



 CC-BY 4.0
CC-BY 4.0
This article is dedicated to the memory of the outstanding virologist Dennis H. Bamford (1948–2025).
1. Introduction
Viruses are best known as agents of human, animal and plant diseases, some of these widespread and devastating. Suffice it to mention numerous epidemics of smallpox prior to the introduction of broad vaccination, the 1918–1920 pandemic of Spanish flu, the catastrophic epidemics of poliomyelitis in the 1930s and 1940s, eventually curtailed by vaccines, and obviously, the recent COVID-19 pandemic (Rochman et al., 2022). Other viruses, such as certain strains of human papillomaviruses or Epstein–Barr virus, are known to cause cancer (zur Hausen, 2002; Shannon-Lowe and Rickinson, 2019). Relatively well known are also viruses causing disease in plants, such as the tobacco mosaic virus, the very first virus that was discovered in the late 19th century (Creager, 2022). However, it is much less commonly appreciated that viruses causing diseases in animals and plants, however numerous and diverse, are but tiny parts of the vast virus world, also known as the virosphere. The myriads of other viruses comprising the virosphere infect virtually all other organisms on the planet, in many cases killing the host, but in many others, causing no harm or even being beneficial.
Viruses are defined as mobile genetic elements (MGE) encoding at least one protein that is a major component of virions (virus particles) encasing the genome of the virus (Koonin, Dolja, Krupovic and Kuhn, 2021). Viruses are obligate intracellular symbionts that reproduce inside cells of all organisms, with the sole possible exception of some of the most drastically reduced intracellular bacterial symbionts of eukaryotes. Moreover, most organisms, from bacteria to humans, are hosts to a broad variety of viruses with a wide range of genome sizes, diverse cycles of genome replication and expression, virion structures and modes of interaction with the hosts. When describing viruses, we purposefully use the umbrella term “symbionts” rather than the more commonly used “parasites” because virus–host interactions span the entire spectrum of symbiotic relationships with the hosts, from aggressive parasitism to commensalism to mutualism (Gonzalez et al., 2020).
The emergence of MGE, including viruses, is an intrinsic, fundamentally inevitable, and essential feature of the evolution of life (Koonin, Wolf, et al., 2017). There are strong theoretical arguments and overwhelming empirical evidence that MGE, including, most likely, bona fide viruses, have been associated with the evolution of cellular life on Earth ever since its earliest stages, that is, for more than 4 billion years (Koonin, Senkevich, et al., 2006; Koonin and Dolja, 2013; Forterre and Prangishvili, 2009; Forterre and Prangishvili, 2013). In themselves, viruses cannot be legitimately considered life forms inasmuch as they are not reproducers, that is, they lack physical continuity across generations, and strictly depend on the host cells for their replication (Moreira and Lopez-Garcia, 2009; Lopez-Garcia, 2012). Instead, viruses are replicators, maintaining genetic continuity and autonomy, and following evolutionary trajectories that are distinct from, even if entangled with, those of the hosts (Koonin, Dolja, Krupovic and Kuhn, 2021).
In the last decade, our understanding of the world of viruses has been transformed through the combination of the explosive growth of metagenomic and metatranscriptomic sequence databases, and sustained efforts on the discovery of novel viruses and the elucidation of the evolutionary relationships within the virosphere (Simmonds, Adams, et al., 2017; Koonin, Dolja, Krupovic, Varsani, et al., 2020; Roux and Coclet, 2026). These developments that take advantage of advanced methods for sequence and protein structure analysis have been codified in the new, comprehensive evolutionary taxonomy of viruses that has been formally adopted by the International Committee on Taxonomy of Viruses (ICTV) (Simmonds, Adriaenssens, Lefkowitz, et al., 2025). In this review article, we undertake to present a synthesis of these revolutionary advances, outlining the global structure of the virosphere along with the concepts, trends and specific scenarios of the evolution of the major groups of viruses. We further explore the connections between viruses and other types of MGE, such as plasmids and transposons, and the position of the virosphere within the replicator space.
2. The dimensions of the virosphere
Before discussing our current understanding of the evolution of the virosphere, we outline its key characteristics and dimensions. Unlike cellular life forms, which all share the same fundamental scheme of genetic information storage and transmission, with genomes consisting of enormous double-stranded (ds) DNA molecules, viruses explore the entire space of possible nucleic acid-based information storage and transmission route cycles (Baltimore, 1971; Agol, 1974; Koonin, Krupovic and Agol, 2021). All forms of DNA and RNA are used as genomes, that is, the information carriers incorporated into virions, by different groups of viruses. The nature of the genome defines the viral replication-expression cycle, and accordingly viruses are traditionally divided into 7 so-called Baltimore classes (first introduced in David Baltimore’s seminal 1971 paper (Baltimore, 1971)) (Figure 1A). Within different Baltimore classes, viral genomes can be either circular or linear and either continuous or segmented. The size of viral genomes also depends on the Baltimore class. Viruses of 6 of the 7 classes, with the exception of class I (dsDNA genomes), have (relatively) small genomes, within the range of 1 to about 60 kilobases (kb). By contrast, viral dsDNA genomes span 3 orders of magnitude from about 5 kb to more than 4 megabases (Mb) (Figure 1B,C). Notably, the largest genomes of the so-called giant viruses exceed in size and the number of genes the genomes of numerous bacteria and archaea (and some parasitic eukaryotes as well), defying the notion of viruses as miniscule entities and obliterating the boundary between viruses and cellular life forms with regard to genetic complexity (Claverie, 2006; Koonin and Yutin, 2019).
The Baltimore classes and realms of viruses. (A) Replication and expression of viral genomes in the 7 Baltimore classes. Each of the classes is defined according to the form of the nucleic acid incorporated into virions (the viral genome). (B) Sankey diagram showing the correspondence between the Baltimore classes and viral realms. (C) Range of genome sizes (kb) in each viral realm. The genome size data for classified representatives of each realm was retrieved from the NCBI GenBank database (Sayers et al., 2025).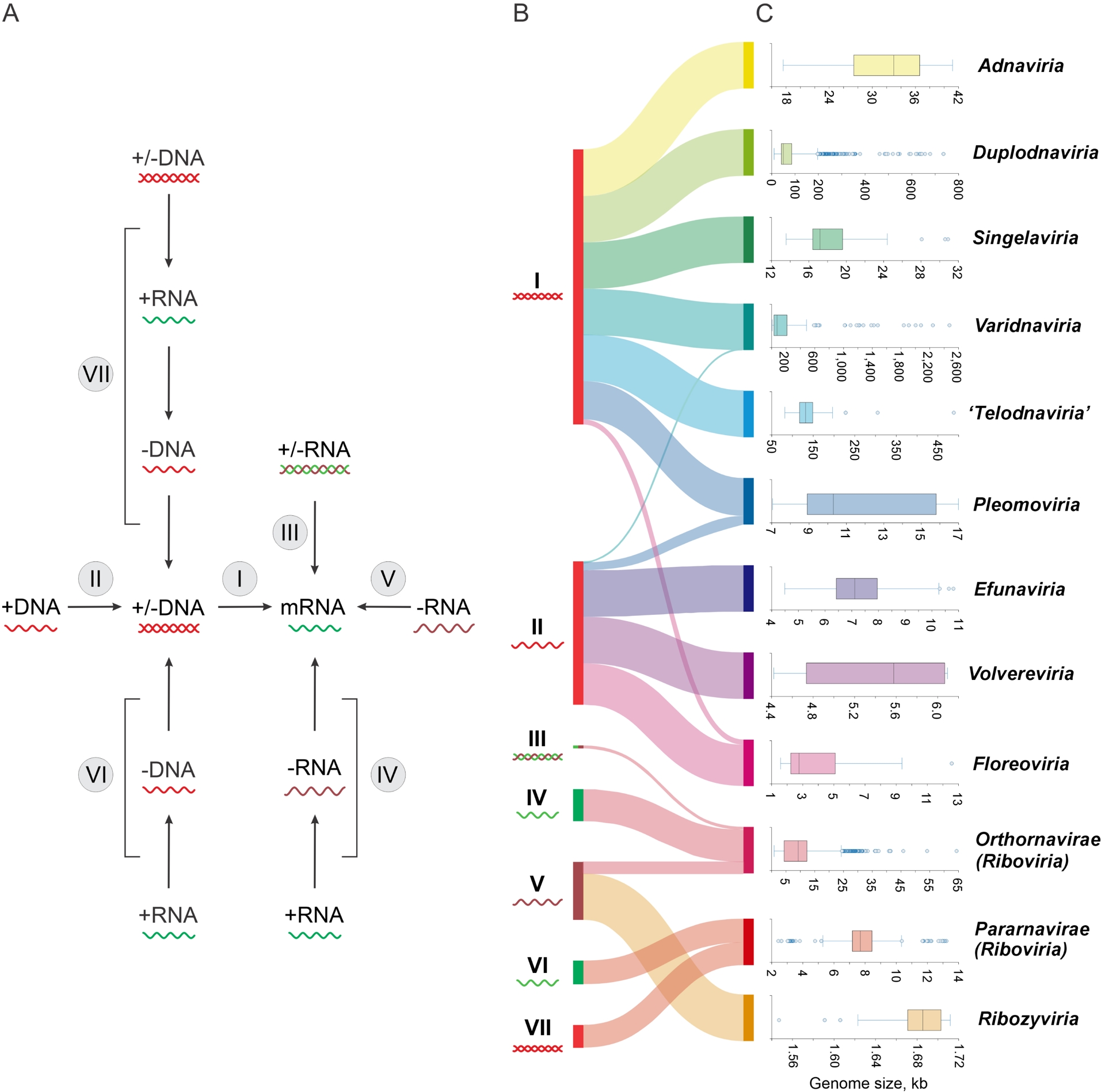
The diversity of the molecular structures and the wide size range of viral genomes are complemented by the diversity of sizes, shapes and molecular organizations of virions. Most virions are small, but like in the case of the genomes, the particles of giant viruses are larger than many bacterial and archaeal cells. In the great majority of virions, the core structure is the capsid, the proteinaceous shell encasing the genome. Most common capsids adopt one of the few shapes, primarily, spherical or filamentous, built on icosahedral or helical symmetry, respectively (Crick and Watson, 1956; Caston and Carrascosa, 2013). There is, however, a broad variety of comparatively rare, odd capsid shapes as well as several groups of viruses that lack a typical capsid while retaining a glycoprotein-rich lipid envelope encasing the genomic nucleic acid (see below). Furthermore, some MGE that are traditionally considered viruses lack genes for structural proteins and do not package their genomes at all, while having clearly evolved from typical, capsid-encoding viruses (Koonin and Dolja, 2014) (see also discussion below). In some viruses, virions are multilayered, consisting of more than one icosahedral shell, whereas in other viruses, the protein capsid is enveloped by a lipid membrane and/or contains an internal membrane surrounding the genome.
A central theme in the study of virus evolution is the origin of functional and structural features by divergence from common ancestors vs. independent, convergent emergence. In the discussion that follows, we shall see that neither the Baltimore classes of viruses nor the basic shapes and organizations of virions are monophyletic, convergence being a major trend in the evolution of the virosphere. To a large extent, evolutionary convergence is underpinned by physical constraints. As a major case in point, the number of basic capsid shapes is limited to only a few, with the great majority of capsids being either spherical or filamentous (often described as rod-shaped in the case of rigid filaments), which are two simple, symmetrical, thermodynamically favorable architectures. A larger variety of odd capsid shapes are rare in the virosphere. Importantly, convergence at the level of capsid shapes or morphologies does not translate to convergence of the actual molecular solutions to capsid building. Indeed, both icosahedral and helical capsids are constructed from capsid proteins with multiple distinct structural folds (Krupovic and Koonin, 2017b). Conversely, there is no evidence that structurally similar capsid proteins convergently evolved from structurally distinct ancestral proteins. Thus, structural similarity among capsid proteins within a particular virus group is interpreted as evidence of common ancestry and is one of the cornerstones of megataxonomy. In a more general context, although viruses are clearly polyphyletic, origin of new types of viruses is rare and is typically followed by extensive evolutionary diversification, as discussed below.
There is a popular meme that virus particles are the most abundant biological entities on Earth. The number of virions present on the planet at any given time has been estimated at the hyper-astronomical value of about 1031, with a virus/microbe ratio (VMR) of 10 or higher (Hendrix, 2003; Mushegian, 2020). However, these striking VMR values have been calculated largely by using epifluorescence microscopy or flow cytometry to count extracellular virions, primarily, in marine environments. Both these techniques are prone, on the one hand, to missing intracellular virions (including lysogens), but on the other hand, to erroneously reporting non-viral particles as virions (Forterre, 2013). More reliable estimates for DNA viruses have been obtained recently by quantification of the major capsid protein genes in metagenomes compared to host hallmark genes (namely, ribosomal protein genes). These analyses yielded a more nuanced picture whereby VMR was generally lower than previously thought, about 2 on average, and broadly varied across environments (Lopez-Garcia, Gutierrez-Preciado, et al., 2023). The highest VMR, 3–4 on average, was observed in metagenomes from aquatic environments, whereas in animal-associated metagenomes, the typical VMR was much lower, with the mean value around 1 (ibid.), in agreement with previous estimates for bacterial (Carding et al., 2017; Shkoporov and Hill, 2019) and archaeal (Baquero, Medvedeva, et al., 2024) viruses. Intermediate VMR values were reported for soils, sediments, and microbial mats. These findings would bring down the estimates of virus abundance in the biosphere by about an order of magnitude, but still support the excess of virus particles over cells on both the planetary scale and in most habitats. The VMR is not a linear function of the microbial density in the given environment. Analysis of thousands of estimates from marine samples demonstrated that the dependence of the VMR on microbial density is best described by power law functions with exponents below unity, that is, the ratio of virions to cells decreases with increased microbial population density (Wigington et al., 2016). At least in part, this dependency is explained by the piggyback-the-winner model for temperate bacteriophages whereby lysogeny becomes advantageous for a virus compared to lytic growth at high host density (B. Knowles et al., 2017).
Although a comprehensive census of the virosphere remains a major challenge for the future, simple, ballpark estimates can give us an idea of the total number of virus species and viral genes in the biosphere (Koonin, Krupovic and Dolja, 2023). The bulk of the diversity in the virosphere is accounted for by tailed bacteriophages with large dsDNA genomes, with the other phages, archaeal and eukaryotic viruses adding relatively little. There are at least 106–107 species of bacteria in the biosphere, with some estimates suggesting orders of magnitude more. A typical bacterial species is the host to multiple viruses. For example, for the Escherichia coli strain K12, over 100 distinct viruses are currently known (Humolli et al., 2025), whereas for a single strain of Mycobacterium smegmatis, more than 2000 viruses have been identified following extensive sampling (Hatfull, 2020; Abad et al., 2023; Hatfull, 2022). In all likelihood, these numbers underestimate the actual diversity of the viromes of the respective bacterial species. Conservatively assuming 10–100 viral species per bacterial host species, the global virome can be estimated to include at least 107–109 virus species. The upper bound on this range should be considered a more realistic estimate, given the conservative assumptions used. Currently, only about 17 000 virus species are formally recognized (Simmonds, Adriaenssens, Lefkowitz, et al., 2025), and even taking into account numerous new ones that are waiting to be established based on the results of metagenome and metatranscriptome mining, only a tiny part of the virosphere has been explicitly described so far. However, as we shall see below, this small sampling is likely to be representative of the large, distinct regions of the virosphere.
Based on the simple calculations above, the genetic diversity of the virosphere, that is, the total number of unique genes in the global virome, can be roughly estimated as well (Koonin, Krupovic and Dolja, 2023). The large dsDNA genomes of typical bacteriophages that quantitatively dominate the virosphere encompass many lineage-specific genes that are mostly involved in counteracting the host antivirus defenses (Yan et al., 2023). Conservatively assuming 10 such unique genes per virus species, the plausible low bound of the virosphere diversity can be estimated at about 1010 genes, which is likely one to two orders of magnitude greater than the number of unique genes in cellular life forms. Thus, the virosphere is the main reservoir of genetic novelty on our planet. These estimates are generally consistent with the more formal attempt to estimate the potential virus genome and protein space by simulating the process of virus discovery in viral metagenomic studies. Using the power function, which was found to best fit the increasing trends of virus diversity, it was estimated that there are at least 8.2 × 108 viral operational taxonomic units and 1.6 × 109 viral protein clusters on Earth (Lu et al., 2025). These estimates suggest that less than 3% of the viral genetic diversity has been uncovered thus far.
