

 CC-BY 4.0
CC-BY 4.0
1. Introduction
More than half a century after the elucidation of the codon table [1], the origin of the genetic code remains the most fascinating question of all biological processes in which products are needed to synthesize the elements of their own synthesis. This conceptual difficulty is accentuated here by the interplay between two distinct classes of molecules, RNA and proteins, whose origins and interactions widen the spectrum of possibilities. From the earliest examinations of the codon table [2, 3, 4] to the most recent results on the chemical details of the translational machinery [5, 6] and the phylogenies of its functional elements [7, 8, 9, 10, 11], the possible origin of the genetic code has been explored from different viewpoints (reviewed in [12]). Three major theories have emerged. In the coevolution theory [13, 14, 15], the primary driving force for the evolution of the genetic code is sought for in the sequential emergence of new amino acids (a.a.s hereafter) within the primordial biochemical systems. It is argued that a.a.s with the simplest side chains or requiring the smallest number of biochemical steps for their biosynthesis, as well as those existing in prebiotic environments [16], were likely the earliest to have entered the coding repertoire. The correlation between the a.a. biosynthetic pathways and the class of tRNA synthetases (aaRS) activating them [17] brings support to this hypothesis. Alternatively, the stereochemical theory postulated long ago [18, 19], which emphasizes the existence of chemical affinity between a.a.s and RNA molecules to explain the codon table, has also received subsequent support [20, 21, 22, 23, 24] and can now be tested by experimental evolution of aptamers [25, 26]. Finally, the error minimization theory posits that the codon table was selected during evolution such as to minimize the effects of mutational and translational errors [27, 28, 29, 30]. This idea, opposed to the original view of a frozen accident [3], has been submitted to recent theoretical evaluations [31, 32, 33, 34, 35], considering also the variant codon tables in diverse organisms [36].
Over the decades, the standard codon table has been subject to numerous investigations searching for the origin of the code in its internal logic. The wobble hypothesis formulated long ago [37] to explain the general coding degeneracy in boxes of two or four codons (exceptionally three) has evolved into exhaustive lists of tRNA species deduced from complete genome sequences and detailed descriptions of their chemical modifications and interactions with the ribosomes to explain the specificity of the third codon position relative to the first two [38, 39, 40]. Rules for codon assignment that have been sought for in the correlations between a.a.s and nucleotide positions in codons at the same early period [41], have resulted in pinpointing the second position of codons as a major determinant in the differentiation between hydrophobic and hydrophilic a.a.s [42]. The observation that all codons with C at the second position encode a.a.s activated by class II aaRS [43], whereas nearly all those with U encode a.a.s activated by class I aaRS (those with G or A are equally mixed) suggested a differentiation model via successive asymmetrical binary choices as the origin of the code [44]. A further distinction of the central codon position relative to the first (and of course the third) is also suggested by the fact that all codons with C at this position belong to fully degenerated 4-codon boxes (unsplit codon families) when all codons with A at this position belong to 2-codon boxes (split codon families). Both types of families are in equal numbers for codons with G or U at the central position. None of the above asymmetries exist for the first codon position.
Recently, a major progress in our vision of the genetic code and the decoding process has been made with the comprehensive reexamination of the thermodynamics of codon–anticodon interactions taking into account known chemical modifications of tRNA molecules and the structural interactions of the codon–anticodon duplexes with the translational apparatus [39]. Three classes of codon families were defined depending on the calculated free energy corresponding to the pairing of the first two codon positions with anticodons. The average values range from −13 kJ/mole for two G–C pairs to −4.2 kJ/mole for two A–U pairs with an intermediate figure of −9.2 kJ/mole for one G–C and one A–U pair. Furthermore, considering the structural characteristics of the tRNA anticodon hairpin in its interactions with the ribosomal decoding site [45], this work showed how specific tRNA modifications determine non-canonical structures at the wobble position to equilibrate the thermodynamic stability between synonymous codon–anticodon pairs. The authors concluded that the code started from an early GC-rich stage, limited to the codon–anticodon interactions of highest free energy, and evolved to its modern form by the gradual incorporation of weaker and weaker codon–anticodon interactions stabilized by the chemical modifications of the tRNA molecules in parallel with the evolution of the translational apparatus. The correlation between the average free energy of codon–anticodon helices (as defined by the nucleotides at first two codon positions) and the degeneracy of codon families (defined by the same two positions) suggests that unsplit codon families (boxes of 4) preceded split codon families (boxes of 2, exceptionally 3) during the evolution of the code. The fact that the known variations of the code [46, 47] are only found within codon families of lowest or intermediate energy and almost always in split families corroborates this view.
