

 CC-BY 4.0
CC-BY 4.0
1. Introduction
A CRISPR-Cas9 screen identifies FHL1 as an essential cell host factor for CHIKV. (A) Schematic of CRISPR-Cas9 genome-wide screen in HAP1 haploid cells (B) Results of the CHIKV screen analyzed by MAGeCK. Each circle represents individual gene. Y -axis represents the significance of sgRNA enrichment of genes in the selected population compared to the non-selected control population. X-axis represents a random distribution of the genes (C) Schematic of FHL1 isoform A protein. Below: organization of the LIM domains. Inset displays the two tandem zinc fingers of a LIM domain.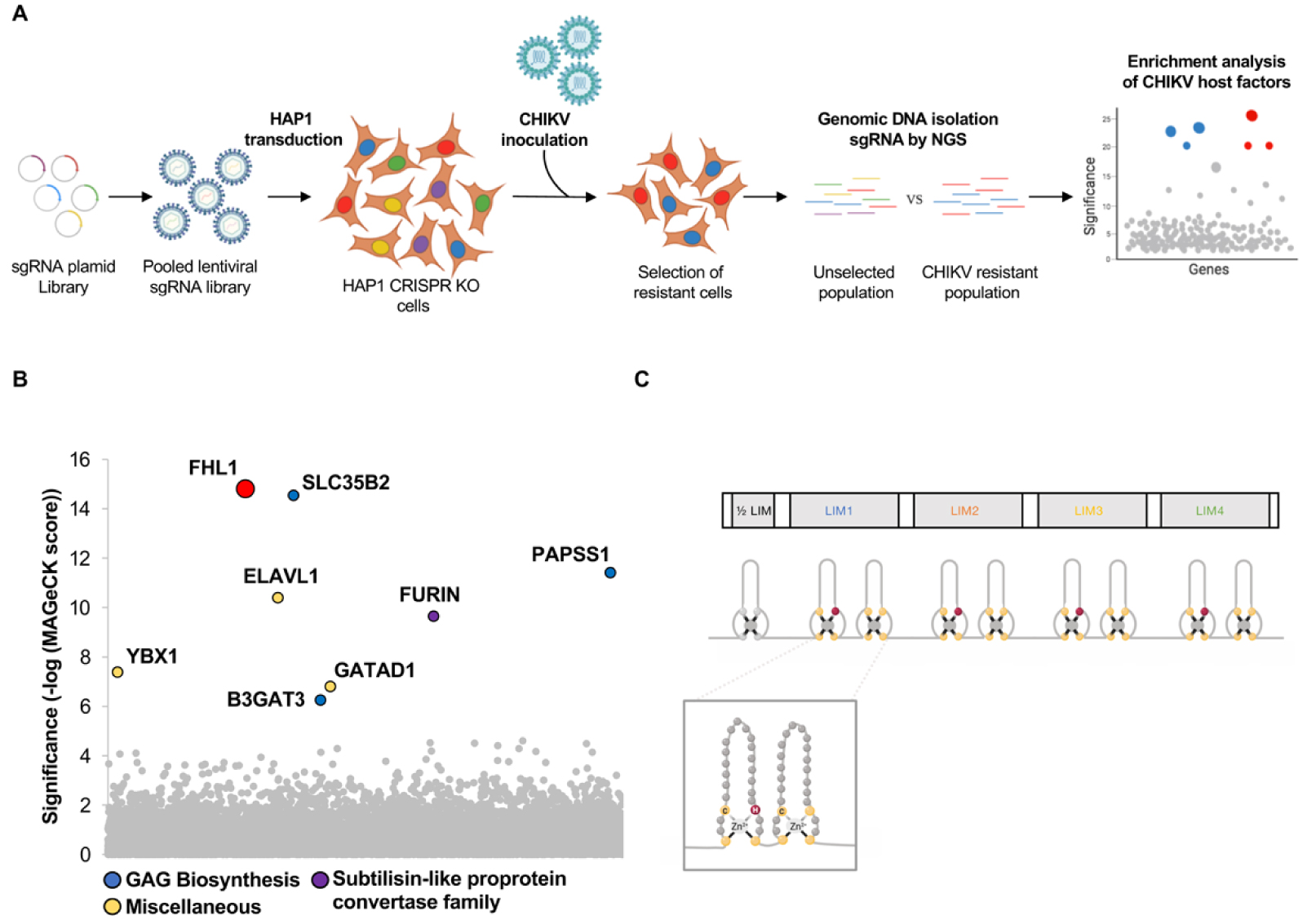
Chikungunya virus (CHIKV) is an arbovirus (Arthropod-borne virus) belonging to the genus Alphavirus of the Togaviviradae family. This pathogen, isolated in 1952 during an outbreak in present-day Tanzania [2], has reemerged in 2000 and spread to new worldwide areas causing millions of infection [3]. CHIKV is the etiological agent of the chikungunya disease, a word from Makonde language that translates as “that which bends up”, referring to the severe muscular and joint pains that impede the mobility and daily activities of infected patients. CHIKV is transmitted to humans through the bite of infected mosquitoes of the Aedes aegypti and albopictus species. Following inoculation in the skin, CHIKV infects dermal cells, mainly resident fibroblasts, then disseminates in the organism to reach preferential sites of replication such as peripheral joints and musculoskeletal tissue [4]. Although it could be asymptomatic, CHIKV infection frequently progresses to high fever and debilitating arthralgia and myalgia that can persist for months. Intensive CHIKV replication in muscle and joints triggers a strong immune response leading to pro-inflammatory chemokines and cytokines release, most certainly responsible for the onset of the disease. Whereas the clinical aspects of the disease are well described, the molecular determinants of the viral tropism and pathogenesis are poorly understood. Like other viruses, CHIKV is an obligate intracellular parasite which relies on host cellular functions to accomplish its replication cycle. Several studies have already identified many host factors exploited by the virus to accomplish its infectious cycle. However, none of them accounts for the preferential tropism of the virus for muscle and joint tissues, nor the clinical onset. This report consists of an overview of the study done by our team that has led to the identification of the protein Four and Half LIM Domain (FHL1) as a crucial host factor for CHIKV infection and pathogenesis. This work was performed in collaboration with teams from the Pasteur Institute and the Hospital La Pitié Salpetrière. It has been recently published in the journal Nature [1].