The general message from this brief survey of the virosphere dimensions is that, although viruses are in general tiny compared to cellular life forms and fully depend on the hosts for their replication, the virosphere is vast and enormously diverse, in some dimensions, exceeding the diversity of cellular organisms. Understanding the evolution of the biosphere without deep exploration of the virosphere would be a hopeless undertaking.
3. The metagenomic revolution and virosphere expansion
In the last decade, the knowledge of virus diversity, which can be translated into reconstruction of the virosphere evolution, took a major leap forward thanks to the advances of metagenomics and metatranscriptomics (Simmonds, Adams, et al., 2017). In the past, the study of viruses required propagation in the laboratory, either in the actual host organisms or in cell culture. Accordingly, the exploration of the virosphere diversity had been limited to the relatively few species of well-studied animals and plants and several model protist, bacterial and archaeal species. However, it is well known from the surveys of microbial diversity by 16S RNA sequencing that only about 0.1% of microbes currently can be grown in the laboratory (Rinke et al., 2013; Solden et al., 2016). Thus, the traditional approaches only scratch the proverbial surface of the global microbiome and, consequently, of the global virome. The fast progress of metagenomic and metatranscriptomic approaches has radically changed this situation (Koonin, Makarova and Wolf, 2021). Indeed, already in 2017, metagenome and metatranscriptome mining became by far the most important source of new virus discovery (Simmonds, Adams, et al., 2017). It is difficult to come up with exact estimates, but clearly, the number of viruses representing putative distinct virus species discovered by metagenome and metatranscriptome mining now exceeds that identified by traditional laboratory methods by several orders of magnitude. The increasingly detailed understanding of the global organization and evolution of the virosphere that we discuss below could not have been possibly attained without extensive analysis of metagenomic and metatranscripotomic sequence databases. Importantly, acknowledging the dominant role of metagenomics and metatranscriptomics in the exploration of the virosphere, the ICTV in 2017 formally recognized the legitimacy of viral species and higher taxa established based solely on sequence analysis, without isolation of the actual virus (Simmonds, Adams, et al., 2017; Adams et al., 2017).
4. The global structure and evolution of the virosphere and viral megataxonomy
4.1. Viral hallmark genes
A fundamental fact that needs to be emphasized before even starting to discuss the structure and evolution of the virosphere is that viruses do not share a single common origin, in sharp contrast to cellular life forms. About 100 genes are strictly universal among cellular organisms and can be used to build the “Tree of life”, individually or more commonly, in different combinations (Koonin, 2003; Ciccarelli et al., 2006). Viruses, however, lack any universal genes, and accordingly, there can be no single tree of viruses, in principle (Koonin, Senkevich, et al., 2006). Put another way, unlike cellular life forms, which all can be traced back to the Last Universal Cellular Ancestor (LUCA), viruses are polyphyletic, that is, had multiple origins. That said, although the total number of independent viral origins is hard to estimate, there are relatively few vast assemblages of viruses for which common ancestry is traceable.
The evolutionary connections between diverse large groups of viruses were traced by comparative and phylogenetic analysis of shared viral hallmark genes (VHG) (Iranzo, Krupovic, et al., 2016; Iranzo, Koonin, et al., 2016; Wolf, Kazlauskas, et al., 2018; Koonin, Dolja and Krupovic, 2022). The VHGs are difficult to define formally because of an apparent circularity inherent in that definition: VHGs are conserved across diverse groups of viruses which are themselves defined through shared VHGs. Informally, however, VHGs are readily identifiable, and their identities are not surprising: these are genes encoding major virion morphogenesis (primarily, capsid) proteins and key components of viral replication machineries (Table 1). In viruses with small genomes, the VHGs represent a large fraction of the genes, and in some cases, all of the genes. By contrast, in viruses with large genomes, the VHGs can comprise less than 1% of the genes. Nevertheless, in all viruses, the VHGs are central both to virus replication and morphogenesis, and to the study of the evolution of the virosphere.
Virus hallmark proteins
| Protein | Structural fold | Function | Distribution in the virosphere |
|---|---|---|---|
| RNA-dependent RNA polymerase | RRM | Replication and expression of RNA genomes | Orthornavirae |
| Reverse transcriptase | RRM | Replication of all RNA and DNA genomes with a reverse transcription stage in the replication cycle | Pararnavirae |
| HUH superfamily rolling circle replication endonuclease | RRM | Initiation of rolling circle DNA replication | Efunaviria, Volvereviria, Floreoviria, Pleomoviria, some Duplodnaviria, Varidnaviria and Adnaviria |
| Family B DNA polymerase | RRM | Replication of dsDNA genomes | Many Duplodnaviria, Varidnaviria, Naldaviricetes, Bidnaviridae, several families of archaeal viruses |
| Archaeo-eukaryotic primase | RRM | Priming of DNA synthesis | Many Duplodnaviria, Varidnaviria, Naldaviricetes |
| Superfamily 3 helicase | P-loop NTPase (AAA+ ATPase class) | Unwinding of RNA and DNA genomes during replication | Many Riboviria, Duplodnaviria, Varidnaviria, Floreoviria, Naldaviricetes |
| FtsK-like ATPase | P-loop NTPase | ATP-dependent genome packaging | Most Varidnaviria, Singelaviria,Efunaviria |
| Terminase, large subunit | P-loop NTPase (RecA class)—RNase H-fold nuclease | ATP-dependent genome packaging and processing | Duplodnaviria |
| Single jelly-roll capsid protein | Jelly-roll (8-stranded β-barrel) | Capsid formation: major and minor capsid proteins | Volvereviria, Floreoviria, Orthornavirae, Singelaviria, Varidnaviria |
| Double jelly-roll capsid protein | Double jelly-roll (2 tandem 8-stranded β-barrels) | Capsidformation: major capsid protein | Majority of Varidnaviria |
| HK97-like capsid protein | HK97-fold | Capsid formation: major capsid protein | Duplodnaviria |
| Portal protein | Unique, mostly α-helical, “portal” fold | Capsid assembly and genome packaging | Duplodnaviria |
The VHGs define the distinct domains of the virosphere that correspond to viral realms, the top rank in virus taxonomy (International Committee on Taxonomy of Viruses Executive Committee, 2020). In accord with the fundamental principles of evolutionary taxonomy, each realm is supposed to be monophyletic, that is, all viruses in a realm are assumed to share a common origin (Koonin, Dolja, Krupovic, Varsani, et al., 2020; Simmonds, Adriaenssens, Zerbini, et al., 2023). In actuality, this common origin can be limited to sharing only a few or even a single VHG. Nevertheless, given the central roles of the VHGs in virus replication, this approach is productive for reconstructing the evolution of the virosphere and delineating higher taxa of viruses to develop the taxonomic system that is informally known as megataxonomy (Koonin, Dolja, Krupovic, Varsani, et al., 2020; Koonin, Kuhn, et al., 2024). By the conventions of taxonomy, any virus can belong to only one realm although, when some viruses combine VHGs characteristic of different realms, an apparent conflict emerges. Below we discuss some such cases.
Initially, four major realms were established, Riboviria, Monodnaviria, Varidnaviria, and Duplodnaviria, each including a vast diversity of viruses, and two much smaller realms were added shortly thereafter. However, subsequent in-depth evolutionary analysis led to the split of Varidnaviria into two realms, and a split of Monodnaviria into four realms, with the new realms approved by ICTV. More splitting of realms based on a stricter approach to monophyly can be expected, as discussed below.
4.2. Riboviria: the expanding viral RNA World
The viruses in the realm Riboviria share a single VHG, namely, the gene encoding RNA-directed RNA polymerases (RdRPs) or RNA-directed DNA polymerases (reverse transcriptases, RTs), the homologous enzymes responsible for viral genome replication and expression (Figure 2A,B). This realm covers 5 Baltimore classes, including all three classes of viruses with RNA genomes that replicate without a DNA intermediate as well as viruses with RNA or DNA genomes whose replication involves a reverse transcription stage. The study of the large-scale evolution of this virus realm is, out of necessity, based on comparison and phylogenetic analysis of RdRP/RT, the only VHG that is conserved across the realm. Notably, no other genes come even close to RdRP/RT with respect to the conservation among ribovirians (ending “virians” refers to all members of a realm (Koonin, Kuhn, et al., 2024)): the most common of these, such as the single jelly-roll (SJR) capsid proteins (Figure 2C), the principal building block of icosahedral ribovirian virions, are shared only by subsets of the viruses in the realm (Wolf, Kazlauskas, et al., 2018). Thus, comparative analysis of these genes can shed light on the evolution of particular groups of ribovirians but not of the entire realm. Indeed, virions of ribovirians are highly diverse with respect to morphology and complexity, and are built from many structurally unrelated major capsid proteins (Figure 2C,D). Thus, if virion morphogenesis proteins of ribovirians were chosen for realm definition, over a dozen realms would have to be created for the classification of all RNA and RT viruses.
Megataxonomy of viruses: realm Riboviria. (A) Taxonomic structure: from realm down to classes. The taxonomic tree was retrieved from the ICTV website (Black et al., 2026). (B) Structural models and topology diagrams of the hallmark proteins of Riboviria, RNA-dependent RNA polymerase (RdRP; PDB: 1ra7) and reverse transcriptase (RT; PDB: 7o0h). (C,D) Virion architectures and structural models of major capsid proteins (CP or CA) and nucleocapsid (N) proteins in the kingdoms Othornavirae (C) and Pararnavirae (D). The proteins were selected to represent the diversity of structural folds in Riboviria. Virion diagrams were obtained from ViralZone (De Castro et al., 2024). The structures are denoted with the corresponding PDB accession numbers. Abbreviations: SJR CP, single jelly-roll capsid protein; C, core protein; PsV-F, Penicillium stoloniferum virus F; TYMV, turnip yellow mosaic virus; VEEV, Venezuelan equine encephalitis virus; WNV, West Nile virus; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; BDV, Borna disease virus; TMV, tobacco mosaic virus; WMV, watermelon mosaic virus; RVFV, Rift Valley fever virus; CCHFV, Crimean-Congo hemorrhagic fever virus; HBV, hepatitis B virus; ACNDV, African cichlid nackednavirus; HIV-1, human immunodeficiency virus 1; PFV, prototype foamy virus.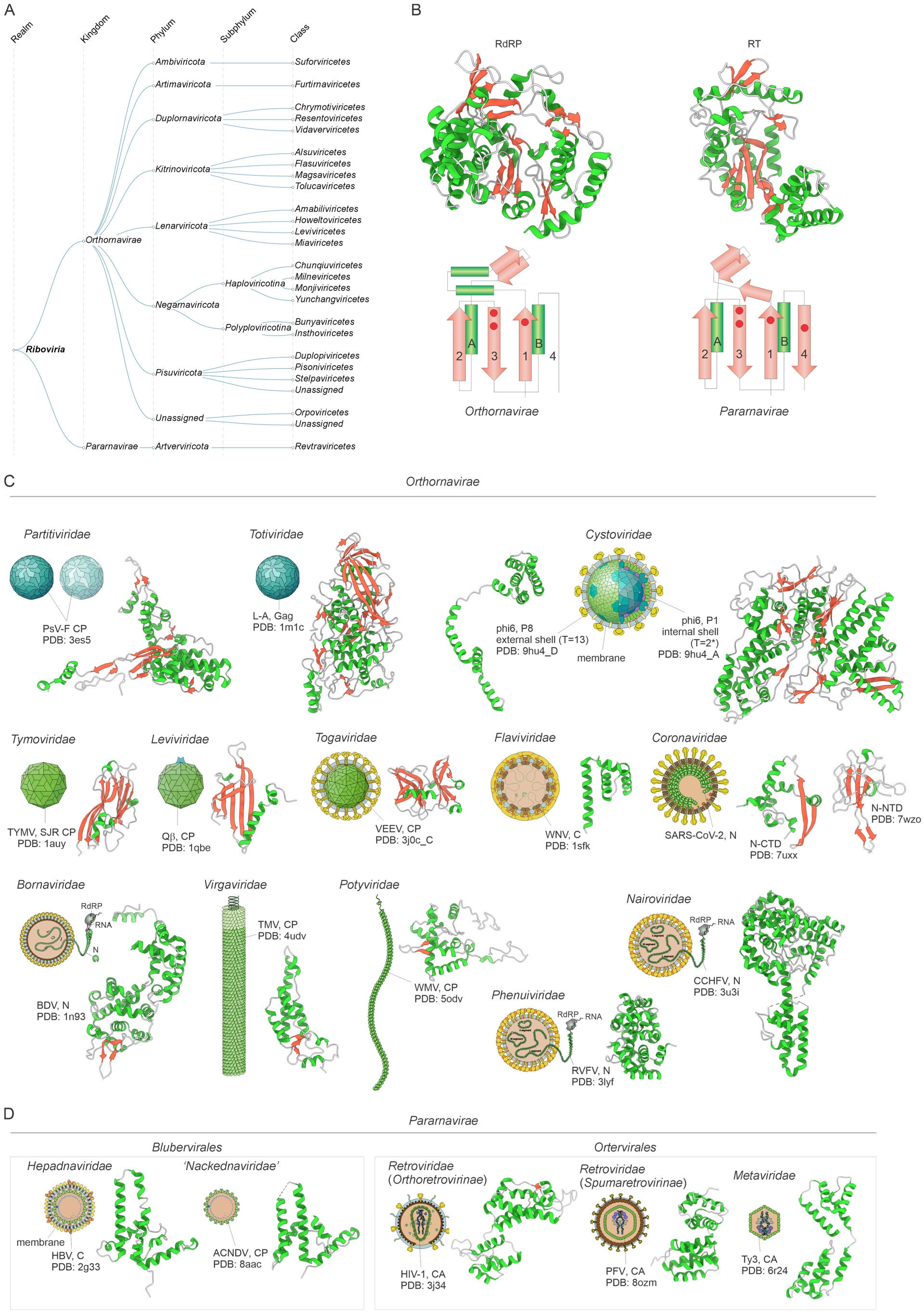
The realm Riboviria is currently divided into two kingdoms, Orthornavirae, viruses that encode RdRP and have no DNA stage in their replication cycles, and Pararnavirae, viruses that encode RTs and replicate via alternating RNA and DNA stages (Figure 2A). The two kingdoms share no conserved genes other than RdRP and RT. All RdRPs and RTs share a common structural fold (Figure 2B), and their sequences can be confidently aligned to produce a single phylogenetic tree that is not fully resolved but does contain major, robust clades (see below). Moreover, among the polymerases sharing a conserved core fold, known as the Palm domain or RNA recognition motif (RRM), RdRPs and RTs are relatively close, forming a single clade (Xiong and Eickbush, 1990). Thus, beyond doubt, all RdRPs and RTs evolved from a single ancestral enzyme that possessed polymerase activity, either RdRP or RT, or both. However, this homology does not actually mean that the two kingdoms share a common viral ancestry. All known RdRP-encoding replicators are viruses or derivatives of viruses that have lost genes for virion proteins (see below). Most likely, the common ancestor of orthornaviraens (ending “viraens” refers to all members of a kingdom (Koonin, Kuhn, et al., 2024)) was a simple virus encoding the RdRP and a SJR capsid protein. Orthornaviraens are ubiquitous in eukaryotes, and as recently demonstrated by metatranscriptome analysis, are also common in bacteria (Neri et al., 2022; Edgar et al., 2022; Zayed et al., 2022; Hou et al., 2024). Thus, the ancestral orthornaviraens most likely infected bacterial hosts, having emerged at an early stage of life evolution. Notably, despite considerable effort, orthornaviraens and more generally ribovirians have not been thus far detected in archaea, the causes of this potential exclusion remaining unclear.
Pararnaviraens have a completely different provenance, apparently, descending from group II introns, which are, in effect, prokaryotic retrotransposons (Gladyshev and Arkhipova, 2011; Toro and Nisa-Martinez, 2014). Pararnaviraens evolved only in eukaryotes as a result of the capture of the (nucleo)capsid proteins (Krupovic, Blomberg, et al., 2018). Thus, there is no evidence of common origin of orthornaviraens and pararnaviraens from a viral ancestor. Furthermore, there are strong indications that two orders within Pararnavirae, Ortervirales and Blubervirales, have evolved from distinct families of transposons by recruiting structurally unrelated major virion proteins (Krupovic, Blomberg, et al., 2018; Gong and G. Z. Han, 2018; Gong and G. Z. Han, 2025) (Figure 2D). Accordingly, following the principle of viral realm monophyly, the current realm Riboviria should be split into at least three separate realms, one including all orthornaviraens and two more for the two independently evolved branches of pararnaviraens.