In the present work, I have deliberately ignored the structural and functional constraints of the protein-synthesizing machinery despite their obvious importance (see Discussion) to focus on the internal logic of triplet interactions assuming: (i) distinct pairing options at each of the three positions of the duplexes and (ii) the existence of hypothetical primitive RNA molecules composed of various sets of nucleobases. The results suggest the possible emergence of a precursor code built on primitive RNA molecules composed of a single purine (G or precursor) and two pyrimidines (C and U or precursors) if G ∙ U pairs were tolerated at positions 1 or 2 of codons. With a maximum of 27 codons, this code could have allowed the incorporation of 5 to 7 distinct a.a.s. in short peptides, depending on the chosen pairing option. Conservation of the same pairing options during expansion of the precursor code at the arrival of A (or precursor) produced a fully-coding hypothetical intermediate in which defined codons were interconnected to one another in a defined network due to their common interactions with some anticodons. Traces of this phenomenon, referred to here as codon intricacy (to distinguish from coding degeneracy) are visible into the standard and variant forms of the modern code. They demonstrate that the early codon–anticodon duplexes were obligately made of a Watson–Crick purine-pyrimidine pair at the central position (explaining its uniqueness) but not at the first and third positions of codons where weaker purine-pyrimidine interactions were initially tolerated. A chronological order of appearance of a.a.s and aaRS can be deduced from this evolutionary scheme, which is remarkably coherent and consistent with independent conclusions.
2. Deconstructing the modern genetic code reveals traces of a possible 27-codon precursor
Beside the specificities of the second position of codons briefly mentioned above and summarized in Supplementary Table S1, the modern genetic code reveals an additional asymmetry if one examines the 27 remaining codons (9 subfamilies of 3 codons each) after assuming an absence of some of the four nucleobases in RNA molecules (Table 1). In absence of C (a code built on hypothetical primitive RNA molecules made of G, A and U only), 2 codon families are unsplit (GGD for Gly and GUD for Val) and 7 are split between two a.a.s and/or stop codons. The absence of G (A, C and U primitive RNA) or U (G, A and C primitive RNA) gives more equilibrated results with, in each case, 5 unsplit families for 4 split ones (note that the AUH subfamily is unsplit contrary to the AUN family because all codons correspond to Ile in the standard code). By contrast, a code built on hypothetical primitive RNA molecules composed of G, C and U only (absence of A) shows 7 unsplit families for only 2 split families (and a total absence of any stop codon). Keeping the hypothesis that unsplit codon families in the modern code are more representative of its ancestral form than the split ones [39], this asymmetry favors the idea of a 27-codon precursor code built on primitive RNA molecules composed of G, C and U only. Note that 7 of the 8 unsplit families of the modern code are present in such a code (only the ACN family coding for Thr is missing, which does not necessarily exclude Thr from the early a.a.s, see below).