FHL1 is essential for CHIKV susceptibility. (A) Primary myoblasts and fibroblasts from FHL1 deficient patients are resistant to CHIKV infection. Quantification of infection in primary myoblasts (PM1, PM2, PM3) and fibroblasts (PF2, PF4) from EDMD patients or healthy donors (CM1, CM2, CF1 et CF2) inoculated with CHIKV. (B) FHL1 ectopic expression in fibroblast from EDMD patient confers susceptibility to CHIKV infection. Quantification of infection in fibroblast from patient (PF2 et PF4) transduced with a lentiviral vector encoding FHL1A or a control vector and then challenged with CHIKV.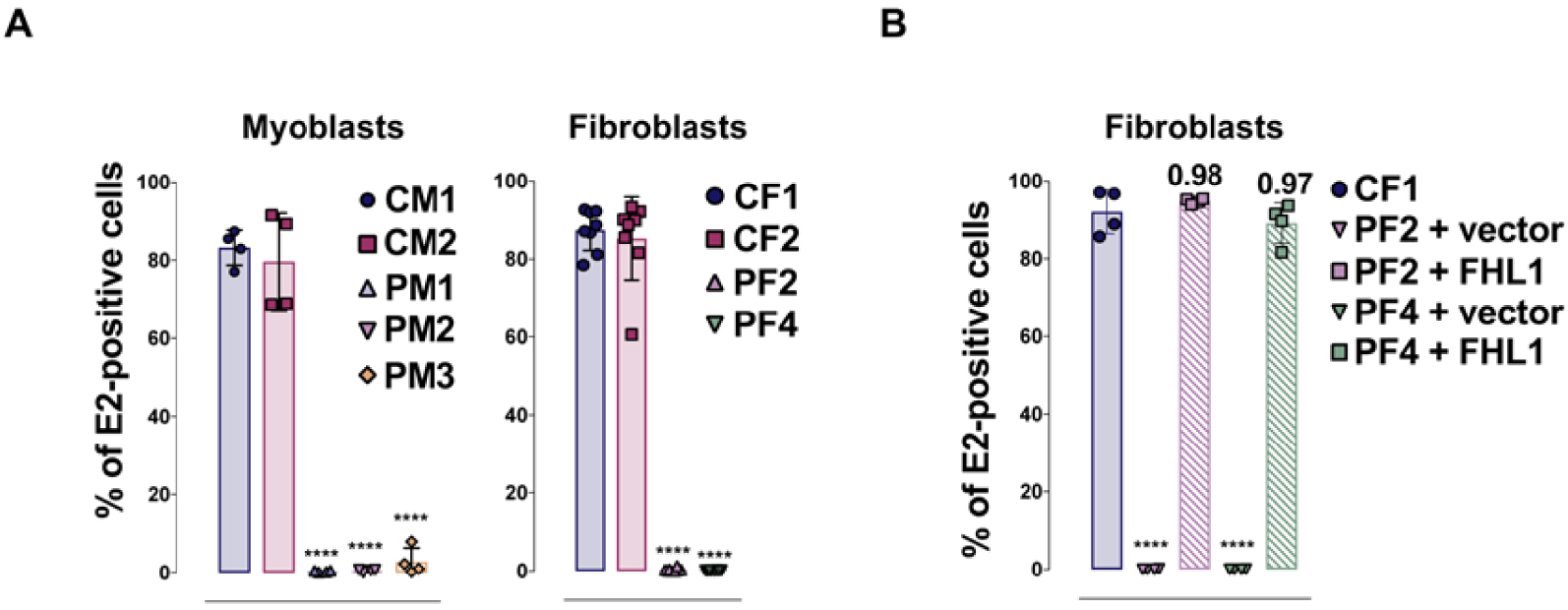
FHL1 interacts with CHIKV nsP3 and is required for CHIKV RNA replication. (A) Control and FHL1-depleted 293T cells were transfected with in vitro transcribed CHIKV RNA expressing luciferase. Luciferase activity was monitored at the indicated time points on the X-axis. (B) Negative stranded viral RNA quantification by qRT-PCR from control and FHL1-depleted 293T cells inoculated with CHIKV, and collected at the indicated time points. (C) (Left panel) Schematic representation of CHIKV nsP3 constructs deleted for the R1, R2, R3 or R4 sequences. (Right panel) 293T cells were transfected with FHL1A-HA and either an empty vector or plasmids encoding FLAG-tagged nsP3 constructs. Cell lysates were immunoprecipitated with anti-FLAG followed by immunoblot analysis with anti-HA to detect FHL1.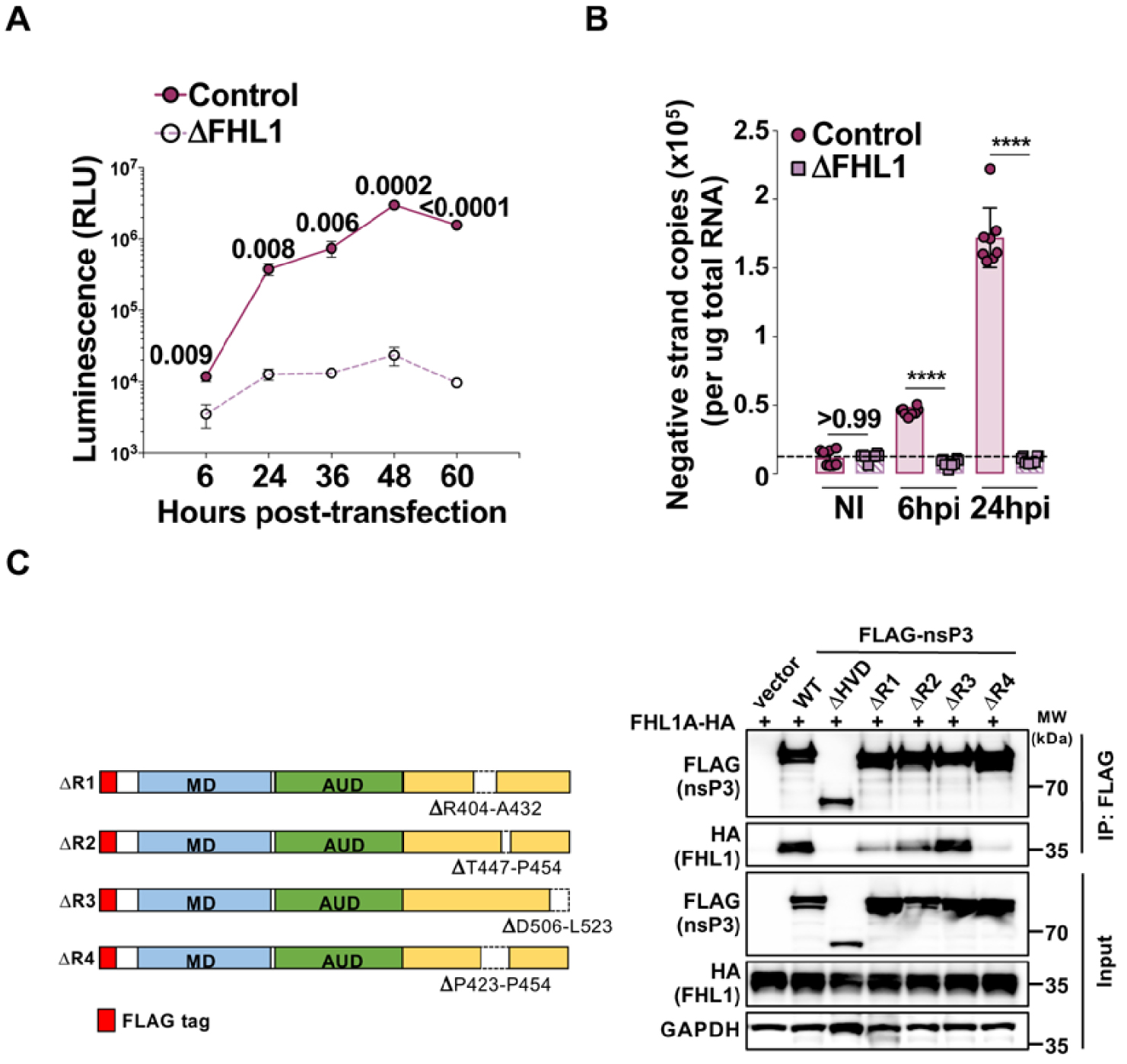
2. Identification of FHL1 as a critical cellular factor for CHIKV infection cycle
To uncover host factors important for CHIKV replication, we performed a systematic genome-wide screen using the CRISPR-Cas9 technology (Figure 1A). This approach involved expressing a library of single guide RNA (sgRNA) delivered as a lentiviral pool (6 sgRNA/gene) into the haploid human cell line HAP1 which is permissive to CHIKV. Then, the transduced-cells were inoculated with a CHIKV strain isolated during the 2005–2006 outbreak in La Reunion island. Since CHIKV infection is cytopathic, only cells that lost a gene essential for viral replication can survive and proliferate. It was then possible to retrieve, by combining next-generation sequencing technology and bioinformatic analysis, the sgRNAs responsible for the “resistant” phenotype, and therefore the identity of the invalidated genes. This experimental approach allowed us to identify the gene FHL1 (Four and a Half LIM domains 