Initial phylogenetic analysis of orthornaviral RdRPs produced a tree with 5 major clades that were designated phyla (Wolf, Kazlauskas, et al., 2018; Wolf, Silas, et al., 2020). Three of the 5 phyla consist of positive-sense RNA viruses (although one phylum, Pisuviricota, also includes some groups of dsRNA viruses), one of negative-sense RNA viruses, and the last one of dsRNA viruses. The monophyly of each phylum is strongly statistically supported but the relationships among them remain largely uncertain (Neri et al., 2022; Edgar et al., 2022; Zayed et al., 2022). However, the basal position of phylum Lenarviricota, which consists of riboviruses infecting bacteria and their direct descendants infecting eukaryotes, is robust in RdRP trees rooted by RT, implying that all the diverse orthornaviraens of eukaryotes evolved from bacterial ancestors.
Metatranscriptome mining led to a manyfold increase in the known diversity of orthornaviraens, including several branches without a close relationship to any of the 5 large phyla that are candidates for new phyla (Neri et al., 2022; Edgar et al., 2022; Zayed et al., 2022; Hou et al., 2024). Two of these that have already been approved by the ICTV are of particular note. The new phylum Artimaviricota includes viruses with genomes consisting of two segments of dsRNA, one of which encodes a distinct polymerase that, as suggested by structural comparison, could be an evolutionary intermediate between RdRP and RT (Urayama, Fukudome, Hirai, et al., 2024). Artimaviricots (ending “viricots” refers to all members of a phylum (Koonin, Kuhn, et al., 2024)) were discovered in metatranscriptomes from hot springs dominated by hyperthermophilic bacteria, which are the likely hosts of these viruses. Prokaryotic hosts are suggested also by the genome organization of artimaviricots, with genomic segments encompassing multiple protein-coding genes preceded by ribosome-binding sites (RBS or Shine-Dalgarno sequences).
More generally, perhaps, the greatest insight from the metatranscriptome mining for ribovirians is the dramatic expansion of the number and diversity of orthornaviraens associated with bacterial hosts, even if in many cases tentative. Apart from the discovery of an unexpected plentitude of previously unknown lenarviricots, multicistronic genome organization, with the telltale RBS preceding each protein-coding region, in addition to artimaviricots, was discovered in picobirnaviruses, paraxenoviruses and several branches of partitiviruses, all viruses with dsRNA genomes in phylum Pisuviricota (Neri et al., 2022; Krishnamurthy and D. Wang, 2018; D. Wang, 2022; Yoshida et al., 2025). Furthermore, it has been shown that many picobirnaviruses, initially thought to infect eukaryotes, encode functional bacterial lysins, strongly supporting the bacterial host for these viruses (Gan and D. Wang, 2023). In addition, several deep branches, representatives of putative new phyla, showed the same features suggestive of prokaryotic hosts. Thus, metatranscrtiptomics shattered the traditional view that riboviruses were primarily characteristic of eukaryotes, showing that RNA bacteriophages are a substantial, diverse component of prokaryotic viromes that has been largely overlooked before large metatranscriptome sequence datasets became available. Moreover, the mixing of viruses with known eukaryotic hosts with those predicted to infect bacteria among the partitiviruses suggests that, in some groups of orthornaviraens, particularly those that, like partitiviruses, harbor two or more genomic segments, each carrying a single gene, the switch from prokaryotic to eukaryotic hosts or vice versa is comparatively easy and occurred on multiple occasions (Neri et al., 2022; Urayama, Fukudome, Hirai, et al., 2024). The remarkable ability of partitiviruses to replicate in highly diverse hosts has been demonstrated experimentally in eukaryotes, where the same virus has been shown to replicate in hosts from three eukaryotic kingdoms, including fungi, plants and animals (Telengech et al., 2024). How such simple viruses can overcome the physical barriers of the host envelope and defense systems in highly distinct cell types is an intriguing question.
Perhaps, an even more startling discovery is the 7th recognized phylum of orthornaviraens, Ambiviricota, which includes fungal parasites with covalently closed circular (ccc) RNA genomes encoding a distinct RdRP without clear affinity with any other viruses and an uncharacterized protein (Kuhn, Botella, et al., 2024). Ambivirus genomes resemble those of viroids in that both are circular and contain ribozymes, RNA segments with distinct secondary structures endowed with catalytic activity (B. D. Lee et al., 2023; Forgia et al., 2023). The mechanistic interplay between the ribozymes and RdRP during the ambiviral replication cycle remains to be elucidated. Ambiviricots combine features of two virus realms, Riboviria and Ribozyviria (see below), but given the presence of the RdRP, the hallmark of Orthornavirae within Riboviria, the realm and kingdom are not in doubt.
Mapping the gene content and genome organization of orthornaviraens to the RdRP tree allowed an approximate reconstruction of evolutionary trajectories, revealing the major trend of genome growth and complexification via accretion of genes captured from hosts and other viruses. The ancestral orthornaviraen is inferred to have had a small genome of perhaps about 3 kb, with two genes only, encoding the RdRP and a capsid protein, most likely, with the SJR fold (Wolf, Kazlauskas, et al., 2018). Subsequent evolution involved a substantial increase in genome size occurring independently in different lineages, up to 64 kb in some nidoviruses (Neuman et al., 2025; Zhong et al., 2025; Dolja, 2025). Acquisitions that occurred in parallel in different lines of descent included RNA helicases that apparently enabled efficient replication of larger RNA genomes, proteases that cleave viral polyproteins into mature, functional proteins and a variety of genes involved in virus–host interaction, particularly inhibition of host defenses and movement proteins in plant viruses.
Pararnaviraens are far less diverse than orthornaviraens, with only one phylum and class, which splits into two orders, Ortervirales and Blubervirales (Krupovic, Blomberg, et al., 2018) (Figure 2A). Ortervirals (ending “virals” refers to all members of an order (Koonin, Kuhn, et al., 2024)) include the numerous animal retroviruses with RNA genomes replicating via a DNA intermediate along with plant caulimovirids (ending “virids” refers to all members of a family (ibid.)) with dsDNA genomes replicating via an RNA intermediate and three families of viruses that are often considered as “long terminal repeat (LTR) retrotransposons”, namely, Metaviridae, Pseudoviridae and Belpaoviridae. Members of the three families share with the animal retrovirids homologous RTs, aspartic proteases and a set of structural proteins, namely, capsid and nucleocapsid proteins, that form virus particles (Krupovic and Koonin, 2017a) (Figure 2D). Thus, according to the virus definition given above, “LTR retrotransposons” are bona fide viruses. Recently, a fourth potential family of ortervirals, described as Troyka “retrotransposons”, has been reported (Kojima, 2025), and additional divergent lineages of RT viruses are likely to be discovered in the near future.
Blubervirales includes human hepatitis B virus and its relatives infecting other vertebrate animals as well as a diverse group of related non-enveloped viruses informally known as nackednaviruses (Figure 2D). More recently, the reach of blubervirals was extended to rotifers, a group of microscopic and near-microscopic pseudocoelomate animals, by a family-level lineage, dubbed proto-nackednaviruses, which in the RT phylogeny form a basal group to hepadnavirids and nackednaviruses (Gong and G. Z. Han, 2025). Blubervirals possess DNA genomes that replicate via an RNA intermediate and their structural proteins are unrelated to those of ortervirals (Wynne et al., 1999) (Figure 2D).
Pararnaviraens are limited in their spread to eukaryotic hosts and apparently evolved from non-viral retroelements, such as group II self-splicing introns and retrotransposons, by acquiring host proteins and exapting them for structural roles in the virions (Krupovic and Koonin, 2017b). Furthermore, the two orders within this kingdom apparently evolved at two independent points of origin from different retrotransposon families (Gong and G. Z. Han, 2018; D. Wang, 2022; Malik and Eickbush, 2001). Whereas the exact ancestor of ortervirals remains obscure, blubervirals have apparently evolved from a distinct group of retroelements known as HEART (Gong and G. Z. Han, 2018).
4.3. Four realms of viruses with ssDNA genomes
Until recently, the vast majority of viruses with ssDNA genomes and two groups of viruses with small dsDNA genomes (papillomavirids and polyomavirids) were classified in a single realm, Monodnaviria. The realm was held together by a single VHG that encodes a distinct endonuclease of the HUH superfamily (or its inactivated derivative) involved in the initiation of the genome replication via the rolling circle (or rolling hairpin) mechanism (Ilyina and Koonin, 1992; Chandler et al., 2013). However, recently, upon considerable expansion of the ssDNA virome, largely through metagenomics, and improved understanding of evolutionary relationships among these viruses, the realm was split into four monophyletic realms, corresponding to the four initially defined kingdoms (Figure 3A).
Megataxonomy of viruses: the four realms of viruses with small ssDNA or dsDNA genomes. (A) Taxonomic structure, from realms down to orders, and structural models of the realm-specific hallmark proteins: major capsid proteins (Efunaviria and Volvereviria), rolling circle replication initiator containing the HUH superfamily endonuclease and superfamily 3 helicase domains (Floreoviria), and membrane fusion protein VP5 (Pleomoviria). The structures are denoted with the corresponding PDB accession numbers. AF3 denotes structures modeled with AlphaFold3. The taxonomic tree was retrieved from the ICTV website (Black et al., 2026) and modified. (B) The three distinct replication mechanisms catalyzed by unrelated proteins and yielding ssDNA genomes, which are encapsidated into virus particles. Note that some members of the realm Pleomoviria encapsidate either dsDNA or ssDNA replicative intermediates. Also shown is the theta mode of genome replication initiated by the inactivated HUH endonuclease—superfamily 3 helicase of polyomavirids and papillomavirids. Abbreviations: BmBDV, Bombyx mori bidensovirus; TP, terminal protein; pPolB, protein-primed family B DNA polymerase; TAg, large T antigen; SV40, simian virus 40; ori, origin of replication; TIR, terminal inverted repeats; Tnp, transposase; PCV2, porcine circovirus 2; MCP, major capsid protein; HRPV-6, Halorubrum pleomorphic virus 6. Virion diagrams were obtained from ViralZone (De Castro et al., 2024).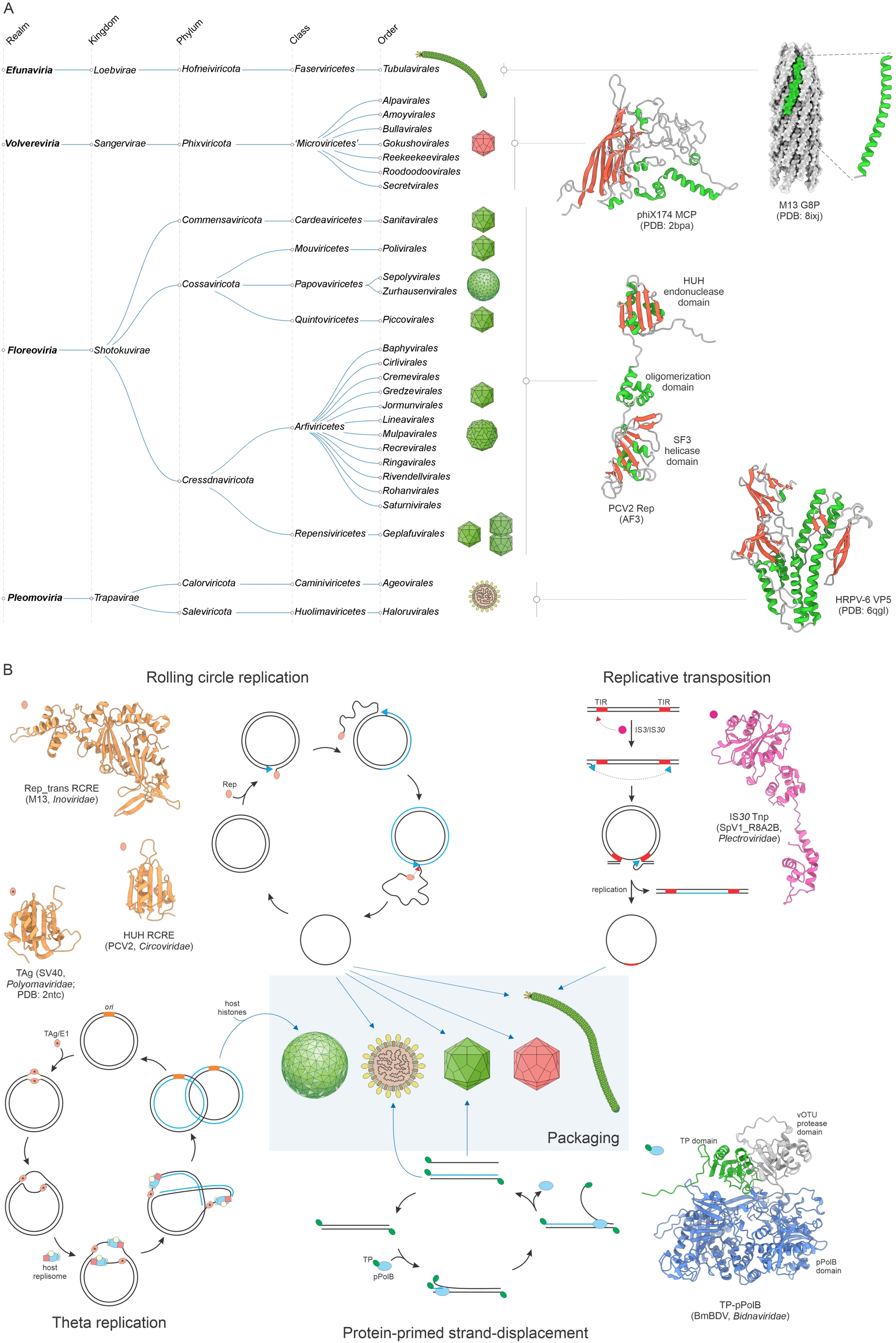
Realm Efunaviria, with the kingdom Loebvirae, consists of filamentous prokaryotic viruses with circular ssDNA genomes, including some well-characterized phages of family Inoviridae such as M13, f1 and fd (Roux, Krupovic, Daly, et al., 2019). The morphogenetic module and the overall virion assembly mechanism of efunavirians are conserved. All efunavirians encode a highly hydrophobic capsid protein consisting of a single α-helix, which coats the circular ssDNA genome upon its extrusion through the cytoplasmic membrane with the aid of the conserved virus-encoded FtsK-family ATPase (Rakonjac et al., 2024) (Figure 3A). By contrast, the genome replication enzymes vary. Although some members of the realm employ HUH endonucleases, the majority relies on a variety of rolling circle replication initiation endonucleases of the Rep_trans superfamily that is not homologous to the HUH superfamily (Roux, Krupovic, Daly, et al., 2019), whereas others use transposases that generate ssDNA molecules as a transposition intermediate (Krupovic and Forterre, 2015) (Figure 3B). Efunavirians infect widely diverse bacterial hosts, suggesting long-term association. However, putative efunavirians were also discovered in some methanogenic archaea (Roux, Krupovic, Daly, et al., 2019), suggesting that the emergence of this virus realm could be more ancient, potentially dating back to the LUCA (Krupovic, Dolja, et al., 2020).
Realm Volvereviria, with the kingdom Sangervirae, represents a relatively uniform group of prokaryotic viruses, exemplified by the iconic bacteriophage phiX174 and other phages of the recently created class Microviricetes (formerly, family Microviridae). Volverevirians form small icosahedral capsids built of a distinct SJR-fold capsid protein (Dokland et al., 1997; H. Lee et al., 2022; Mietzsch, Kailasan, et al., 2024; Bardy et al., 2025) (Figure 3A), which is commonly used as a marker for the discovery and classification of volverivirians (Roux, Krupovic, Poulet, et al., 2012; Kirchberger, Martinez and Ochman, 2022; Olo Ndela et al., 2023; Lund et al., 2024). These viruses have circular ssDNA genomes replicated by the rolling circle mechanism and uniformly (thus far) encode the HUH endonuclease (Doore and Fane, 2016; Kirchberger and Ochman, 2023) (Figure 3B). Volverevirians can be either strictly lytic, typically lysing the host by inhibiting peptidoglycan synthesis (Chamakura and Young, 2020), or temperate, integrating into the bacterial genome using the host recombination machinery (Krupovic and Forterre, 2011; Kirchberger and Ochman, 2020). All experimentally characterized members of this realm are phages, but for the majority of volverevirians identified via metagenomics, the hosts remain unknown, so it cannot be excluded that some of them replicate in archaea.