Deconstruction of the modern genetic code
| Composition of primitive RNA molecules | Missing nucleotide | Significance of remaining codons in the modern code | |
|---|---|---|---|
| Unsplit families | Split families | ||
| G + A + U | C | GAD > Asp + Glu | |
| AGD > Ser + Arg (Gly) | |||
| GGD > Gly | AAD > Asn + Lys | ||
| GUD > Val | AUD > Ile + Met | ||
| UGD > Cys + Trp + stop | |||
| UAD > Tyr + stop (Gln) | |||
| UUD > Phe + Leu | |||
| A + C + U | G | ACH > Thr | AAH > Asn + Lys |
| AUH$ > Ile | CAH > His + Gln | ||
| CCH > Pro | UAH > Tyr + stop (Gln) | ||
| CUH > Leu | UUH > Phe + Leu | ||
| UCH > Ser | |||
| G + A + C | U | GGV > Gly | GAV > Asp + Glu |
| GCV > Ala | AGV > Ser + Arg (Gly) | ||
| ACV > Thr | AAV > Asn + Lys | ||
| CGV > Arg | CAV > His+ Gln | ||
| CCV > Pro | |||
| G + C + U | A | GGB > Gly | |
| GCB > Ala | |||
| GUB > Val | UGB > Cys + Trp | ||
| CGB > Arg | UUB > Phe + Leu | ||
| CCB > Pro | |||
| CUB > Leu | |||
| UCB > Ser | |||
Starting from the modern code, the table indicates the remaining codon subfamilies if one of the four nucleotides were missing in hypothetical primitive RNA molecules. In all cases, 9 subfamilies of 3 codons remain with either D (A, G or U), H (A, C or U), V (A, G or C) or B (G, C or U) in codon position 3. Considering the significance of each codon in the modern code, the families are either unsplit (all three codons with identical significance) or split (distinct significance). Corresponding a.a.s are indicated in blue if activated by a class I aaRS or red if activated by a class II aaRS (note that Lys can be activated by an aaRS of either class depending on organisms, [48]). Brackets indicate encoded a.a.s in the variant forms of the code used in this work (Gln replaces stop codons in the UAN family, and Gly replaces Arg in the AGN family, see text). $ Contrary to the AUN family that encode both Ile and Met in the standard and many variant codes, the AUH subfamily is not split if one adopts its significance in the standard code (encodes Ile only) but remains split between Ile and Met in many variant forms of the code.
The G, C and U composition of primitive RNA molecules (absence of A) also appears the most favorable of the four possibilities in terms of the formation of RNA secondary structures because it allows the formation of one high energy pair (G–C) and one weak energy pair (G ∙ U). The possibility of forming two pairs of distinct free energies also exists for the G, A and U composition (absence of C) but with a lower energetic differentiation (A–U and G ∙ U). This possibility does not exist for the two other hypothetical compositions (absence of G or C). Further deconstruction of the modern genetic code assuming even more primitive RNA molecules made of only two nucleobases gives no more conclusive results, except that G and C could have been the earliest nucleobases as already proposed [39, 49, 50, 51].
3. Rationale of the present investigation
The above observation prompted me to further examine the possibility that the genetic code could have evolved from a 27-codon precursor that started as soon as primitive RNA molecules became able to form two distinct purine-pyrimidine pairs, differentiated by their free energies, i.e. contained three distinct nucleobases. This could have been achieved with a single purine able to pair with two pyrimidines (as in the G, C and U hypothesis above) or with a single pyrimidine able to pair with two purines (as with the G, A and U hypothesis above). Both possibilities have been examined but, to facilitate reading, the second one will be reserved for the Discussion. For the same reason, the possibility that some ancient nucleobases in the hypothetical primitive RNA molecules were not identical to the modern G, A, C and U (for example, presence of hypoxanthine (I) offers an interesting possibility) will only be examined in the Discussion.
The starting point of this work is the exhaustive examination of all pairwise interactions between all possible codons and anticodons formed in hypothetical RNA molecules composed of selected sets of purine (R) and pyrimidine (Y) nucleobases, assuming triplets of nucleotides and independent pairing options at each position of the triplet duplexes. This strategy was first applied to hypothetical 3-nucleobase primitive RNA molecules (composed of either 1R/2Y or 2R/1Y), forming 27 triplets generating 729 possible pairwise interactions. It was then extended to 4-nucleobase RNA molecules (composed of 2R/2Y), forming 64 triplets generating 4096 possible pairwise interactions. In each case, R–Y facing is demanded at each position of the triplets, reducing the number of pairwise interactions to examine to only 64 (4 × 4 × 4) or 512 (8 × 8 × 8) for 3 or 4 nucleobases, respectively. For each of these interactions, 3 pairing options, illustrated in Supplementary Figure S1A, were considered. In option 1, it is assumed that a high-energy R–Y pair (Watson–Crick type) is obligatory at position 2 of codons whereas a weaker pair (G ∙ U type) is also tolerated at position 1. In option 2, the high-energy pair is obligatory at position 1 of codons but a weaker pair is also tolerated at position 2. Finally, in option 1–2, it is assumed that a weaker pair is tolerated at position either 1 or 2 but not at both simultaneously (note that results of option 1–2 are equivalent to the sum of results of option 1 and option 2). In all options, all R–Y pairs are tolerated at position 3 of codons.