1), which has the most enriched targeting-sgRNAs during the phenotypic selection (Figure 1B). This gene localizes on the X chromosome and encodes the FHL1 protein for which three isoforms exist (A, B et C). Isoform FHL1A is predominantly expressed in striated muscle and fibroblasts [5], the preferential cellular targets of CHIKV. FHL1A contains 4 and a half LIM domains that are two tandemly arranged zinc finger domains (Figure 1C) [6]. FHL1A is cytoplasmic protein that plays an important role in muscle development and assembly of large structural complexes. Mutations in the FHL1 gene leading to the absence of FHL1 protein have been described in X-linked myopathies such as the Emery–Dreifuss muscular dystrophy (EDMD) [7].
3. FHL1 is a cellular factor essential for CHIKV replication
Since FHL1A expression correlates with the natural tropism of the virus and that this protein was never previously reported as a host factor involved in viral infection, we have focused our study on understanding its role during CHIKV infection. We have first assessed the susceptibility to CHIKV in different cellular models for which FHL1 expression was invalidated by CRISPR-Cas9. In these cells, FHL1 depletion greatly decreased infection by different primary strains of CHIKV responsible for outbreaks. Interestingly, FHL1 poorly affected a CHIKV strain maintained in a sylvatic cycle between Aedes mosquitoes and African monkeys. Secondly, we have assessed the importance of FHL1 on the infectious cycle of other viruses. We showed that FHL1 is also selectively used by O’nyong-nyong virus (ONNV), an alphavirus phylogenetically closely related to CHIKV. In contrast, FHL1 is dispensable for infection by other related alphavirus or other arborviruses such as Dengue or ZIKA virus. Finally, the importance of FHL1 in CHIKV biology was strengthened by our observations showing that the virus was unable to replicate in cells isolated from patients with a rare genetic disease, the Emery–Dreifuss muscular dystrophy (EDMD). In these patients, the muscular pathology results from mutations in the FHL1 gene responsible for FHL1 protein degradation. We have shown that primary myoblasts and fibroblasts from EDMD patients lack FHL1 expression and are naturally refractory to CHIKV infection (Figure 2A). In these cells, ectopic expression of FHL1 confers permissiveness to CHIKV, highlighting the essential role of FHL1 in CHIKV tropism (Figure 2B).