Realm Floreoviria, with the kingdom Shotokuvirae, encompasses a highly diverse group of eukaryotic viruses with small ssDNA or dsDNA genomes, which in most members are circular, but can also be linear (e.g., families Parvoviridae, Bidnaviridae, Oomyviridae). Floreovirians form icosahedral capsids from SJR capsid proteins (Mietzsch, Bennett, et al., 2025) which, however, appear to have a distinct origin from the SJR capsid proteins of volverevirians (Krupovic and Koonin, 2017b). Furthermore, in some lineages (e.g., Bacilladnaviridae, Naryaviridae, Nenyaviridae, and cruciviruses), the ancestral SJR capsid protein has been replaced by distinct SJR capsid protein variants from RNA viruses, allowing for the formation of larger (T = 3) capsids (Roux, Enault, et al., 2013; Kazlauskas, Dayaram, et al., 2017; de la Higuera et al., 2020; Krupovic and Varsani, 2022; Gebhard et al., 2025; Munke et al., 2022). A characteristic feature of viruses in this realm is the two-domain replication protein, consisting of the N-terminal HUH endonuclease and C-terminal superfamily 3 helicase domains (Kazlauskas, Varsani, et al., 2019) (Figure 3A). The ancestor of floreovirians, in all likelihood, encoded the two-domain HUH superfamily endonuclease and replicated via the rolling circle mechanism, which is employed by the majority of the members of this realm (Figure 3B). However, in some descendants, this gene has been either inactivated (polyomavirids and papillomavirids), concomitant with a switch from the rolling circle to the theta-like replication mechanism (Iyer, Koonin, et al., 2005), or replaced by a protein-primed family B DNA polymerase (bidnavirids) (Krupovic and Koonin, 2014) (Figure 3B). An even more dramatic departure from the canonical gene combination is observed in members of the family Anelloviridae (phylum Commensaviricota, order Sanitavirales). Anellovirids retain the SJR capsid protein, albeit with major modifications not observed in other floreovirians (Butkovic, Kraberger, et al., 2023; Liou et al., 2024; De Koch et al., 2025), but the ancestral replication initiation gene has been lost, with the genome replication apparently fully depending on the host replication machinery (Boisvert et al., 2025). Although the exact mechanism remains to be elucidated, it has been suggested that anellovirids employ recombination-dependent replication by recruiting the host DNA polymerase alpha and BTR (Bloom’s syndrome helicase (BLM), topoisomerase IIIα, RMI1, and RMI2) complexes (ibid.), with the circular ssDNA genomes being produced by a process that resembles the formation of extratelomeric C-circles (J. Lee, J. Lee, et al., 2024; J. Lee, Sohn, et al., 2025).
Finally, the realm Pleomoviria, with the kingdom Trapavirae, includes several families of archaeal viruses with enveloped pleomorphic virions (Figure 3A). The virions resemble membrane vesicles with two major structural proteins, a spike protein with a unique structural fold responsible for host recognition and membrane fusion, and a matrix protein embedded in the viral envelope (El Omari et al., 2019; Y. Liu, Dyall-Smith, et al., 2022; Bignon et al., 2022). The membrane fusion protein is a signature of pleomovirians (Figure 3A). Initially, Trapavirae included a single family, Pleolipoviridae, of viruses infecting extremely halophilic archaea (Y. Liu, Dyall-Smith, et al., 2022; Alarcon-Schumacher et al., 2023). However, more recently, related viruses were also identified in hyperthermophilic, methanogenic and nano-sized hyperhalophilic archaea (Medvedeva, Borrel, et al., 2023; Baquero, Bignon, et al., 2024; Zhou et al., 2025), expanding the genetic diversity and host range of this virus group. The genomes of pleomovirians can be ssDNA or dsDNA, circular or linear. Accordingly, the replication mechanisms and the corresponding enzymes are highly variable, including several non-orthologous and even non-homologous rolling circle replication endonucleases (Kazlauskas, Varsani, et al., 2019; Baquero, Bignon, et al., 2024; Roine et al., 2010; Y. Wang et al., 2026), primases (Alarcon-Schumacher et al., 2023) or protein-primed family B DNA polymerases (Y. Liu, Dyall-Smith, et al., 2022) (Figure 3B). Thus, it is the shared morphogenetic rather than genome replication module that holds this realm together.
Viruses of the four realms, Efunaviria, Volvereviria, Floreoviria and Pleomoviria, formerly joined under Monodnaviria typically have small genomes (2–10 kb; Figure 1C) and their evolution appears to be tightly intertwined with that of small plasmids, largely replicating via the rolling circle mechanism, and apparently, ancestors of the four realms evolved from different families of such plasmids (Kazlauskas, Varsani, et al., 2019). In all cases of independent origins of viruses with small ssDNA genomes, the capsid protein genes were captured by the emerging viruses independently from other viruses, such as orthornaviraens, or from host genes.
4.4. Realms Varidnaviria and Singelaviria: the epitome of viral diversity
Realm Varidnaviria consists of an enormous diversity of dsDNA viruses infecting bacteria, archaea, and eukaryotes (Figure 4A) that typically have an icosahedral capsid built of double jelly-roll (DJR) major capsid protein (MCP), the principal VHG that holds the realm together (Krupovic and Bamford, 2008), and SJR minor capsid protein (penton), which form hexagonal and pentagonal capsomers, respectively (N. G. Abrescia et al., 2004) (Figure 4B,C). Most varidnavirians also share another VHG encoding a genome packaging ATPase of the FtsK-HerA superfamily (Iyer, Makarova, et al., 2004). The varidnavirians display a remarkable variation in virion sizes, morphologies and complexity. Most icosahedral virions in this realm contain an internal lipid membrane, sandwiched between the protein capsid and the genome, which likely represents the ancestral trait of Varidnaviria (Figure 4D). However, in some lineages (e.g., adenovirids), the internal membrane was lost, whereas in others, an additional external membrane was added (e.g., iridovirids), and/or a second internal icosahedral shell built from unrelated capsid proteins has been added (e.g., asfarvirids and faustoviruses) (Figure 4D). Even more dramatic deviations from the canonical structural layout occurred in several groups of varidnavirians, e.g., pandoraviruses and pithoviruses, where the DJR MCP was replaced by unrelated proteins, resulting in odd capsid shapes (Philippe et al., 2013; Legendre, Bartoli, et al., 2014; Souza et al., 2019; Krupovic, Yutin and Kooninet al., 2020; Queiroz et al., 2023) (Figure 4D). Other groups of viruses within Varidnaviria, such as poxvirids, encode a homolog of the DJR MCP but incorporate it only into intermediates of the virion morphogenesis, whereas the odd-shaped mature capsids consist of unrelated viral proteins (Coulibaly, 2024).
Megataxonomy of viruses: realm Varidnaviria. (A) Taxonomic structure, from realms down to orders. The taxonomic tree was retrieved from the ICTV website (Black et al., 2026). (B) Structural models of the double jelly-roll major capsid protein (DJR-MCP; top) and single jelly-roll penton protein (bottom) of bacteriophage PRD1 (Tectiviridae). The structures are denoted with the corresponding PDB accession numbers. (C) Structural models of the MCP trimer and penton pentamer, which form hexagonal and pentagonal capsomers. In tectivirids and adenovirids, the two types of capsomers are arrayed to form icosahedral capsids with a pseudo T = 25. (D) Diversity of virion organizations and complexity among varidnavirians and proposed evolutionary scenarios. Virion diagrams were obtained from ViralZone (De Castro et al., 2024).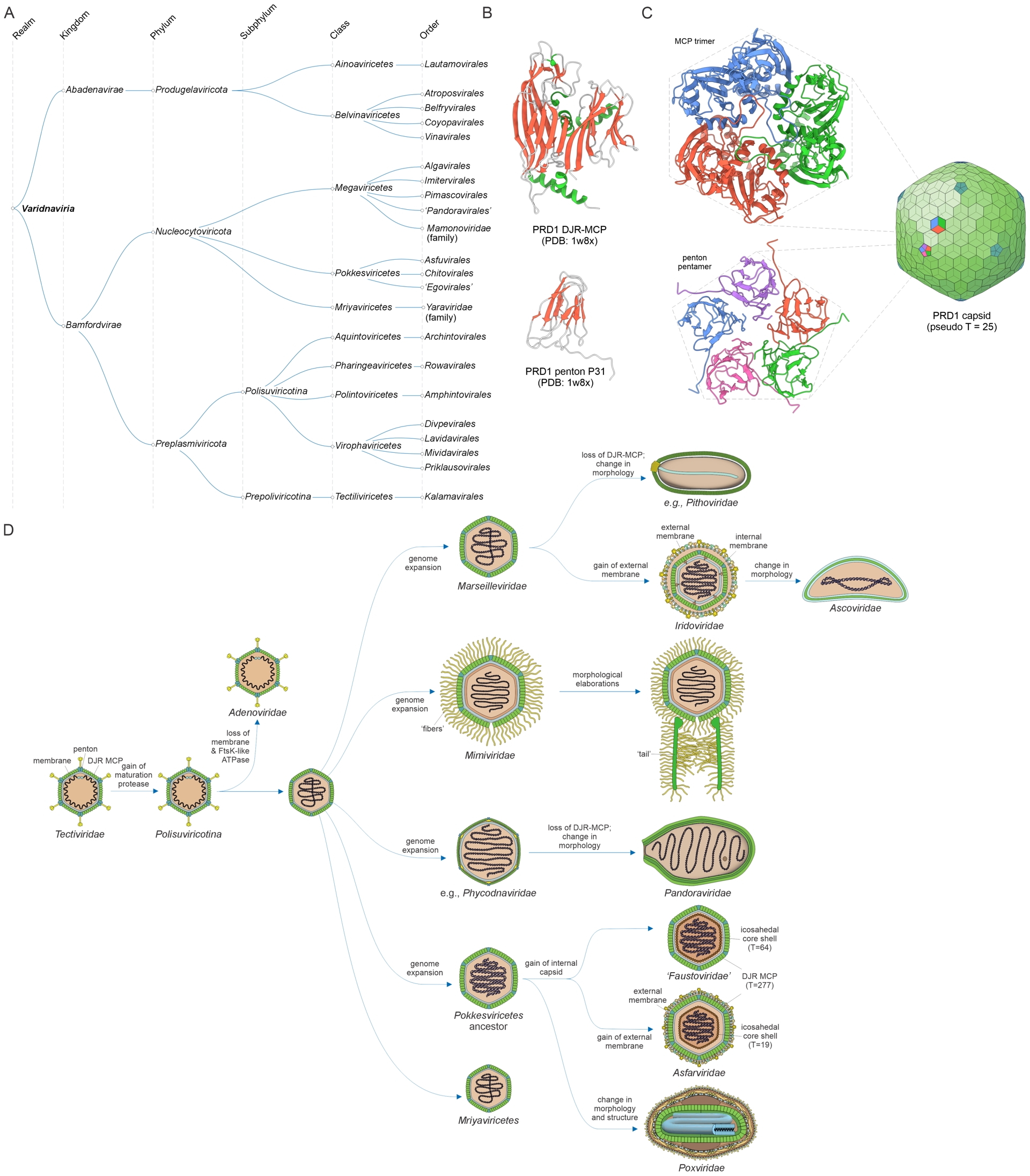
Varidnaviria consists of two kingdoms, Bamfordvirae and Abadenavirae (Koonin, Fischer, et al., 2024). The kingdom Abadenavirae includes all bacterial and archaeal viruses with DJR MCP and relatively small dsDNA or ssDNA genomes (e.g., finnlakevirids), except for one family of bacterial viruses, Tectiviridae. The kingdom Bamfordvirae includes tectivirids and a broad diversity of eukaryotic viruses, characterized by highly diverse genome and virion sizes, including megagenomoviruses with genomes in excess of 4 Mb (Buscaglia et al., 2025) (Figure 4A).
The evolutionary scenario for Varidnaviria derives the entire, striking diversity of eukaryotic varidnavirians from a tectivirid ancestor (Figure 4D), possibly, passed to the emerging eukaryote by the mitochondrial endosymbiont (Krupovic and Koonin, 2015; Krupovic, Kuhn, Fischer, et al., 2024). Notably, members of a diverse assemblage of bamfordviraens with relatively small genomes (15–35 kb) were originally recognized as DNA transposons known as polintons (after Polymerase and Integrase, two proteins encoded in their genomes; alternatively, called mavericks) that are integrated in genomes of a broad variety of eukaryotes, in some cases, in numerous copies (Kapitonov and Jurka, 2006; Pritham et al., 2007; Barreat and Katzourakis, 2021; Jeong et al., 2023). Subsequent analyses have shown, however, that polintons encode a complete morphogenetic module comprising typical DJR-MCP, penton protein with the SJR fold, capsid maturation protease and the packaging ATPase, strongly suggesting that polintons are bona fide varidnavirians (Krupovic, Bamford and Koonin, 2014). Currently, polintons and numerous polinton-like viruses, of which many but not all encode integrases and are known or predicted to integrate into the host genomes, are classified into subphylum Polisuviricotina of phylum Preplasmiviricota that also includes the tectivirids (Koonin, Fischer, et al., 2024). Many polisuviricotins are virophages, that is, symbionts (parasites or commensals) of large viruses of phylum Nucleocytoviricota (La Scola et al., 2008; Fischer, 2021; Roux, Fischer, et al., 2023; Roitman et al., 2023).
Similar to viruses with smaller DNA genomes, varidnavirians employ a variety of genome replication strategies and, accordingly, encode a range of non-homologous replication proteins, ranging from Rep_trans and HUH endonucleases for rolling circle replication (e.g., finnlakevirids and corticovirids, respectively (Laanto et al., 2017; Krupovic and Bamford, 2007)) to protein-primed family B DNA polymerases and their various derivatives (e.g., tectivirids and adenovirids (Krupovic, Kuhn, Fischer, et al., 2024)) to family A DNA polymerases (e.g., sputnivirovirids (Iyer, Abhiman, et al., 2008)) to the complete or near-complete replisome in most nucleocytoviricots (Koonin and Yutin, 2019; Koonin and Yutin, 2018). Notably, major variations are observed within Nucleocytoviricota: members of the class Mriyaviricetes, the nucleocytoviricots with the smallest genomes that apparently comprise the basal branch of this phylum, encode an HUH endonuclease but no DNA polymerase and are predicted to replicate via the rolling circle mechanism (Yutin, Mutz, et al., 2024). Even relatively closely related viruses (e.g., those from the same order) may encode unrelated replication modules. Furthermore, the presence of the conserved HUH endonuclease of Mriyaviricetes formally connects this viral class to two of the realms of viruses with ssDNA genome, for which this protein is a hallmark. However, the overall phylogenomic analysis leaves no doubt about the assignment of mriyaviricetes to Nucleocytoviricota.
Realm Singelaviria includes bacterial and archaeal viruses with linear or circular dsDNA genomes and icosahedral capsids built from SJR MCPs (Figure 5A,B). All structurally characterized, formally classified singelavirians encode two paralogous SJR MCPs, which form two types of heterohexameric capsomers (Figure 5B), resembling the homotrimeric capsomers of varidnavirians (Figure 4C). Furthermore, similar to most varidnavirians, the icosahedral virions of singelavirians contain an internal membrane (Demina et al., 2023). Thus, initially, it has been assumed that fusion of the two singelavirian SJR MCP genes yielded the DJR MCP (Rissanen et al., 2013; Santos-Perez et al., 2019; De Colibus et al., 2019). This scenario was further supported by the fact that, like varidnavirians, singelavirians encode an FtsK-HerA superfamily genome packaging ATPase (Strömsten et al., 2005). Accordingly, singelavirians were included in the realm Varidnaviria as kingdom Helvetiavirae. However, more recently, a detailed comparison of protein structures showed that MCPs of singelavirians and varidnavirians evolved from distinct cellular proteins, an SJR carbohydrate-binding protein and DJR glycoside hydrolase, respectively (Krupovic, Makarova, et al., 2022). Accordingly, kingdom Helvetiavirae has been reclassified as a separate realm.
