loci et Genome Wide Association Study). Une autre approche consiste à identifier les polymorphismes qui ont été sélectionnés au cours de la domestication et de la sélection des espèces cultivées. Suivant cette méthode, par exemple dans le cas du blé, le reséquençage des génomes (plus précisément des exomes) de près de 500 génotypes couvrant les espèces de blé cultivées de nos jours au niveau mondial a dévoilé 5089 régions génomiques et polymorphismes qui ont été sélectionnés au cours des deux derniers siècles d’amélioration du blé et dont l’impact sur les phénotypes ou les processus biologiques demeure encore inconnu, ouvrant de nouvelles voies dans l’utilisation de ces informations sous-exploitées pour la sélection des blés modernes [8].
Pour étudier avec précision le gain phénotypique obtenu à partir du gène et de l’allèle associés résultant d’une approche de recherche translationnelle à partir d’espèces modèles ou pivots, il faut pouvoir évaluer au champ le gain phénotypique de manière précise et pertinente et ceci à haut débit. En complément des conditions standardisées (serres, chambres de cultures), offrant une reproductibilité maximale des conditions expérimentales, des plateformes de phénotypage au champ existent pour refléter des conditions réelles de combinaisons de conditions environnementales ou de stress fluctuants [37].
6. Conclusion
Le transfert efficace d’informations génomiques (telles que la séquence, l’annotation et les fonctions des gènes) d’une espèce (considérée comme modèle ou pivot) vers un taxon moins étudié et, en fin de compte, vers les espèces cultivées (légumineuses, fruitiers et céréales), nécessite l’identification robuste des gènes qui sont fonctionnellement conservés entre les espèces, on parle de « phénologues » (c’est-à-dire les gènes contrôlant le même phénotype/trait entre les espèces). L’une des méthodes les plus précises pour identifier les phénologues putatifs entre espèces implique la reconstruction des génomes ancestraux et l’identification de gènes précisément conservés ou spécifiques à une lignée. Fournir les ancêtres des grandes familles botaniques ainsi qu’à la base des angiospermes, livrant l’ensemble des gènes conservés entre les monocotylédones et les eudicotylédones, est l’un des outils dont dispose actuellement la communauté scientifique pour le transfert efficace des gènes et, plus généralement, des contextes/loci génomiques entre angiospermes, de modèles aux espèces cultivées. Au-delà de la montée en puissance des travaux de recherche translationnelle chez les plantes des modèles ou des pivots aux espèces cultivées, cette approche peut échouer notamment pour les traits qui ont une histoire évolutive récente et qui ne sont pas conservés entre espèces (par exemple la tolérance à des maladies très spécifiques d’espèces ou de lignées) ou pour les traits qui sont hautement polygéniques et qui sont déterminés par une interaction complexe entre plusieurs gènes (par exemple le rendement). Cependant, l’approche de recherche translationnelle proposée, fondée sur la paléogénomique, demeure un outil prometteur pour les applications en biotechnologie et en sélection, afin de développer des variétés à haut rendement et tolérantes au stress chez un large éventail d’espèces végétales importantes sur le plan agronomique.
Conflit d’intérêt
L’auteur n’a aucun conflit d’intérêt à déclarer.