4. FHL1 regulates the viral replication step by interacting with the viral protein nsP3
FHL1 is critical for CHIKV pathogenesis in mice. (A, B) Wild-type (WT) and Fhl1 KO (FHL1-null) newborn mice were injected with CHIKV by intradermal route and sacrificed 7-day post-inoculation. (A) Quantification of viral titers in tissues of WT and Fhl1 KO mice. (B) Hematoxylin and eosin staining of transversal section of skeletal muscle in CHIKV-infected mice.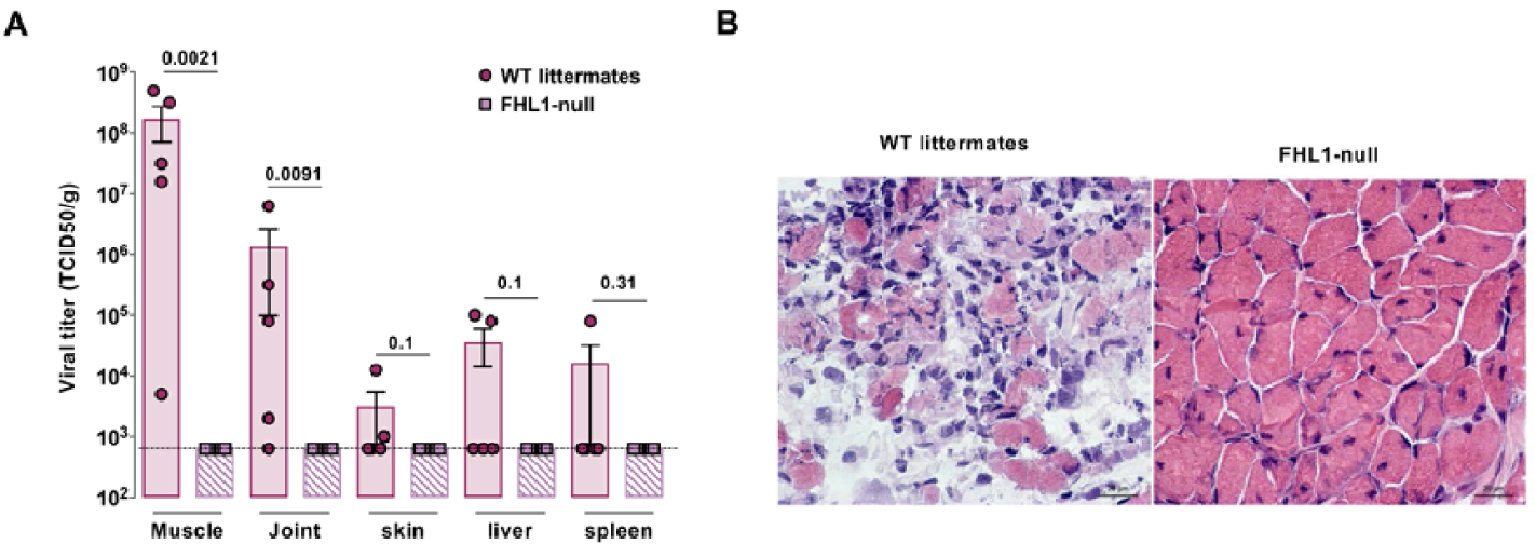
We have used several molecular virology and cellular biology tools to identify which step of CHIKV infectious cycle is impacted by FHL1. Our results revealed that FHL1 is critical for the replication of viral RNA. Indeed, in cells lacking FHL1 and infected by CHIKV, we have observed a lack of viral genome synthesis both at the level of neosynthesized genomic viral RNA (gvRNA) (Figure 3A) and negative strand intermediates, which serve within the replication complex as matrix for gvRNA synthesis (Figure 3B). In line with these data, our electronic microscopy studies showed that FHL1 is involved in the formation of plasma-membrane-associated spherules as well as the cytoplasmic vacuolar membrane structures, which are CHIKV-induced viral replication platforms [8]. We have also shown that FHL1 is recruited at these structures by the viral protein nsP3, a key player in viral RNA synthesis. FHL1 interacts directly with nsP3 C-terminal hypervariable domain, an intrinsically disordered domain known to mediate recruitment of cellular proteins [9]. By comparing the HVD sequence of several alphaviruses, we have identified a common region between CHIKV and ONNV strains that is essential for the interaction with FHL1 (Figure 3C). Deleting this region abolishes the interaction between FHL1 and nsP3, and viruses lacking the FHL1-interacting sequence showed a strong defect in viral replication when compared to their wildtype counterpart.
5. Role of FHL1 in pathophysiology of CHIKV in mice
To assess in vivo the role of FHL1 in CHIKV pathogenesis, Fhl1 heterozygous female mice were crossed with wild-type males carrying the same genetic background. FHL1-deficient male mice were identified and 9-day-old newborn mice were injected with CHIKV by intradermal route and then sacrificed 7 days after inoculation. Our results, obtained in collaboration with Marc Lecuit’s team at Pasteur Institute, showed high viral loads in muscle (108 TCID 50/g) and joints (order of magnitude of 106 TCID 50/g) of wild-type littermates, whereas viral loads in FHL1-deficient mice remain below the threshold of detection of the technique (Figure 4A). Moreover, histology and immunohistochemistry analyses showed necrotizing myositis with massive infiltrates and necrosis of the muscle fibers in skeletal muscle of WT littermates, while FHL1-null mouse muscle showed no detectable pathology (Figure 4B). Altogether these results showed that FHL1-deficient mice are resistant to CHIKV and that FHL1 contributes to CHIKV pathogenesis in mice.
6. Conclusions and perspectives
Our study identifies for the first time a cellular factor specific of CHIKV and essential for its replication cycle. High FHL1 expression in muscle and fibroblasts suggests that FHL1 is a key player of CHIKV tropism and pathogenesis. In normal conditions, FHL1 participates in healthy muscle homeostasis. During infection, FHL1 is highjacked from its physiological function by the CHIKV-nsP3 protein, likely to build the replication complex essential for viral genome amplification. The molecular mechanisms by which FHL1 accomplishes its pro-viral function are currently unknown and remain to be elucidated. Our works on FHL1 open new therapeutic opportunities to treat CHIKV. Identification of small-molecule inhibitors able to bind nsP3 and to disrupt its interaction with FHL1, could constitute a new therapeutic strategy against chikungunya disease.
French version
1. Introduction sur le virus chikungunya
Le virus chikungunya (CHIKV) est un arbovirus (Arthropod-borne virus) appartenant au genre Alphavirus de la famille des Togaviviradae. Ce pathogène, isolé en 1952 lors d’une épidémie dans l’actuelle Tanzanie [2], a ré-émergé ces dernières années et s’est propagé dans de nombreuses nouvelles régions du globe terrestre, causant des millions d’infections [3]. CHIKV est l’agent étiologique de la maladie chikungunya qui signifie en Makondé « celui qui marche courbé », en référence aux douleurs musculaires et articulaires sévères dont souffrent les patients infectés, les empêchant de se déplacer normalement et de mener à bien leurs activités quotidiennes. CHIKV est transmis à l’homme par la piqure des moustiques des genres Aedes aegypti et albopictus. Suite à l’inoculation du virus dans la peau, CHIKV infecte les cellules dermiques, notamment les fibroblastes résidents puis se dissémine dans l’organisme jusqu’aux sites préférentiels de réplication que sont les articulations périphériques et le tissu musculosquelettique [4]. Bien que pouvant être asymptomatique, l’infection par CHIKV évolue fréquemment vers de fortes fièvres, ainsi que des douleurs articulaires et musculaires intenses pouvant persister plusieurs mois. L’intense réplication du virus dans les tissus musculaires et articulaires déclenche ainsi une forte réponse immunitaire conduisant à la libération de chimiokines et cytokines pro-inflammatoires certainement à l’origine des symptômes de la maladie. Si les aspects cliniques de la maladie chikungunya sont bien décrits, les déterminants moléculaires du tropisme et de la pathogénèse virale sont encore très mal compris. Comme tous les virus, CHIKV est un parasite obligatoire intracellulaire dont le déroulement du cycle réplicatif est totalement dépendant des fonctions de la cellule hôte. Plusieurs travaux avaient déjà identifié de nombreux facteurs cellulaires exploités par le virus pour accomplir son cycle infectieux. Cependant, aucun ne peut expliquer le tropisme préférentiel du virus pour les cellules des tissus musculaires et articulaires, ainsi que les signes cliniques observés. Cet article fait la synthèse de l’étude conduite par notre équipe et aboutissant à l’identification de la protéine Four and Half LIM Domain (FHL1) comme facteur cellulaire jouant un rôle crucial dans le cycle infectieux et la pathogénèse du CHIKV. Ces travaux réalisés en collaboration avec des équipes de l’Institut Pasteur et de l’Hôpital Pitié Salpetrière, ont récemment été publiés dans la revue Nature [1].