The resulting codon–anticodon pairing matrices harbor two intrinsic and fundamental properties: (i) a decoding ambiguityi.e. a same codon can be recognized by more than one anticodon and (ii) a codon intricacyi.e. distinct codons can be recognized by the same anticodon (Supplementary Figure S1B). The codons and anticodons affected by these properties differ between the pairing option chosen, but always remain precisely defined. This distinction is instrumental to compare the theoretical predictions with the modern code (see below).
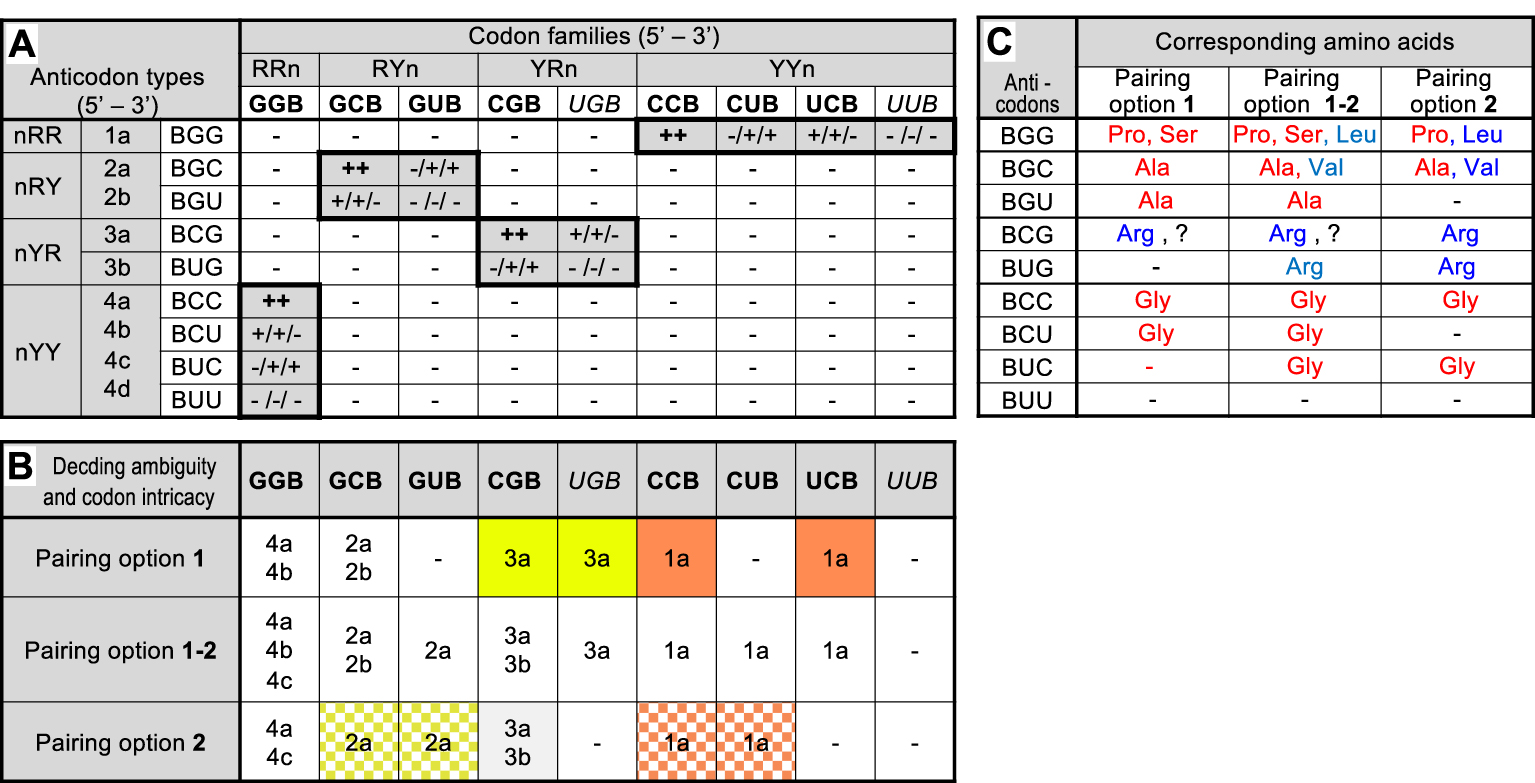
Codon–anticodon interaction matrix in the hypothetical 27-codon G, C, U precursor code and consequences. Part A: Interaction matrix between the 9 codon families (third line) and the 9 anticodon types (third column) that can be formed in primitive RNA molecules composed of the 3 nucleotides G, C and U if one ignores the third position of codon/first position of anticodon (B = not A). Codons and anticodons were classified according to their 5′ to 3′ sequences (R: purine, Y: pyrimidine, n: any nucleotide) and anticodon types have been arbitrarily numbered (second column). Codons in bold or italicized type correspond, respectively, to unsplit or split families in the modern code. Interactions in which all 3 positions of codon–anticodon duplexes involve a purine-pyrimidine pair are highlighted by shadowed boxes surrounded by thick lines. Note that a purine-pyrimidine pair is always assumed at the third position of codon/first position of anticodon i.e. the presented matrix is actually the sum of two independent but identical matrices respecting this condition. Predicted result of each pairwise interaction in terms of formation of an active codon–anticodon duplex are symbolized by ++ active pairing independent of chosen option (two G–C or C–G pairs at positions 1 and 2 of codons): + active pairing dependent of chosen option (one G–C or C–G pair and one G ∙ U or U ∙ G pair at positions 1 and 2 of codons); results are presented in the left, right or center for option 1, option 2 or option 1–2, respectively ; − no pairing (any other combination). Part B: Summary of predicted decoding ambiguity and codon family intricacy corresponding to each pairing option (see Supplementary Figure S1 for an example). The table indicates all anticodon type(s) (indicated by their numbers) predicted to read each codon family under each pairing option. Families with more than 1 anticodon type are potentially ambiguous. Families with no anticodon type (−) are potentially non-coding. Families sharing the same anticodon type are highlighted by similar color backgrounds (ignored for clarity for pairing option 1–2). Part C: Tentative association of a.a.s to anticodon types as deduced from the significance of codons of unsplit families in the standard form of the modern code (?: significance of codons in a split family). Amino acid color relates to the class of their respective aaRS (blue: class I, red: class II). − : inactive anticodon type (absence of cognate codon under the pairing option selected). Masquer