Megataxonomy of viruses: realm Singelaviria. (A) Taxonomic structure, from realms down to families. Family Portogloboviridae is connected to the realm by a dashed line because it is currently not formally included in Singelaviria, but is likely to represent the ancestral state of the realm. (B) Structural models of the single jelly-roll fold major capsid proteins (MCP) and their oligomers. Some members of the realm, e.g., Sulfolobus polyhedral virus 1 (SPV1; Portogloboviridae), encode a single MCP VP4 (red) which forms homohexameric capsomers, whereas others, e.g., Haloarcula hispanica icosahedral virus 2 (HHIV-2; Sphaerolipoviridae), encode two paralogous MCPs, VP7 (red) and VP4 (blue), which form two types of heterohexameric capsomers, hexamers 1 and 2, respectively. The structures are denoted with the corresponding PDB accession numbers.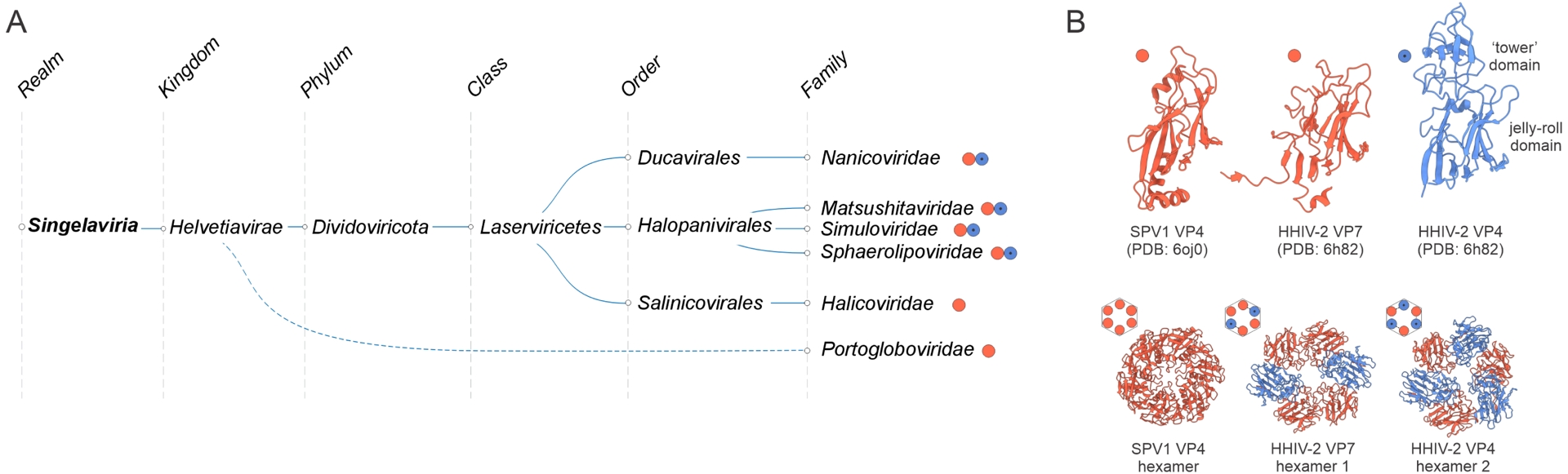
Further exploration of virus diversity led to the identification of relatively close singelavirian relatives that encode the FtsK-HerA genome packaging ATPase and a single SJR MCP (e.g., halicovirids), implying homohexameric capsomers (Zhou et al., 2025; Makarova et al., 2014). Finally, archaeal viruses of the Portogloboviridae family (Y. Liu, Ishino, et al., 2017; Prangishvili et al., 2021), currently not formally assigned to Singelaviria (Figure 5A), might even more closely resemble the ancestral state. Portoglobovirids lack the genome packaging ATPase and encode a single SJR MCP, which closely resembles MCPs of singelavirians and forms homohexamers (Figure 5B), as predicted for halicovirids (F. Wang, Y. Liu, et al., 2019).
4.5. Realm Duplodnaviria, the champions of the viral world
Realm Duplodnaviria includes the most abundant viruses on Earth, namely, the tailed dsDNA bacterial and archaeal viruses (class Caudoviricetes) (Dion et al., 2020; Cai et al., 2021; Adriaenssens et al., 2025), the recently discovered “mirusviruses” associated with unicellular eukaryotic hosts (putative new phylum “Mirusviricota”) (Gaia et al., 2023), and animal viruses of the order Herpesvirales (Dotto-Maurel et al., 2025) (Figure 6A). All these viruses share a distinct morphogenetic module that consists of four VHGs encoding a HK97-fold MCP, a genome packaging ATPase-nuclease (known as large terminase subunit) that is distinct from the functional counterpart in varidnavirians, a portal protein, and a distinct capsid maturation protease (Iranzo, Krupovic, et al., 2016) (Figure 6B). The four VHGs play key roles in capsid assembly, maturation and genome packaging (Figure 6C) and are highly conserved throughout Duplodnaviria. The assembly pathways following the genome packaging differ in a host-dependent manner. In prokaryote-infecting Caudoviricetes, the packaged capsid is combined with a preassembled tail (Dion et al., 2020; Baquero, Y. Liu, et al., 2020), whereas in eukaryote-infecting herpesvirals and mirusviruses, the capsid is enveloped with a lipid membrane (Crump, 2018; Chung et al., 2025) (Figure 6C).
Megataxonomy of viruses: realm Duplodnaviria. (A) Taxonomic structure: from realm down to orders. The taxonomic tree was retrieved from the ICTV website (Black et al., 2026) and modified to include the proposed phylum “Mirusviricota”. (B) Structural models of the hallmark proteins comprising the morphogenetic module conserved in duplodnavirians, exemplified by the major capsid protein (MCP), large subunit of the terminase (TerL), portal protein, and capsid maturation protease of bacteriophage HK97. (C) Virion morphogenesis in the three phyla of duplodnavirians. The assembly pathway was depicted using the modified virion diagrams obtained from ViralZone (De Castro et al., 2024).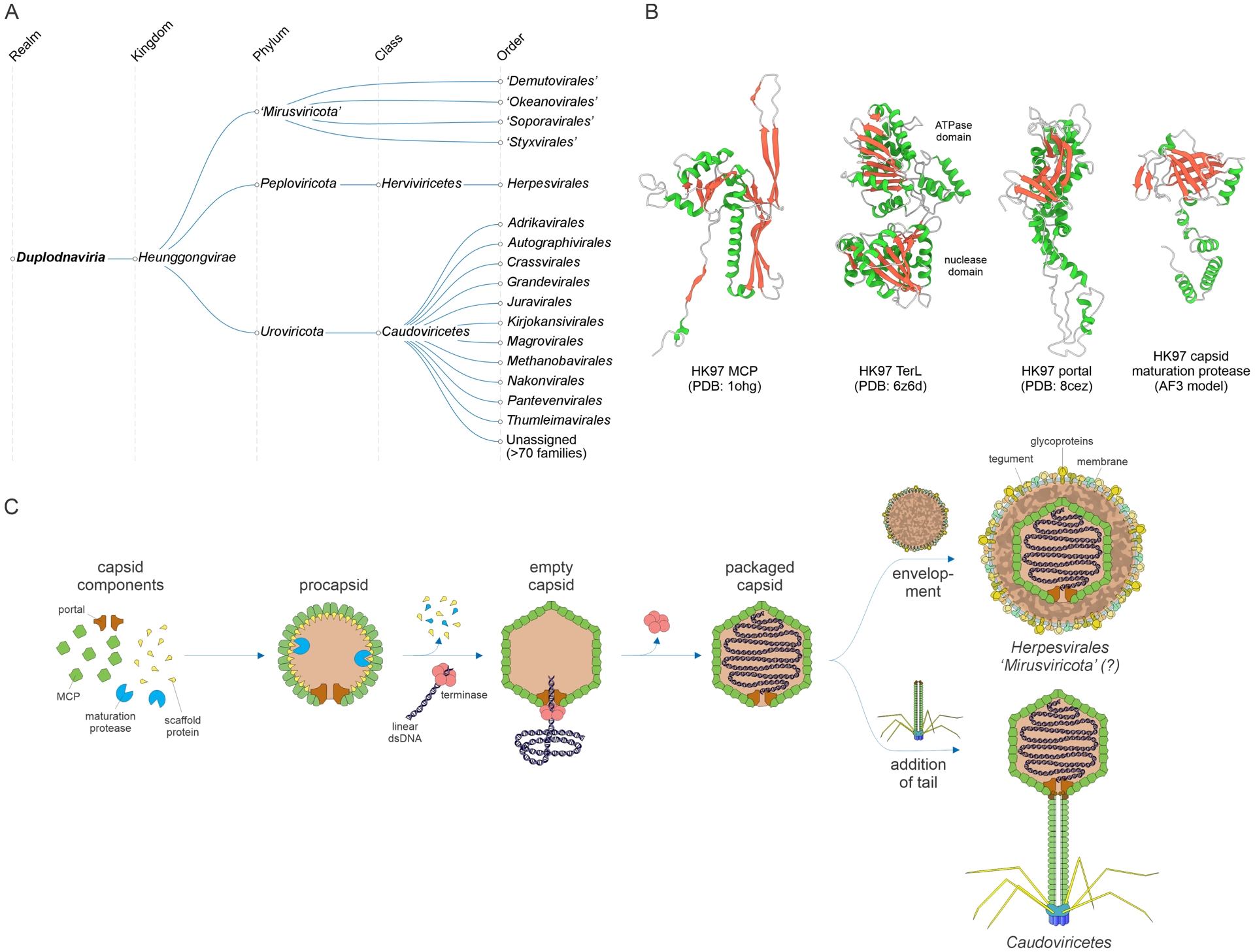
Realm Duplodnaviria has a simple taxonomic structure including a single kingdom and three phyla (Figure 6A), Uroviricota (caudoviricetes), Peploviricota (herpesvirals) and the yet to be formally recognized “Mirusviricota” (mirusviricots). Notwithstanding this uniformity at the top ranks of the taxonomy, the diversity of uroviricots is enormous, resulting in a constant flux of taxonomy reorganization (Adriaenssens et al., 2025; Y. Liu, Demina, et al., 2021; Turner et al., 2023).
The range of genome size and complexity of duplodnavirians nearly matches that of varidnavirians, spanning from about 10 kb to about 750 kb, all within Uroviricota (Luque et al., 2020; Al-Shayeb, Sachdeva, et al., 2020). Until the recent discovery of “Mirusviricota”, the evolutionary history of this realm appeared enigmatic, with a broad gulf separating tailed viruses of prokaryotes (bacteria and archaea) from herpesviruses that have been only identified in animals. However, “Mirusviricota”, a vast phylum of viruses apparently associated with a broad variety of unicellular eukaryotic hosts, fill this gap, in all likelihood, representing the original diversity of duplodnavirians in eukaryotes and the ancestors of herpesvirals (Gaia et al., 2023; Medvedeva, Guyet, et al., 2026).
As in the case of other virus groups discussed above, duplodnavirians display a remarkable diversity of genome replication strategies, especially among bacterial and archaeal caudoviricetes (ending “viricetes” refers to all members of a class (Koonin, Kuhn, et al., 2024)), which employ all conceivable DNA replication strategies and encode suites of replication factors ranging from single-protein initiators to complete replisomes (Weigel and H. Seitz, 2006; Krupovic and Bamford, 2010; Kazlauskas, Krupovic and Venclovas, 2016; Philosof et al., 2017). Notable variations also exist in the replication modules of eukaryotic duplodnavirians. For instance, in the order Herpesvirales, family B DNA polymerases of orthoherpesvirids are not orthologous to those of malacoherpesvirids and alloherpesvirids (Kazlauskas, Krupovic, Guglielmini, et al., 2020), whereas many mirusviricots lack DNA polymerases altogether (Medvedeva, Guyet, et al., 2026).
4.6. Small realms Adnaviria and Ribozyviria
Realm Adnaviria consists of archaeal viruses with rigid or flexible filamentous virions that can be either non-enveloped or contain an external lipid membrane (Krupovic, Kuhn, F. Wang, et al., 2021; Krupovic, F. Wang, et al., 2025) (Figure 7A). The remarkable feature of these viruses is that their linear dsDNA genome is stored in the virions in the A conformation (Baquero, Y. Liu, et al., 2020; DiMaio et al., 2015). Adnavirians lack conserved VHGs although members of some families do encode them, e.g., HUH endonuclease that is conserved in rudivirids (Oke et al., 2011). However, among themselves, adnavirians share several conserved genes, most notably, the MCP with a unique α-helical fold (Kasson et al., 2017; Y. Liu, Osinski, et al., 2018; F. Wang, Baquero, L. C. Beltran, et al., 2020; F. Wang, Baquero, Su, et al., 2020), first described in the rudivirid SIRV2 (DiMaio et al., 2015). Although some adnavirians encode a single MCP, many encode two paralogous MCPs; these bind dsDNA as homodimers and heterodimers, respectively (Figure 7B). Adnavirians were initially detected in hyperthermophilic archaea of the phylum Thermoproteota, but subsequent metagenomics surveys showed that these viruses are also associated with hosts from the other archaeal phyla (Laso-Perez et al., 2023; Tamarit et al., 2022).
Megataxonomy of viruses: three small realms. (A) Adnaviria, taxonomic structure from realm down to families. (B) Virion architecture and structural model of the homodimeric and heterodimeric major capsid proteins of adnavirians, Saccharolobus islandicus rod-shaped virus 2 (SIRV2) and Acidianus filamentous virus 1 (AFV1), respectively. The structures are denoted with the corresponding PDB accession numbers. (C) Ribozyviria, taxonomic structure from realm down to families. (D) Virion architecture and structural model of the nucleocapsid protein (Delta antigen) octamer of the prototype ribozyvirian, hepatitis delta virus. The structural model was generated using AlphaFold3 (AF3). S-DAg and L-DAg, small and large delta antigens. S, M, L: hepatitis B virus glycoproteins. (E) “Telodnaviria”, taxonomic structure from realm down to families. “Telodnaviria” has not yet been formally recognized by the ICTV. (F) Virion architecture and structural model of the major capsid protein dimer of a well-characterized telodnavirian, Autographa californica multiple nucleopolyhedrosis virus (AcMNPV). The PDB accession number is indicated. AmFV, Apis mellifera filamentous virus.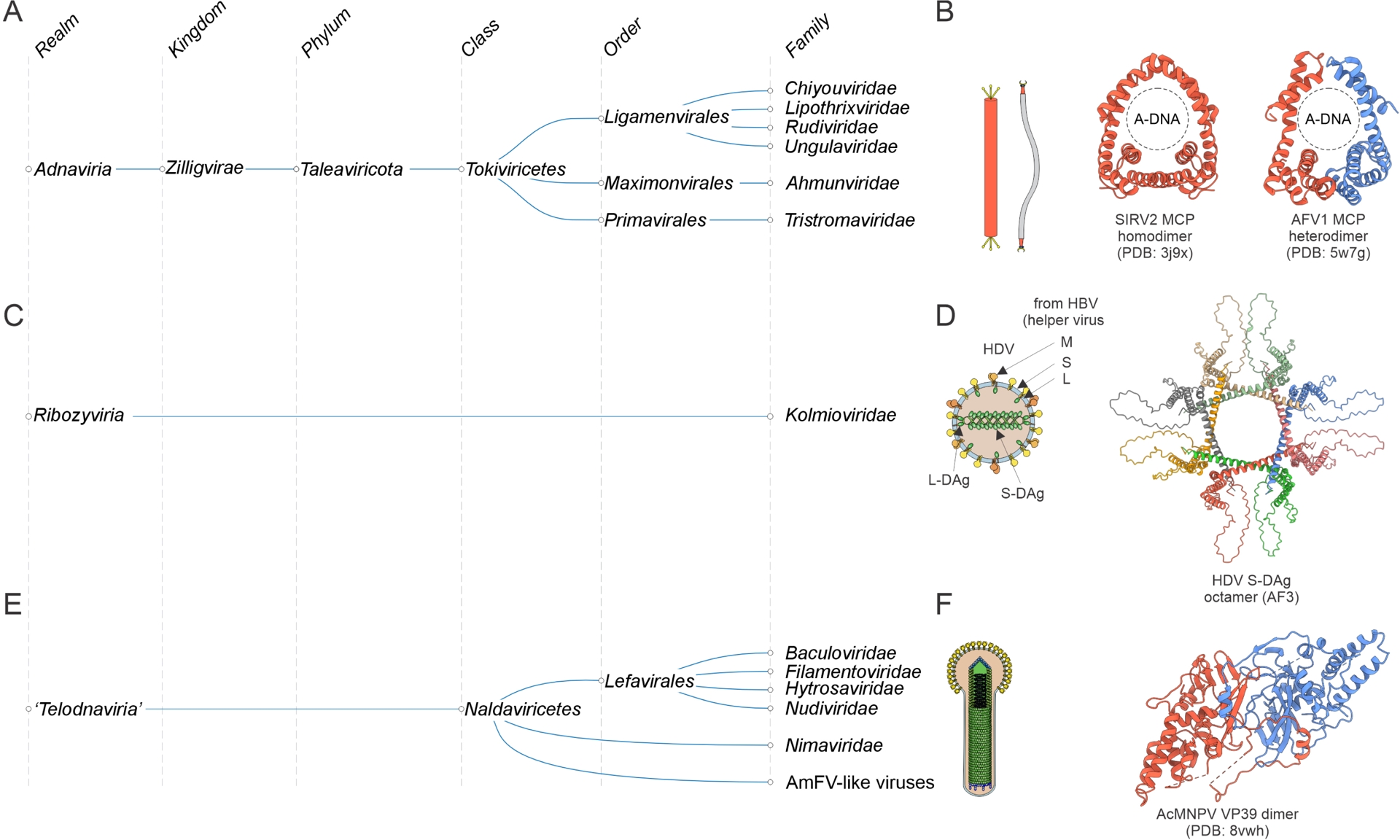
The small realm Ribozyviria (Hepojoki et al., 2021; Walker et al., 2022) currently includes a single family, Kolmioviridae (Kuhn, Babaian, et al., 2024), which encompasses human hepatitis D viruses 1–8 (genus Deltavirus) and their relatives discovered in other animals (Wille et al., 2018; Chang et al., 2019; Paraskevopoulou et al., 2020; Bergner et al., 2021; Perez-Vargas et al., 2021) (Figure 7C). Ribozyvirians are viroid-like covalently closed circular (ccc) RNA replicons encoding a nucleocapsid protein (delta antigen, DAg). Similar to viroids, these viruses hijack the cellular transcription machinery for their genome replication and depend on other viruses (bluberviral hepatitis B virus in the case of deltaviruses, arenavirids in case of daletviruses, unknown for most of the others) for the formation of infectious enveloped virions (Khalfi et al., 2023; Szirovicza et al., 2020). DAg adopts a unique α-helical fold and forms octamers (Figure 7D) (Cornillez-Ty and Lazinski, 2003). The viral pseudo-dsRNA is thought to wrap around the DAg octamers that mimic nucleosomes and facilitate the recruitment of the host RNA polymerase II and the associated chromatin remodeling complexes for viral RNA replication (Abeywickrama-Samarakoon et al., 2020; Yang et al., 2025). DAg also induces packaging of the kolmiovirid ribonucleoprotein complex into progeny virions. Recent evidence suggests that this complex is directly incorporated into the enveloped virions of their helper viruses (McKellar et al., 2026). No autonomous ribozyvirians have been described to date, suggesting that all members of this realm are satellites of other viruses. Metagenome mining led to the discovery of a substantial variety of ribozy-like cccRNAs encoding distant homologs of the deltavirus nucleocapsid protein and most likely replicating in unicellular eukaryotes (Edgar et al., 2022; B. D. Lee et al., 2023). Thus, although Ribozyviria will probably remain a small viral realm, its true diversity appears to be much greater than reflected in the current taxonomy.