Un crible CRISPR-Cas9 identifie FHL1 comme facteur cellulaire essentiel de CHIKV. (A) Représentation schématique du crible CRISPR-Cas9 dans les cellules HAP1 transduites avec une librairie de sgRNA à l’échelle du génome. (B) Analyse par le logiciel MAGeCK de l’enrichissement des gènes du crible CRISPR-Cas9. L’axe Y montre l’importance de l’enrichissement des sgRNA dans la population résistante par rapport à la population non sélectionnée. L’abscisse représente une distribution aléatoire des gènes. (C) Représentation schématique de l’isoforme A de la protéine FHL1. En dessous sont représentés les motifs en doigt de zinc des domaines LIM. Un tandem de doigts de Zinc est présenté dans l’encart.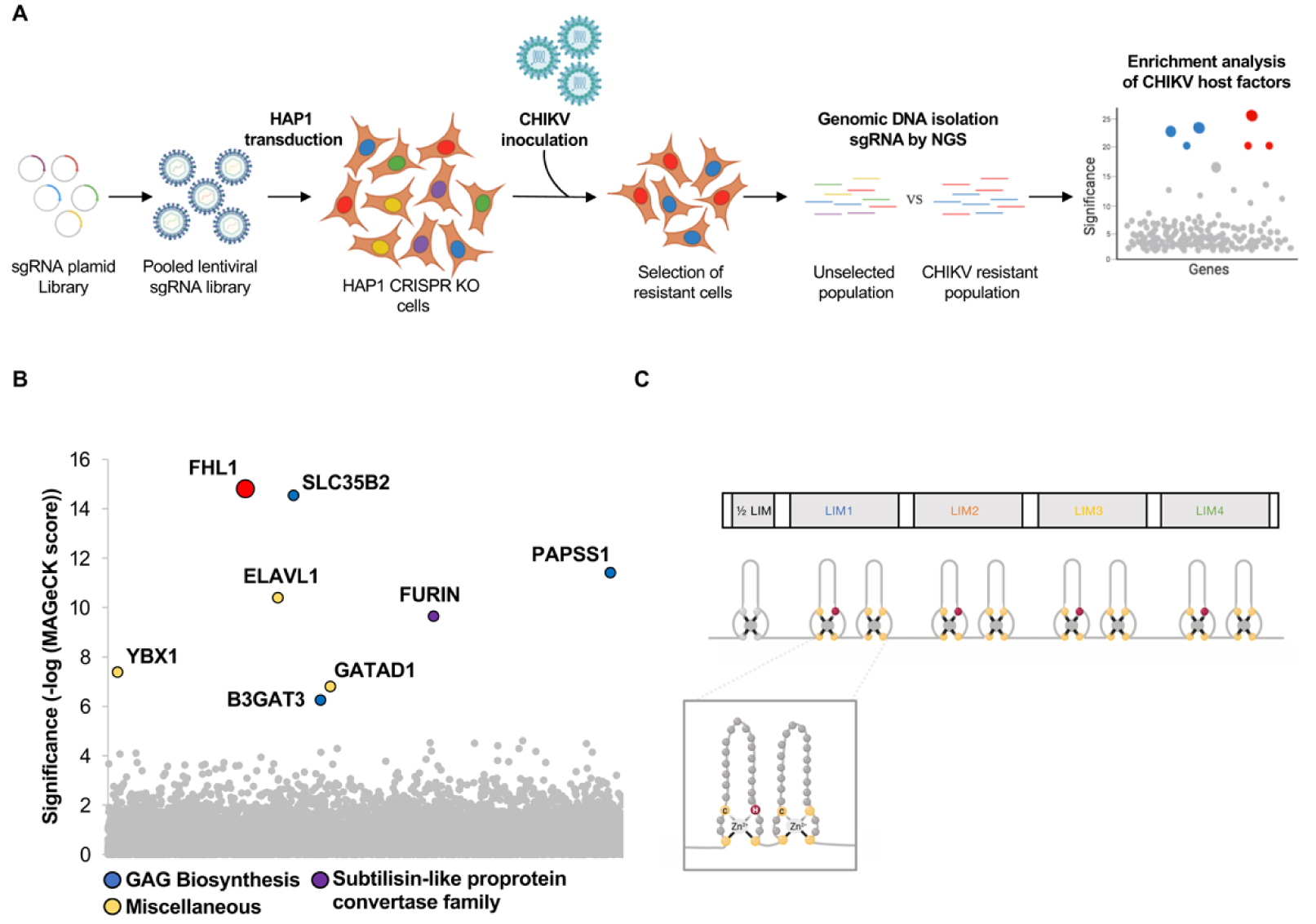
FHL1 est essentielle à la réplication virale de CHIKV. (A) Les myoblastes et fibroblastes de patients EDMD sont réfractaires à l’infection par CHIKV. Quantification de l’infection dans les myoblastes (PM1, PM2, PM3) et fibroblastes (PF2, PF4) de patients EDMD ou de donneurs sains (CM1, CM2, CF1 et CF2) inoculés avec CHIKV. (B) L’expression ectopique de FHL1 confère la susceptibilité à l’infection par CHIKV aux fibroblastes de patients EDMD. Quantification de l’infection dans les fibroblastes de patients (PF2 et PF4) transduits avec un plasmide contrôle ou codant pour FHL1, et inoculés avec CHIKV.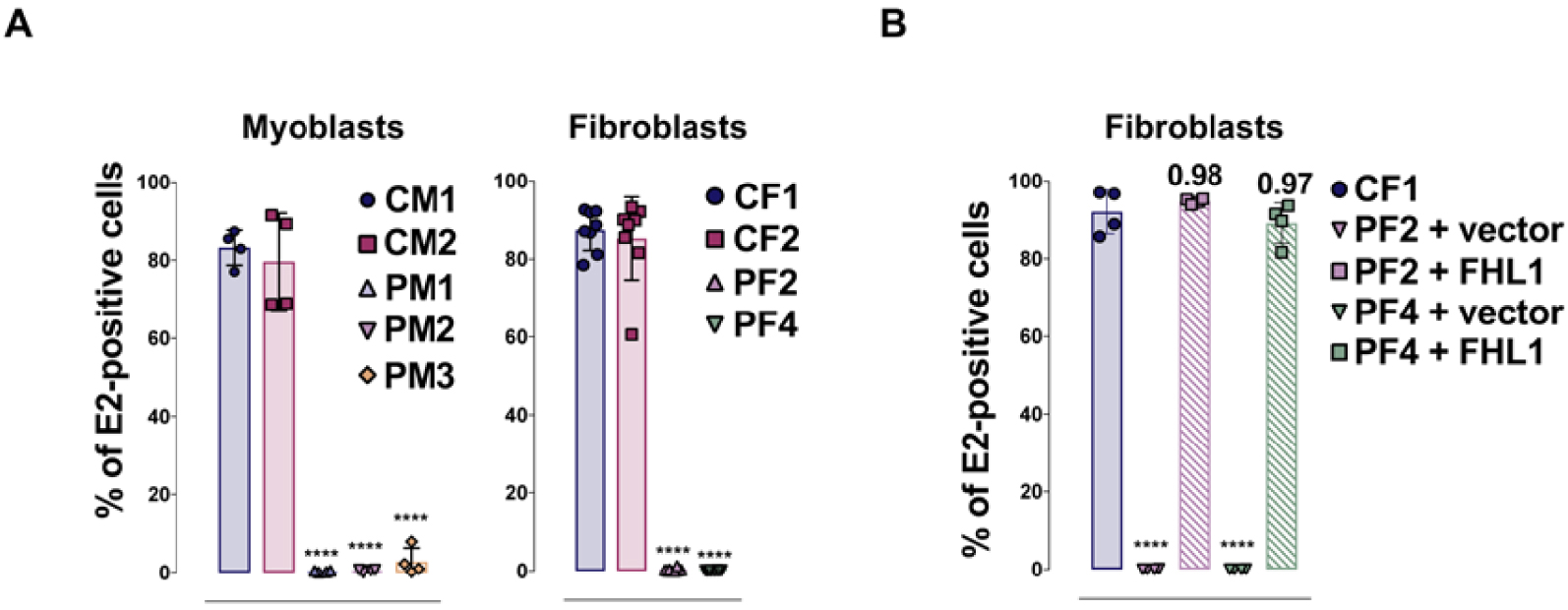
FHL1 est requise pour la réplication de l’ARN viral via son interaction avec le HVD de nsp3. (A) Les cellules HEK-293T et HEK-293T déplétées pour FHL1 ont été transfectées avec un ARN viral transcrit in vitro et exprimant la luciférase. L’activité luciférase a été quantifiée aux différents temps indiqués en abscisse. (B) Quantification par RT-qPCR du brin d’ARN viral de polarité négative à différent temps après inoculation des cellules HEK-293T et HEK-293T déplétées pour FHL1. (C) Panel de gauche : représentation schématique des différentes constructions de la protéine nsp3 de CHIKV dans lesquelles les séquences R1, R2, R3 ou R4 ont été délétées. Panel de droite : Les différentes constructions de nsp3 portant une étiquette FLAG ont été cotransfectées avec la protéine HA-FHL1. Immunoprécipitation des nsp3 avec un anti-FLAG puis immunoblot avec un anti-HA pour détecter FHL1.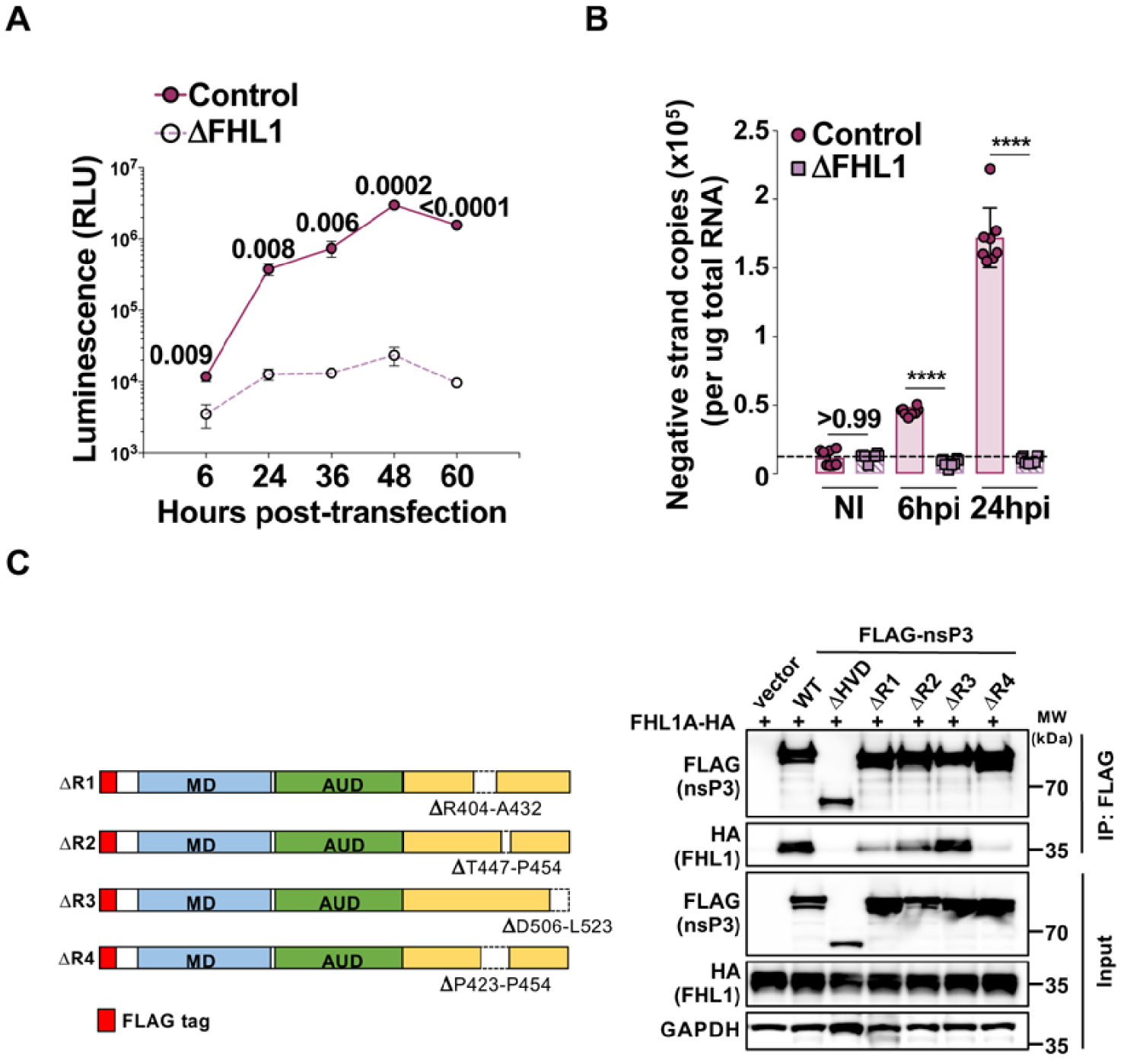
2. Identification de FHL1 comme facteur cellulaire important du cycle infectieux de CHIKV
Afin d’identifier les facteurs de l’hôte nécessaires à la réplication du CHIKV, nous avons effectué un criblage systématique du génome de cellules humaines par la technologie dite de CRISPR-Cas9 (Figure 1A). Cette approche a consisté-à exprimer une librairie de petits ARN guides (sgRNA) délivrée sous forme d’un pool lentiviral (6 sgRNA/gène) dans des cellules haploïdes HAP1 permissives à CHIKV. Les cellules ont été ensuite infectées par une souche de CHIKV isolée durant l’épidémie sur l’Ile de la Réunion en 2005. Le CHIKV étant cytopathique, seules les cellules pour lesquelles le système CRISPR-Cas9 aboutit à l’ablation d’un gène essentiel au déroulement du cycle viral, peuvent survivre et proliférer. Il est ensuite possible de retrouver, en combinant séquençage à haut débit avec des analyses bio-informatiques, les sgRNA responsables de ce phénotype « résistance » et par conséquent l’identité des gènes invalidés. Cette approche expérimentale nous a permis d’identifier le gène FHL1 (Four and a Half LIM domains 1) pour lequel les sgRNAs ont été les plus enrichis durant la sélection phénotypique (Figure 1B). Le gène FHL1 est localisé sur le chromosome X et code pour les protéines FHL1 dont il existe trois isoformes (A, B et C). L’isoforme FHL1A est majoritairement exprimée dans le muscle strié et dans les fibroblastes [5], les cibles cellulaires préférentielles de CHIKV. FHL1A contiennent 4 et un demi-domaines LIM, chacun constitué d’un tandem de motifs en doigt de zinc (Figure 1C) [6]. FHL1A, qui est cytoplasmique, joue un rôle essentiel dans le développement musculaire et assure un rôle d’échafaudage dans la mise en place de larges complexes structuraux. Des mutations dans le gène FHL1 conduisant à l’absence de la protéine FHL1 ont été décrites dans des formes de myopathies liées à X comme la dystrophie musculaire d’Emery–Dreifuss (EDMD) [7].