4.7. Are there more viral realms to be established?
The 10 realms described above encompass ∼95% of the currently recognized virus families (Simmonds, Adriaenssens, Lefkowitz, et al., 2025). What about the remaining 5% that are currently not assigned to any realm? It is highly likely that some of the unassigned families will be unified into additional realms. For instance, viruses with large dsDNA genomes currently classified into the class Naldaviricetes (van Oers et al., 2023; Guinet et al., 2024) were suggested to represent a separate realm, “Telodnaviria” (Johnstone et al., 2024) (Figure 7E). Viruses of this group, of which the best-known ones are baculoviruses due to their wide use as biocontrol agents in agriculture and as tools in molecular biology (Pidre et al., 2022), infect diverse arthropods, including insects and crustaceans, and form complex helical virions. The baculovirus MCP has a unique fold, unrelated to those of viruses from the other realms (Johnstone et al., 2024; Benning et al., 2024) (Figure 7F). Furthermore, members of the Naldaviricetes share a core gene set, including genes involved in genome replication and transcription as well as per os infectivity factors (PIF) (van Oers et al., 2023), which are believed to form a distinct attachment and membrane fusion complex (Johnstone et al., 2024).
Another realm is likely to be created for archaeal viruses with dsDNA genomes and spindle-shaped or lemon-shaped virions. Spindle-shaped viruses are ubiquitous in archaea, associated with phylogenetically diverse hosts (Zhou et al., 2025; Krupovic, Quemin, et al., 2014). Due to their broad distribution in archaea, spindle-shaped viruses are thought to have been coevolving and diversifying with archaea ever since the emergence of the last archaeal common ancestor from the LUCA (Krupovic, Dolja, et al., 2020). Spindle-shaped viruses encode a distinct MCP which adopts a simple α-helical hairpin fold (F. Wang, Cvirkaite-Krupovic, et al., 2022; Z. Han et al., 2022). The highly hydrophobic MCP subunits polymerize into a helical assembly, which has a larger diameter in the central region where the genome is located (F. Wang, Cvirkaite-Krupovic, et al., 2022). Spindle-shaped viruses are highly diverse in terms of gene content and genome replication strategies. Some have linear genomes replicated by virus-encoded protein-primed family B DNA polymerase (Tamarit et al., 2022; Bath et al., 2006; Kim et al., 2019; Medvedeva, Sun, et al., 2022), whereas others have circular genomes and encode either HUH superfamily endonucleases (Zhou et al., 2025; Medvedeva, Sun, et al., 2022) or homologs of various host replisome components, such as RNA-primed family B DNA polymerase (Laso-Perez et al., 2023) or MCM helicase (Gorlas et al., 2012), or lack recognizable replication proteins. Due to their unique architecture and the fact that spindle-shaped viruses do not share conserved VHGs with other virus realms, they are likely to have an independent origin and thus qualify as a separate realm.
Several additional groups of archaeal viruses with enigmatic gene contents and unusual virion morphologies, such as bottle-shaped, droplet-shaped or globular viruses (Baquero, Y. Liu, et al., 2020), and bacterial viruses of the family Plasmaviridae (Krupovic and ICTV Report C, 2018), could represent additional realms. A variety of protein-coding cccRNAs have been discovered in metagenomes, and some of these could be independently originated viruses that would qualify as separate realms (B. D. Lee et al., 2023). However, the available information on the virion organization, diversity and evolution of all these (putative) viruses remains scarce and thus their higher-level taxonomic assignment appears premature.
Can we expect to discover viruses representing additional new realms? It is likely that all major, widespread virus realms have been already sampled, a monumental achievement from a century of culture-dependent and more recent, culture-independent virus discovery efforts. However, it is hard to put an upper bound on the number of independently evolving smaller virus groups restricted to particular hosts or environments. We predict that sampling of the less explored host groups, such as archaea or protists, as well as extreme environments, will continue to expose previously unseen viruses, some of which will represent new realms. Classical, culture-dependent virus isolation will play a key role in this endeavor, whereas metagenomics and metatranscriptomics will be instrumental both in the preceding discovery phase, to identify candidates for new realms, and in the subsequent stage, for determining the extent of diversity and distribution of these new virus groups.
5. The key trends and processes of virus evolution
5.1. Pronounced modularity of viruses
Phylogenomic analysis of the rapidly growing collection of viral genomes allows us to infer some general trends and patterns of virus evolution (Koonin, Dolja and Krupovic, 2022). Any viral genome, with the exception of some satellite viruses and some derived viruses that have lost structural proteins, combines at least two functional modules, those encoding proteins involved in virion morphogenesis and those involved in genome replication and expression. Each of these modules can be represented by only a single gene (or even a ribozyme as in ribozyvirians) or can be highly complex, including numerous genes. For instance, many viruses of the realm Floreoviria, with small ssDNA genomes, e.g., circovirids, encode one capsid protein and one replication-associated protein (Rep) (Varsani et al., 2024), whereas large dsDNA viruses of both Varidnaviria and Duplodnaviria often encode a (nearly) complete DNA replisome (Kazlauskas, Krupovic and Venclovas, 2016), a variety of proteins involved in transcription and translation (Grimm et al., 2019; Hillen et al., 2019; R. A. L. Rodrigues, da Silva, et al., 2020), and scores of structural proteins and virion assembly factors (Shao et al., 2022).
Nearly two decades ago, Dennis Bamford proposed the concept of “viral self”, that is, an evolutionary stable core of vertically inherited genes that define viral lineages (Krupovic and Bamford, 2008; Bamford et al., 2003). In the context of the assemblage of viruses now known as Varidnaviria, the self was defined as the DJR-MCP and the packaging ATPase. The concept has stood the test of time: today, we can indeed trace vertical evolution of viral realms through the phylogenies of VHGs that comprise the viral self. Notably, however, the types of VHGs comprising the self differ across the realms. In viruses of the realms Riboviria, with their small genomes, the self consists of the VHGs involved in replication, namely, RdRP and RT. By contrast, in the realms of viruses with DNA genomes, it is the morphogenesis module that qualifies as the self. There is a simple functional and evolutionary logic to this distinction. Viruses with RNA genomes cannot fully rely on the host replication machinery because RNA replication is not a normal cellular process. Therefore, these viruses must employ dedicated replication enzymes that cannot be replaced by cellular counterparts and thus are consistently inherited throughout the evolution of the viral realm (as always in biology, there are exceptions: ribozyvirians and possibly other viroid-like viruses do not encode any replication proteins, instead hijacking the host transcription apparatus; these unusual viruses, however, encompass ribozymes required for replication). Conversely, RNA viruses often exchange and occasionally reinvent genes for structural proteins (e.g., capsid protein of togavirids or nucleocapsid protein of ortervirals (Koonin, Dolja and Krupovic, 2022)) so that these genes do not qualify as the viral self. In principle, the same logic could be applied to ssDNA viruses because cellular DNA replication mechanisms do not produce ssDNA intermediates and thus the virus must provide its own solution. However, unlike in the case of RNA and RT viruses, there are multiple unrelated enzymes that can yield ssDNA intermediates during replication, including several families of HUH endonucleases, Rep_trans endonucleases, protein-primed family B DNA polymerases and a few others, and all of these are interchangeably used by ssDNA viruses from different realms (Figure 3B), as discussed above. Furthermore, in viruses of the realm Pleomoviria, there is certain flexibility with regard to which replicative intermediate, ssDNA or dsDNA, is packaged into the virion (Roine et al., 2010), which licenses exploration of a broader range of replicative strategies. Of the four realms formerly included in the Monodnaviria, only in Floreoviria, the replication module comprises the self and is used for tracing the evolutionary relationships between the member viruses although some of these viruses (e.g., annelovirids and bidnavirids) lack the signature two-domain HUH endonuclease (Kazlauskas, Varsani, et al., 2019; Krupovic, Varsani, et al., 2020). By contrast, viruses with dsDNA genomes can use the cellular replication machinery, and accordingly, these viruses widely differ in their repertoires of replication genes, from retaining the entire replication machinery to shedding it all. Furthermore, replication genes of these viruses, for example, DNA polymerases, are occasionally exchanged with cellular or non-orthologous or even non-homologous viral counterparts (Yutin, Tolstoy, et al., 2024). Therefore, the self of the respective realms can be only identified with genes encoding structural and morphogenetic proteins. A transitional state between viruses that require a specialized replication machinery and those that can exploit the host replication apparatus is represented by polyomavirids and papillomavirids. After switching from ssDNA to dsDNA genomes, these viruses lost the ancestral capacity to replicate via RCR due to mutational inactivation of the HUH endonuclease and switched to the theta replication mechanism typical of host DNA (Figure 3B), while retaining the inactivated endonuclease as a DNA-binding domain involved in the replisome function (Enemark et al., 2000; D. Li et al., 2003; Bochkareva et al., 2006).
5.2. Complexification and functional convergence: major trends in viral evolution
Reconstruction of virus genome evolution by mapping gene repertoires on phylogenetic trees of VHGs revealed the predominant trend of complexification by gene accretion although reductive evolution also appears to have occurred in some lineages (Koonin and Yutin, 2019; Wolf, Kazlauskas, et al., 2018; Koonin and Yutin, 2018). Remarkably, this evolutionary trend is typical of both viruses with small genomes, such as ribovirians, and those with the largest known genomes, such as members of phylum Nucleocytoviricota in realm Varidnaviria, as well as caudoviricetes. Many virus lineages, however, appear to maintain relative stasis with respect to genome size and complexity across long evolutionary spans, exchanging and replacing many genes while maintaining nearly constant genome size. This evolutionary regime is clearly observed, for example, in phylum Preplasmiviricota within Varidnaviria, and could be linked to the physical constraints, namely, internal volume and packing capacity of the virions.
The great majority of the genes acquired by viruses come from the hosts, although notable cases of viral gene duplication followed by diversification are known as well (Senkevich et al., 2021), whereas some viral genes might have evolved de novo from non-coding sequences (Legendre, Fabre, et al., 2018; Legendre, Alempic, et al., 2019). The genes acquired by viruses encode four broad functional categories of proteins: (i) components of replication and expression systems enabling the relative autonomy of viruses from the respective cellular processes, (ii) structural proteins that initially become minor components of the virions but eventually can replace hallmark MCPs, for example, in pandoraviruses (Krupovic, Yutin and Koonin, 2016), (iii) metabolic enzymes that supplement and/or modulate the respective metabolic pathways of the host cells, (iv) proteins involved in various types of virus–host interactions, in particular, inhibitors of host immune systems. Because the benefits conferred by these functions are common to many if not all viruses, there is extensive convergence among the suites of genes captured by viruses of distant lineages, including different realms, especially in the first of the above functional categories. A notable case in point is the independent acquisition of helicases of three different superfamilies by orthornaviraens of different phyla (Wolf, Kazlauskas, et al., 2018). Apparently, helicases enable replication of (relatively) large RNA genomes, as indicated by the fact that viruses with positive-sense RNA genomes larger than 6 kb encode a helicase of one or another superfamily (Gorbalenya and Koonin, 1989), and thus open up the path of evolution toward genome complexification (again, there is a notable exception, a flavi-like ribovirus with a nearly 12 kb genome lacking any helicase (Dahan et al., 2022)). A further increase of the RNA genome size, beyond the ∼25 kb threshold, apparently required the acquisition of an enzymatic complex involved in RNA proofreading, which occurred in coronavirids and other nidovirals, the giants of the RNA domain of the virosphere (Neuman et al., 2025; Zhong et al., 2025; Gorbalenya, Enjuanes, et al., 2006). Similarly, many lineages of orthornaviraens independently acquired proteases of different families involved in virus polyprotein processing, yielding mature viral proteins which is one of the main strategies for expression in viruses of eukaryotes where initiation of translation typically occurs at the 5′-end of an mRNA (Koonin, Dolja and Krupovic, 2022; Koonin and Dolja, 1993).
A case that has been widely discussed in scientific and even popular literature, giving rise to provocative ideas, is the presence of genes encoding multiple components of the translation system, both proteins and tRNAs, in some viruses of phylum Nucleocytoviricota, in particular, mimivirids (R. A. L. Rodrigues, da Silva, et al., 2020). One of the mimivirids, tupanvirus, encodes the entire suite of translation system components, except for those of the ribosome (Abrahao et al., 2018; R. A. L. Rodrigues, T. S. Arantes, et al., 2019). Given that translation is often considered a quintessential cellular functionality of which viruses are incapable, these findings triggered ideas on the evolution of the “giant” viruses from cells, possibly, even from a hypothetical fourth domain of life (Claverie, 2006; Raoult, Audic, et al., 2004; Colson, Gimenez, et al., 2011; Colson, de Lamballerie, et al., 2012; Colson, Levasseur, et al., 2018; Nasir et al., 2012; Nasir et al., 2017) (see also discussion below). Detailed phylogenetic analyses, however, unequivocally indicate that translation system components have been independently acquired by nucleocytoviricetes from eukaryotic hosts at different stages of evolution (Moreira and Brochier-Armanet, 2008; Williams et al., 2011; Yutin, Wolf, et al., 2014; Moreira and Lopez-Garcia, 2015). Indeed, some of these genes encoding orthologous proteins with the same function apparently have been captured independently by distantly related viruses (Yutin, Wolf, et al., 2014). Scattered genes for translation system components are present also in some bacterial and archaeal caudoviricetes, including multiple tRNAs (up to 62), tRNA synthetases and even ribosomal proteins, clearly, convergent acquisitions (Y. Liu, Demina, et al., 2021; Al-Shayeb, Sachdeva, et al., 2020; Sencilo et al., 2013; Mizuno et al., 2019; L. X. Chen et al., 2022). The roles of virus-encoded translation system components in virus replication are not well understood, but it appears likely that viruses reprogram the host translation system to different degrees depending on the virus–host combinations, including the localization of the translation of viral mRNAs to a distinct compartment within the infected cell (R. Zhang et al., 2026). In addition, the role of certain viral tRNAs in countering diverse tRNA-targeting defense systems has also been demonstrated (van den Berg and Brouns, 2025).