3. FHL1 est un facteur cellulaire essentiel à la réplication de CHIKV
Étant donné que l’expression de FHL1A corrèle avec le tropisme naturel du virus et que le gène FHL1 n’a jamais été rapporté au préalable comme un facteur cellulaire impliqué dans des infections virales, nous avons focalisé notre étude sur la compréhension du rôle de ce facteur dans l’infection par CHIKV. Nous avons dans un premier temps évalué la susceptibilité à CHIKV dans différents modèles cellulaires dont l’expression de FHL1 a été invalidée par CRISPR-Cas9. Dans ces cellules, la déplétion de FHL1 diminuait fortement l’infection des cellules par différentes souches de CHIKV primaires et responsables d’épidémies. De façon intéressante, FHL1 est faiblement exploitée par la souche sylvatique de CHIKV qui a circulé uniquement entre le moustique Aedes et les singes d’Afrique sans être passée par l’homme. Nous avons dans un deuxième temps évalué l’impact de FHL1 sur le cycle infectieux d’autres virus. Nous avons montré que FHL1 était sélectivement utilisée par le virus O’nyong-nyong virus (ONNV), un alphavirus phylogénétiquement très proche de CHIKV. En revanche, FHL1 n’est pas requis pour l’infection par d’autres alphavirus apparentés ou d’autres arbovirus comme les virus de la Dengue ou Zika. Finalement, l’importance de FHL1 dans la biologie de CHIKV a été renforcée par nos observations montrant que le virus était incapable de se multiplier dans des cellules isolées de patients souffrant d’une pathologie génétique rare, la dystrophie musculaire d’Emery–Dreifuss. Chez ces sujets, la pathologie musculaire résulte de mutations du gène FHL1 responsables de la dégradation de la protéine FHL1. Nous avons montré que les myoblastes et les fibroblastes primaires issus de patients EDMD, présentant un défaut d’expression de FHL1, sont naturellement réfractaires à l’infection par CHIKV (Figure 2A). Dans ces cellules, l’expression ectopique de l’isoforme FHL1A confère la permissivité à CHIKV, mettant en évidence le rôle essentiel de FHL1 dans le tropisme de CHIKV (Figure 2B).
FHL1 joue un rôle important dans la physiopathologie de CHIKV chez les souris. (A, B) Les souriceaux sauvages (WT) et Fhl1 KO (FHL1-null) sont injectés par voie intradermique avec CHIKV et sacrifiés 7 jours post-inoculation. (A) Quantification du titre viral dans les différents tissus de souriceaux sauvages et Fhl1 KO. (B) Marquage à l’Hématoxyline et l’éosine des coupes transversales de tissus musculaires squelettiques de souriceaux inoculés avec CHIKV.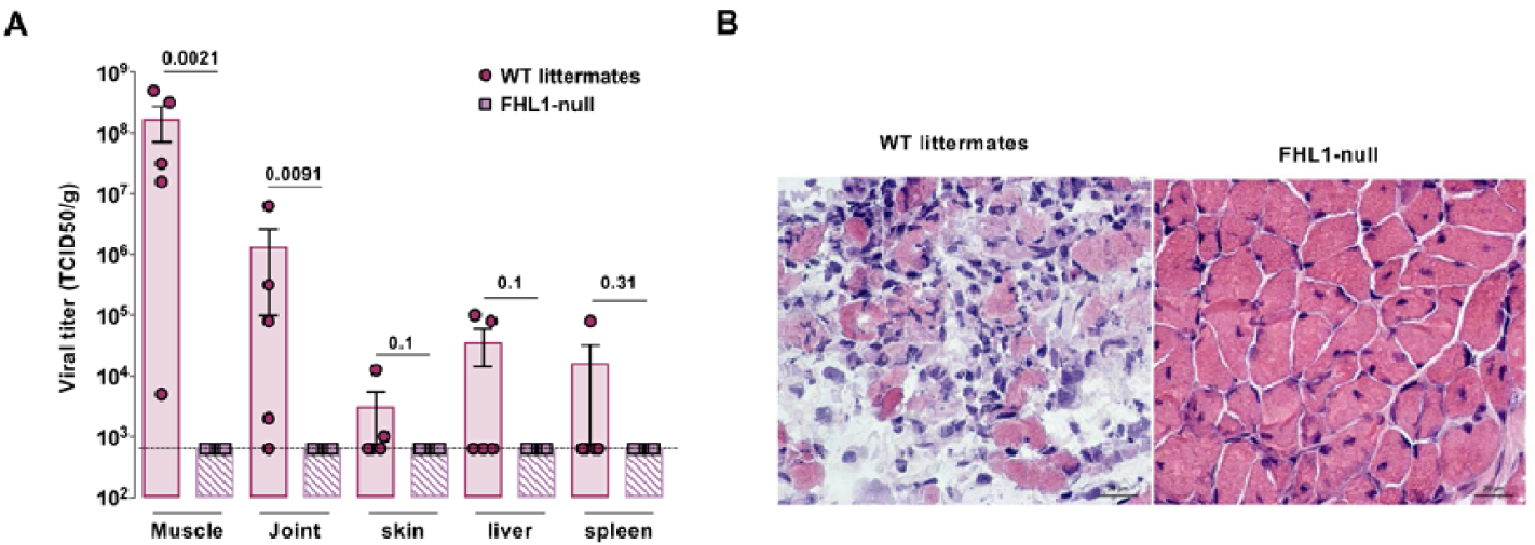
4. FHL1 régule l’étape de réplication virale en interagissant avec la protéine virale nsP3
Nous avons utilisé de nombreux outils de virologie moléculaire et biologie cellulaire pour identifier l’étape du cycle infectieux de CHIKV impactée par FHL1. Nos résultats montrent que FHL1 est importante pour la réplication de l’ARN viral. En effet, dans les cellules dépourvues de FHL1 et infectées par CHIKV, nous avons observé un défaut de synthèse des différents produits de la réplication du génome virale tant au niveau de l’ARN génomique viral (ARNgv) néosynthétisé (Figure 3A) que dans la production d’intermédiaires ARN viraux de polarités négatifs, qui servent au sein du complexe de réplication de matrices pour la synthèse de l’ARNgv (Figure 3B). En accord avec ces données, nos analyses en microscopie électronique ont montré que FHL1 régule la formation des sphérules associées à la membrane plasmatique ainsi que des structures membranaires vacuolaires cytoplasmiques qui sont des plateformes cellulaires de réplication virale induites par CHIKV pour amplifier son génome [8]. Nous avons également montré que FHL1 est recrutée au niveau de ces structures par l’intermédiaire de la protéine virale nsP3, un acteur essentiel de la synthèse de l’ARN viral. FHL1 interagit directement avec le domaine C-terminal hypervariable (HVD) de nsP3, une région intrinsèquement désordonnée connue pour agir comme une plateforme de recrutement de nombreuses protéines cellulaires [9]. En comparant les séquences des HVD de différents alphavirus, nous avons identifié un motif commun à CHIKV et ONNV indispensable pour l’interaction avec FHL1 (Figure 3C). La délétion de cette région abolit l’interaction de FHL1 avec nsP3 et des virus dépourvus du site de fixation de FHL1 présentent un défaut de réplication par rapport au virus sauvage.
5. Rôle de FHL1 dans la physiopathologie de CHIKV
Pour évaluer l’importance de FHL1 dans la pathogénèse de CHIKV in vivo, des souris femelles hétérozygotes pour le gène FHL1 ont été croisées avec des souris mâles de fond génétique identique. Les souriceaux mâles déficients pour l’expression de FHL1 ont été identifiés puis infectés avec CHIKV par voie intradermique 9 jours après leur naissance et sacrifiés après l’inoculation. Nos résultats, obtenus en collaboration avec l’équipe de Marc Lecuit à l’Institut Pasteur, ont montré dans les souris sauvages issues des mêmes portées des charges virales très importantes au niveau du tissu musculaire (108 TCID 50/g) et des articulations (de l’ordre de 106 TCID 50/g), contrairement aux souris déficientes en FHL1 dont les charges virales sont en dessous du seuil de détection de la technique (Figure 4A). D’autre part, les analyses histologiques et immunohistochimiques du muscle de souris contrôles ont montré une myosite nécrosante avec de nombreux infiltrats et une nécrose musculaire tandis que le muscle de souris invalidées pour FHL1 ne présente aucun signe anatomopathologique (Figure 4B). L’ensemble de ces résultats a montré que les souris déficientes en FHL1 sont résistantes au CHIKV et indique que FHL1 contribue à la pathogénèse de CHIKV chez la souris.
6. Conclusions et perspectives
Nos travaux identifient pour la première fois un facteur cellulaire spécifique à CHIKV et indispensable à son cycle réplicatif. La forte expression de FHL1 dans le tissu musculaire et dans les cellules fibroblastes suggère que FHL1 joue un rôle majeur dans le tropisme et la pathogénèse de CHIKV. En temps normal, FHL1 participe au fonctionnement du muscle sain. Durant l’infection, FHL1 serait détournée de sa fonction physiologique par la protéine nsP3 de CHIKV pour former le complexe de réplication essentiel à l’amplification de son génome. Les mécanismes moléculaires par lesquels FHL1 accomplit cette fonction pro-virale sont inconnus à ce jour et restent à élucider. Nos travaux sur FHL1 ouvrent de nouvelles perspectives thérapeutiques pour traiter CHIKV. L’identification de petites molécules inhibitrices se fixant sur nsP3 et capables de bloquer sa liaison avec FHL1 pourrait constituer une stratégie thérapeutique novatrice pour lutter contre la maladie chikungunya.