In the second functional category, metabolic enzymes acquired by viruses, a notable case of convergence is the independent capture by viruses with large dsDNA genomes within both Varidnaviria and Duplodnaviria of enzymes and entire metabolic pathways for the biosynthesis of nucleotides, for example, thymidine kinase, thymidylate synthases of two unrelated families, and ribonucleotide reductases (Koonin, Dolja and Krupovic, 2022). Other metabolic pathways are limited to specific viral lineages, for example, the glycosphingolipid biosynthesis pathway in Coccolithoviridae, a family within Nucleocytoviricota (Vardi et al., 2009; Nissimov et al., 2019).
Which genes are retained by viruses upon accidental acquisition, presumably, depends on the host biology and response to virus infection, particularly in the case of metabolic enzymes. In some well-studied cases, the selection factors driving the fixation of the acquired genes in viruses are clear as, for example, for phages infecting cyanobacteria (Puxty et al., 2015). Many of these cyanophages encode the entire photosystem I or II, and some even both photosystems (Sharon et al., 2009; Fridman et al., 2017). Upon infection, the viral photosystems supplement the host ones, augmenting their activity so that the host cells remain energy-rich and conducive to massive replication of the cyanophages. In most cases, however, the connection with the host biology remains elusive, remaining to be elucidated through direct experiments with the respective virus–host systems, which are typically technically challenging if feasible at all.
The viral genes directly involved in virus–host interactions are even more specifically linked to the host biology. A striking example is the movement proteins (MP) encoded by a broad variety of plant viruses including diverse orthornaviraens, pararnaviraens and cressdnaviricots (realm Floreoviria), and enabling intercellular movement of viruses in plants through the plasmodesmata (Mushegian and Elena, 2015). The MP originally evolved via a duplication of the SJR-MCP in an orthornaviraen, and then, spread extremely widely among plant viruses via HGT (Butkovic, Dolja, et al., 2023). Analogously, in animals, one of the principal mechanisms of virus entry into the host cells is membrane fusion, and accordingly, fusion proteins have spread across a broad variety of animal RNA and DNA viruses (Kielian and Rey, 2006; Guardado-Calvo and Rey, 2021).
The great majority (most likely, all) viruses with large genomes, that is, those of realms Varidnaviria, Duplodnaviria, and Adnaviria, and even some viruses with small genomes in Riboviria and some groups of ssDNA viruses encode proteins and often entire systems involved in the inhibition of host immunity (Crespo-Bellido and Duffy, 2023; S. Zhang et al., 2025). In large viruses, these genes can comprise a major fraction of the gene repertoire, and this part of the genome is highly variable, rarely being conserved beyond a viral family. The counter-defense genes can be conceptually divided into two large classes: (i) repurposed components of immune systems that function as dominant-negative inhibitors according to the “guns for hire principle” (Koonin, Makarova, Wolf and Krupovic, 2020), that is, the use of homologous components for both defense and counter-defense, and (ii) dedicated inhibitors, often without detectable homologs, some of which also employ the dominant-negative mechanism. The typical case in the first category are poxviruses, large animal viruses within Nucleocytoviricota which encode a broad variety of homologs of host proteins involved in immunity and related signal transduction pathways. Examples include homologs of tumor necrosis factor, interleukin 18, MHC Class I and apoptotic factor Bcl2, all of these represented by families of paralogs in poxvirus genomes (Senkevich et al., 2021). Although not all the mechanisms have been studied in detail, these proteins appear to act as dominant negative inhibitors of the cognate host immune pathways. In addition, poxviruses encode a family of proteins of the second class of counter-defense factors containing a chemokine-binding domain that appears to have no cellular counterpart and thus seems to have evolved within poxviruses themselves, possibly, via a radical rearrangement of a pre-existing ancestral domain (C. A. Nelson et al., 2015). In other viruses of eukaryotes, both nucleocytoviricots, and mirusviricots and herpesvirals, the counter-defense genes are not well characterized, in particular, because few if any homologs of host immune proteins that could act as decoys have been identified. However, numerous genes of these viruses (with the partial exception of othoherpesvirids) remain orphans without detectable homologs, and many of these are likely to be involved in counter-defense. The difficulty with the identification of those is, in large part, that the hosts of many of these viruses remain unknown, and even for known unicellular eukaryotic hosts, the immune systems remain largely uncharacterized.
The counter-defense landscape of caudoviricetes as well as adnavirians is better known. It appears that these viruses employ primarily distinct counter-defense proteins, most without detectable homologs outside narrow groups of viruses, rather than mimics of host immune proteins. Most of these counter-defense proteins, in particular, anti-CRISPR proteins that have been investigated in detail in the last few years as part of the CRISPR boom (Pawluk et al., 2018; Bondy-Denomy, Maxwell, et al., 2023) are quite small (about or less than 100 amino acids) and often are not predicted to adopt a globular conformation or predicted to adopt a unique fold (Sahakyan et al., 2023), suggesting the possibility of their emergence de novo from non-coding sequences. Most of these small counter-defense proteins are highly specific towards particular bacterial or archaeal defense systems, binding to unique sites of immune proteins, such as, for example, CRISPR effector nucleases, including Cas9 and others. A notable exception to this high specificity are DNA-mimicking counter-defense proteins, small, negatively charged proteins that can inhibit host immune systems promiscuously by blocking their DNA-binding domains, that is, employing a distinct variant of the decoy strategy (H. C. Wang et al., 2019; S. Li et al., 2024). Although unique counter-defense proteins prevail in large viruses of prokaryotes, the guns for hire principle is employed as well. Thus, some phages, in particular jumbo phages with their large genomes, have coopted complete, functional CRISPR systems targeting host defense systems or other MGE (Seed et al., 2013; Al-Shayeb, Skopintsev, et al., 2022). Even more common in the genomes of phages and archaeal viruses are CRISPR microarrays exploiting the host Cas proteins primarily to target other viruses (Faure et al., 2019; Medvedeva, Y. Liu, et al., 2019) and small anti-CRISPR RNAs that are copies of single CRISPR repeat and form non-productive complexes with Cas proteins, acting therefore as CRISPR-RNA decoys (Faure et al., 2019; Camara-Wilpert et al., 2023; Hayes et al., 2025).
Among viruses with small genomes, only some, apparently, a minority encode dedicated counter-defense proteins, again in connection with the host biology. Thus, diverse plant viruses as well as certain animal viruses, some with very small genomes, encode silencing suppressors, small proteins, possibly, emerging de novo, that inhibit the powerful RNA interference systems of the hosts (Baulcombe, 2022; W. X. Li and Ding, 2022).
To summarize this discussion of the general trends in the evolution of the virosphere, all viruses have a small core (self) that consists of a few, and in many cases, only one VHG(s) essential for viral replication and/or morphogenesis. These viral selves evolve vertically through eons, for millions or even billions of years, providing for the identification of viral realms, kingdoms and phyla. The rest of the viral genomes is highly dynamic and malleable, evolving through numerous gene gains, losses and exchanges, the exact history of which is often hard to reconstruct. There is, however, a lot of convergence and parallelism in the evolution of genes contributing to viral genome replication and expression due to shared requirements of diverse viruses. There is also some but lesser convergence among viral metabolic genes, whereas genes encoding virion components as well as those involved in counter-defense are an epitome of diversity and dynamism.
A consequence of the convergence in the acquisition of functionally analogous and often homologous host genes by diverse viruses is that, although viral realms, by definition, represent assemblages of viruses of independent origins, different realms are loosely connected by patchy sharing of homologous proteins. Prominent examples include the aforementioned RNA and DNA helicases (in particular, superfamily 3 helicases), DNA polymerases and DNA-dependent RNA polymerases, chymotrypsin-like and papain-like proteases, movement proteins of plant viruses, and more (Koonin, Dolja, Krupovic, Varsani, et al., 2020).
6. Viruses as symbionts of cellular life forms
The pre-eminent immunologist Sir Peter Medawar famously said in his 1960 Nobel lecture that “No virus is known to do good. It has been well said that a virus is a piece of bad news wrapped up in protein” (Medawar, 1960). However, the more deeply is the virosphere explored, the clearer it becomes that the news conveyed by viral genomes is not necessarily bad, but perhaps, more often, is neutral or even good. Put another way, viruses are ubiquitous symbionts of cellular life forms, and their relationships with the hosts span the entire symbiotic spectrum, from parasitism to commensalism to mutualism. For obvious reasons, parasitic viruses that lyse host cells (thus, by definition, killing a unicellular host, and often, a multicellular one as well) have been discovered and studied first. However, already the early discovery of temperate (lysogenic) phages that can be vertically transmitted across many generations of bacteria as prophages showed that viruses are not invariably deleterious to the host (Lwoff, 1953). Indeed, prophages often encode defense systems and can have beneficial effects on the host fitness by protecting bacteria from infection by other, virulent phages (superinfection exclusion) (Bondy-Denomy, Qian, et al., 2016; Kirchberger, Martinez, Luker, et al., 2021; Brenes and Laub, 2025). There can be other benefits as well, notably transduction, that is, HGT for which phages serve as vehicles (E. Harrison and Brockhurst, 2017). The genes transferred by phages include defense systems, as per the “guns for hire” concept, antibiotic resistance genes as well as genes encoding enzymes of various metabolic pathways (Chevallereau and Westra, 2022; Y. Zhang et al., 2022; Fillol-Salom et al., 2022; Y. Zhao et al., 2025). Moreover, some bacteria and archaea have domesticated phages, turning them into Gene Transfer Agents (GTA), which are defective phages that incorporate into virions random host genes rather than the phage genome and function as dedicated, stress-induced HGT vehicles (Lang et al., 2012; Kogay and Zhaxybayeva, 2024). Notably, GTAs can rescue recipient cells from DNA damage by providing templates for homologous recombination (Gozzi et al., 2022). Conceptually similar exaptation of large DNA viruses for the transfer of immunosupression genes took place in parasitoid wasps, apparently on several independent occasions, leading to the emergence of viriforms, virus derivatives carrying host DNA (Drezen et al., 2022; Petersen et al., 2022; Kuhn and Koonin, 2023). Recently, it has been shown that a variety of phages considered virulent can accumulate in bacterial cells without either lysing them or integrating (Perfilyev et al., 2026; Dougherty et al., 2026), suggesting that viral commensalism is far more widespread than previously suspected.
A notable case of different types of symbiosis is presented by virophages, the viral symbionts of nucleocytoviricots (Koonin, Fischer, et al., 2024). Some of the virophages efficiently inhibit the propagation of the cognate large viruses, in effect functioning as adaptive immunity systems—and hence mutualists—for the unicellular eukaryotic host (Fischer and Hackl, 2016; Koslova et al., 2024). Other virophages have little if any effect on the supporting large virus and appear to be commensals. More generally, intervirus symbiosis and coevolution in complex virus–host systems are an important, currently poorly understood aspect of the evolution of the virosphere (Del Arco et al., 2024).
Every organism apparently supports a “healthy” virome, that is, commensal and mutualist viruses (Koonin, Dolja and Krupovic, 2021). For example, exploration of plant and fungal transcriptomes reveals an increasing number of cryptic viruses that cause no symptoms and have been largely missed by traditional virus identification approaches (Roossinck and Bazan, 2017; K. B. Gilbert et al., 2019). Similarly, animals, including humans, harbor a variety of viral commensals, for example, anellovirids with tiny ssDNA genomes that are ubiquitous and abundant in humans but have not been associated with any pathology (Butkovic, Kraberger, et al., 2023; Kaczorowska and van der Hoek, 2020; Kraberger et al., 2026). Both mammals and insects also host true viral mutualists, such as endogenous retroviruses (often denoted LTR retrotransposons) integrated in the genome, some of which form particles transferring RNA between host cells and apparently contributing to human physiology, in particular, that of the nervous system (Ashley et al., 2018; Pastuzyn et al., 2018; Shepherd, 2018; Segel et al., 2021).
In general, the scale of viral commensalism and mutualism in the biosphere remains to be elucidated. Infected cell lysis and host killing are not per se targets of selection in the evolution of viruses—rather, the maximum level of virus propagation is. Some viruses achieve this by killing the host, but for many, perhaps the majority of viruses, this is not the case.
7. The origins of viruses: genetic parasites as an intrinsic feature of life, the primordial replicator pool and exaptation of cellular genes
As emphasized above, viruses are literally ubiquitous in the biosphere. Apparently, all organisms, with the possible exception of some intracellular symbiotic bacteria with highly reduced genomes, are infected by multiple viruses, often highly diverse ones, representing different realms. Humans, for example, are hosts to numerous viruses of both kingdoms of Riboviria, Floreoviria (parvovirids, anellovirids, papillomavirids and others), Varidnaviria (poxvirids) and Duplodnaviria (orthoherpesvirids). The empirical data on the ubiquity of viruses are complemented by a theoretical argument on the inevitability of the emergence of genetic parasites in any replicator system (Koonin, Wolf, et al., 2017). This argument has been developed in thermodynamic terms, based on the entropy increase associated with the emergence of genetic parasites. In qualitative terms coming from game theory, the inevitability of the emergence of genetic parasites can be explained quite simply: as soon as, in a replicator system, there is an alienable resource that can be appropriated, such as a replicase, cheaters will emerge that will use that resource without making it. These theoretical considerations are, in turn, compatible with the reconstruction of the virome of the LUCA that is estimated to have existed about 4 billion years ago (Moody et al., 2024) and is the earliest point in the history of life for which such a reconstruction is attainable. This reconstruction indicates that the virome of the LUCA was already highly complex, comparable with the extant bacterial viromes, that is, probably included representatives of all the major viral realms (Krupovic, Dolja, et al., 2020).
The LUCA was certainly not the first life form to appear on earth but rather a product of extensive evolution from primordial protocells that likely existed within the framework of the RNA world, where RNA molecules performed both template and catalytic functions (W. Gilbert, 1986; Joyce, 2002). The inevitability of genetic parasites implies that they emerged concomitantly with the very first replicators. Furthermore, whereas the primordial replicator pool is thought to initially have consisted of RNA molecules only, small and then larger DNA replicators must have evolved at subsequent stages of evolution, long before the time of the LUCA. Thus, replicators corresponding to all Baltimore classes of extant viruses were most likely already parts of the primordial pool (Koonin, Senkevich, et al., 2006). Comparative analysis of the essential proteins involved in viral genome replication is compatible with the very early origin of the viral replication machineries (Krupovic, Dolja, et al., 2019). Indeed, behind the diversity of viral replication enzymes, there is remarkable, deep unity. The replication enzymes of most viruses from different realms, including RdRP, RT, HUH endonuclease, archaeo-eukaryotic primases and family B and A DNAPs, all contain homologous core RNA Recognition Motif (RRM) fold domains. The RRM is one of the most common nucleic acid binding domains in nature, found in an enormous variety of proteins, most of them devoid of enzymatic activity (Clery et al., 2008). The parsimonious explanation of the presence of catalytically active RRMs in viral replication enzymes is that they all radiated from a non-enzymatic ancestral RRM that was likely one of the earliest protein domains to evolve and could have served as a cofactor to ribozyme polymerases at the exit from the RNA world stage (Krupovic, Dolja, et al., 2019) (Figure 8). The second widespread structural scaffold found in diverse RNA and DNA polymerases is the double psi-beta barrel domain that has been suggested to represent the ancestral replicase of cellular organisms (Sauguet, 2019; Koonin, Krupovic, Ishino et al., 2020).
Origin of viruses. The depicted scenario combines origin of viral genomes and replication machinery from the primordial (pre-cellular) pool of replicators with subsequent exaptation of cellular proteins, such as carbohydrate-binding jelly-roll proteins, as capsid proteins, resulting in the emergence of bona fide viruses. Replicative genes are rendered in green, and structural genes, in red and blue. The two major types of primordial replicases based on the RNA recognition motif (RRM) and double psi-beta barrel (DPBB) domain are hypothesized to have evolved in pre-viral and pre-cellular replicators, respectively.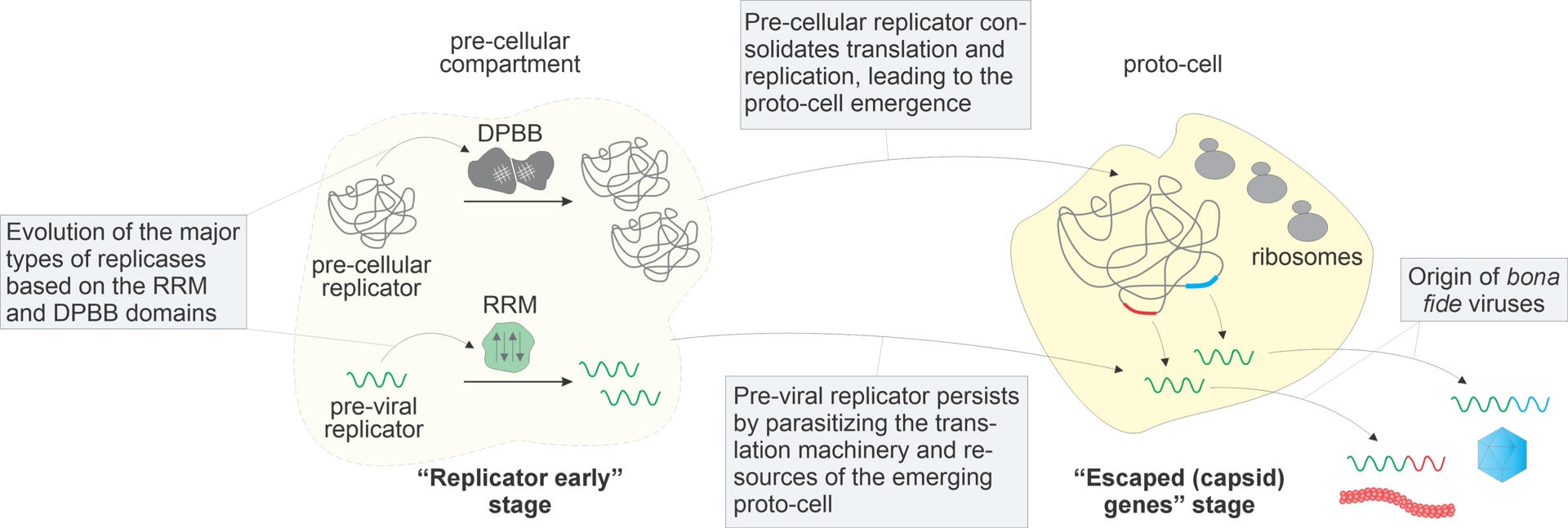
Thus, all types of replicators, including parasitic ones, in all likelihood, evolved very early in the evolution of life, prior to the advent of modern-type cells (Figure 8). However, parasitic replicators are not (yet) viruses, and bona fide viruses hardly could have existed before full-fledged cells emerged. Indeed, for major virion proteins, cellular ancestors have been identified, the most prominent cases in point being the origin of SJR-MCP and DJR-MCP from distinct families of sugar-binding proteins and glycoside hydrolases, respectively (Krupovic and Koonin, 2017b; Krupovic, Makarova, et al., 2022), suggesting viruses appeared on the scene at a relatively advanced stage of evolution, after multiple protein families have already diversified, even if long before the LUCA (Figure 8).
For decades, the origin of viruses has been discussed in terms of three distinct scenarios: (i) viruses early, preceding the origin of cells, (ii) viruses late, via reductive evolution of cellular parasites, (iii) escaped genes, that is, another version of viruses late, deriving viruses from “regular” cellular genes attaining replicative autonomy (Koonin, Senkevich, et al., 2006; Forterre and Prangishvili, 2009; Luria and Darnell, 1967; Forterre, 2006). The evolutionary scenario outlined above is a hybrid of “viruses early” and “escaped genes”: here, the viral replication machinery derives from the primordial pool of replicators (viruses early) whereas the virions evolve later from cellular proteins (escaped genes) (Figure 8). Importantly, the origin of viruses from non-viral symbiotic replicators, in particular, plasmids, is not limited to the hypothetical scenario of viral origin. Rather, this path of evolution was recapitulated on multiple occasions, in particular, at the origins of several groups of viruses with ssDNA genomes (Kazlauskas, Varsani, et al., 2019) as well as the origin of the expansive orthornaviraen family Botourmiaviridae from capsid-less narnavirids (Wolf, Kazlauskas, et al., 2018).
8. Origins of the eukaryotic virome
The origin of eukaryotes (eukaryogenesis) is the second pivotal major transition in the evolution of life, after the origin of cells (Maynard Smith and Szathmary, 1995; Szathmary, 2015). According to the latest, best supported scenario, the eukaryotic cell evolved as a result of engulfment by an Asgard archaeon related to the order Hodarchaeales of an alphaproteobacterium that became the protomitochondrial endosymbiont (Eme et al., 2023; Tobiasson et al., 2026) (Figure 9A). The reconstruction of the virome of the Last Eukaryotic Common Ancestor (LECA) showed, not unexpectedly, that this ancestral virome was highly complex, including representatives of all the larger realms, and likely, most of the phyla of the known viruses of eukaryotes (Krupovic, Dolja, et al., 2023). The unexpected outcome of this reconstruction, however, was the apparent origin of all major groups of viruses of eukaryotes from bacterial rather than archaeal ancestors (Figure 9A,B). In particular, all viruses of Asgard archaea identified to date are typical archaeal viruses without traceable links to viruses of eukaryotes (Tamarit et al., 2022; Medvedeva, Sun, et al., 2022; Rambo et al., 2022). Given that viruses are genetic parasites and the eukaryotic systems for information storage and transmission are of Asgard archaeal descent almost in their entirety (Tobiasson et al., 2026), this finding appeared puzzling and even paradoxical. The best possible explanation seems to be the major difference between the structures of archaeal and bacterial membranes (Lombard et al., 2012), and likely membrane-embedded viral receptors that could have led to the exclusion of archaeal viruses from the evolving eukaryotic cells upon the replacement of the archaeal membranes (and cell walls) with bacterial ones (Krupovic, Dolja, et al., 2023). Under more complex scenarios of eukaryogenesis—such as the syntrophy model, which postulates the initial engulfment of an archaeon by a bacterium, preserving the continuity of bacterial membranes throughout eukaryogenesis (Moreira and Lopez-Garcia, 1998; Lopez-Garcia and Moreira, 2020)—exclusion of archaeal viruses could have occurred at the initial symbiosis stage (Figure 9B).
Eukaryogenesis and the origin of the eukaryotic virome. Two alternative scenarios of eukaryogenesis are shown. (A) Symbiogenetic scenario whereby an Asgard archaeon with a complex cellular organization engulfed an alphaproteobacterium, the proto-mitochondrial endosymbiont, with subsequent replacement of the archaeal membrane (red outline) by the bacterial one (black outline). (B) Syntrophy scenario including two endosymbioses whereby a bacterium (possibly, a deltaproteobacterium) engulfed an Asgard archaeon, resulting in the dissolution of the archaeal membrane and followed by a secondary engulfment of an alphaproteobacterium. In this scenario, the continuity of the bacterial membrane is preserved throughout. Under both scenarios, archaeal viruses are excluded from the emerging eukaryote, and as a result, the eukaryotic virome is seeded by bacterial viruses. FECA, SECA and LECA: first, second and last eukaryotic common ancestor, respectively.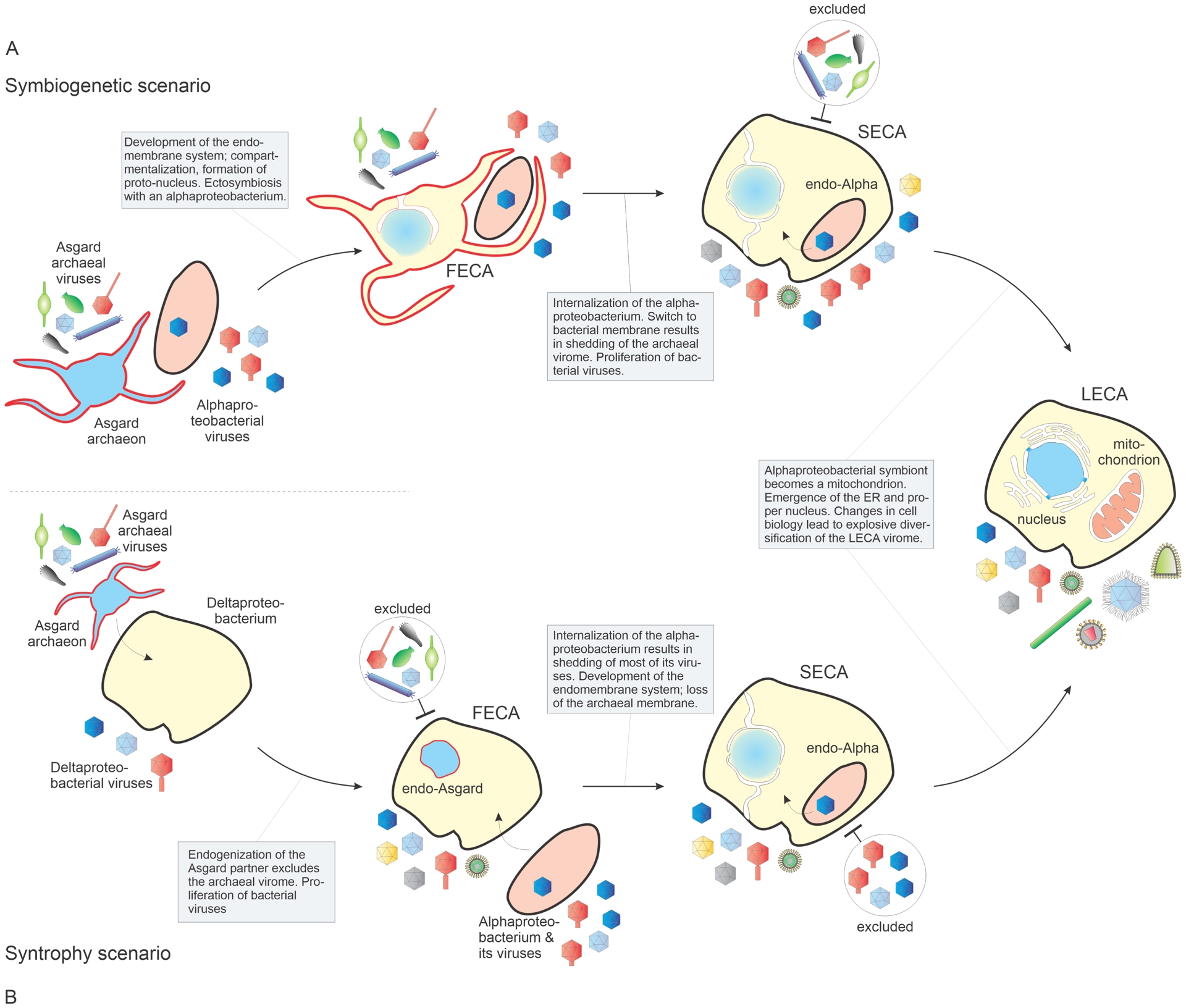
9. Reproducers and replicators, fundamental virus-cell divide and the place of the virosphere within the replicator space
Evolving biological entities belong to two fundamentally distinct types: reproducers and replicators (Szathmary and Maynard Smith, 1997). Reproducers are, essentially, analog devices that retain physical continuity throughout the course of evolution. All cellular life forms possess the properties of reproducers as reflected in the famous formula of Rudolf Virchow: Omnia cellula e cellula. Replicators, in contrast, are digital devices, so that physical continuity is not necessary for their propagation and evolution; information contained in the nucleotide sequence of the genome is sufficient. Clearly, all MGEs, viruses in particular, are replicators (the propagation of many viruses requires not only the genomic nucleic acid but also some virion protein(s) or even macromolecular structures to enter the cell; however, such structures are not transmitted to the next generation). Genomes of cellular life forms in themselves are replicators as well, so that organisms actually represent a union of reproducers and replicators. The establishment of mutualistic symbiosis between primordial reproducers and replicators might have been a pivotal point in the origin of life (Babajanyan et al., 2023).
The split between reproducers and replicators appears to be the most fundamental divide in biology. There is no evidence that the barrier between these two types of propagating, evolving entities has ever been crossed in the more than 4 billion years of the evolution of life, and given this perpetual but not blending symbiosis, this barrier appears to be fundamentally impenetrable. Attempts to define viruses as biological entities distinct from cells on the basis of simple criteria such as size have been hopelessly compromised by the latest findings, in particular the discovery of giant viruses (Koonin, Dolja, Krupovic and Kuhn, 2021). The definition of viruses as capsid-encoding organisms, in contrast to cellular life forms, which are ribosome-encoding organisms (Raoult and Forterre, 2008), fares better (regardless of whether or not it is appropriate to call viruses organisms). However, the discovery of viruses that encode almost all components of the translation system apart from the ribosome or multiple ribosomal proteins (see above) puts even this definition into doubt. What if a virus is discovered that actually encodes its own ribosome? Will this eliminate or blur the distinction between viruses and cells, as it has been repeatedly suggested giant viruses do? In our strong opinion, no such blurring will occur because even a ribosome-encoding virus will remain a replicator, not a reproducer. Thus, the best definition of a virus may be “a replicator encoding at least one protein encasing the viral genome or an MGE demonstrably derived from such a replicator” (Koonin, Dolja, Krupovic and Kuhn, 2021).
The definition of viruses as a distinct type of replicators prompts the question on the position of the virosphere within the space of replicators (ibid.). Apart from typical viruses that meet the above definition and comprise the “orthovirosphere”, a broad variety of virus-like MGE that can be derived from viruses or ancestral to viruses inhabit the “perivirosphere” (Figure 10). The denizens of the perivirosphere include capsid-less derivatives of regular viruses (for example, narnaviruses, mitoviruses, umbraviruses, endornaviruses and others within Riboviria), various satellite nucleic acids, viroids and viroid-like cccRNA replicators, and other MGE, such as viriforms. The orthovirosphere and perivirosphere are embedded in the greater replicator space that encompasses “non-virus-like” replicators such as typical transposons, integrating conjugative elements, plasmids as well as genomes of cellular life forms (Figure 10). Crucially, the boundaries between the different domains of the replicator space are porous, and evolutionary interconversions between different types of elements abound. In sharp contrast, as emphasized above, the wall separating the replicator space from reproducers appears to be impenetrable.
The replicators space and its three domains. The replicator space is represented as consisting of three domains: orthovirosphere including the 11 recognized realms of viruses; perivirosphere including virus-like replicators; and the outer replicator space including all other replicators, in particular, the genomes of cellular life forms. The boundaries between the domains are shown by dashed lines to emphasize multiple evolutionary interconversions between different types of replicators. Obelisks are cccRNA replicators encoding an uncharacterized protein and replicating in bacterial cells (Zheludev et al., 2024; Lopez-Simon et al., 2025; Urayama, Fukudome, Mutz, et al., 2026). Transpovirons are small dsDNA plasmids that are commensals of viruses in the phylum Nucleocytoviricota (Krupovic, Yutin and Koonin, 2016; Desnues et al., 2012).
10. Concluding remarks
Obviously, in this rather long review article, we could only superficially cover most aspects of the organization and evolution of the vast world of viruses. The diversity of known viruses is now growing more rapidly than ever, primarily, through metagenome and metatranscriptome mining, often revealing previously unsuspected features, for example, viruses with genomes in excess of 4 mb or diverse (putative) viruses with cccRNA genomes. Nevertheless, however astonishing this might be, it appears that we already know the global organization of the virosphere and the main structure of the megataxonomy of viruses. At the same time, it should be emphasized that this structure is an inherently moving target, as demonstrated, in particular, by the ongoing splitting of some of the viral realms. From the structuring of the virosphere and extensive phylogenomic analyses, general principles of virus evolution are emerging and new research frontiers are opening up. Some of the important directions of future research include the study of different facets of virus–host symbioses as well as coevolution of viruses in complex systems such as those including satellite viruses. There is no doubt that new, fascinating features of the virosphere and its evolution will be uncovered.
Acknowledgements
EVK is supported by the Intramural Research Program of the National Institutes of Health of the USA.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.