Codon–anticodon interaction matrix in the hypothetical 27-codon G, C, U precursor code and consequences. Part A: Interaction matrix between the 9 codon families (third line) and the 9 anticodon types (third column) that can be formed in primitive RNA molecules ... Lire la suite
4. Remarkable properties of the codon–anticodon pairing matrix of the 27-codon precursor code
The 27 possible triplets originating from the random assembly of nucleotides in primitive RNA molecules composed of G, C and U generate 729 pairwise interactions which can be simplified into 81 combinations if one ignores position 3 of codons (the 27 codons can be classified into 9 families of 3 codons each) and the first position of anticodons (in the 5′–3′ orientation). The resulting codon–anticodon interaction matrix (Figure 1A) exhibits 16 combinations where G and Y face each other at both positions 1 and 2 of codons (actually two such matrices exist if one also considers a required G–Y facing at position 3 of codons but both matrices have identical structures and need not be detailed here). Depending upon the pairing option selected for positions 1 and 2 of codons (here, the strong pair is G–C and the optional weak pair is G ∙ U), 6 (options 1 or 2) or 8 (option 1–2) of the 9 codon families are readable by the set of anticodons (UUB is always excluded, B = not A). The 6 families are GGB, GCB, CGB, UGB, CCB, and UCB under option 1 or GGB, GCB, GUB, CGB, CCB, and CUB under option 2. In both cases, the three remaining codon families (those with U in the second or first position, respectively) cannot form duplexes with any anticodon and are, therefore, predicted to be non-coding. The precursor code imagined here is, therefore, predicted to be potentially ca. 67% coding if options 1 or 2 are retained and ca. 89% coding if option 1–2 is retained, consistent with the synthesis of short peptides. Assuming that the 3 nucleobases are in equimolar amounts, such primitive RNA molecules would have had a reading continuity of 6 to 8 codons on average (depending on actual pairing option) i.e. could have been sufficient for the synthesis of the simplest peptide domains. Similarly, the pairing matrix shows that some of the 9 possible anticodon types remain unable to form duplex with any codon. They are BUG, BUC and BUU under pairing option 1, BGU, BCU and BUU under pairing option 2 and BUU alone under option 1–2. Such useless anticodons in the 27-codon precursor code may have formed a useful reservoir during subsequent code expansion (see below).
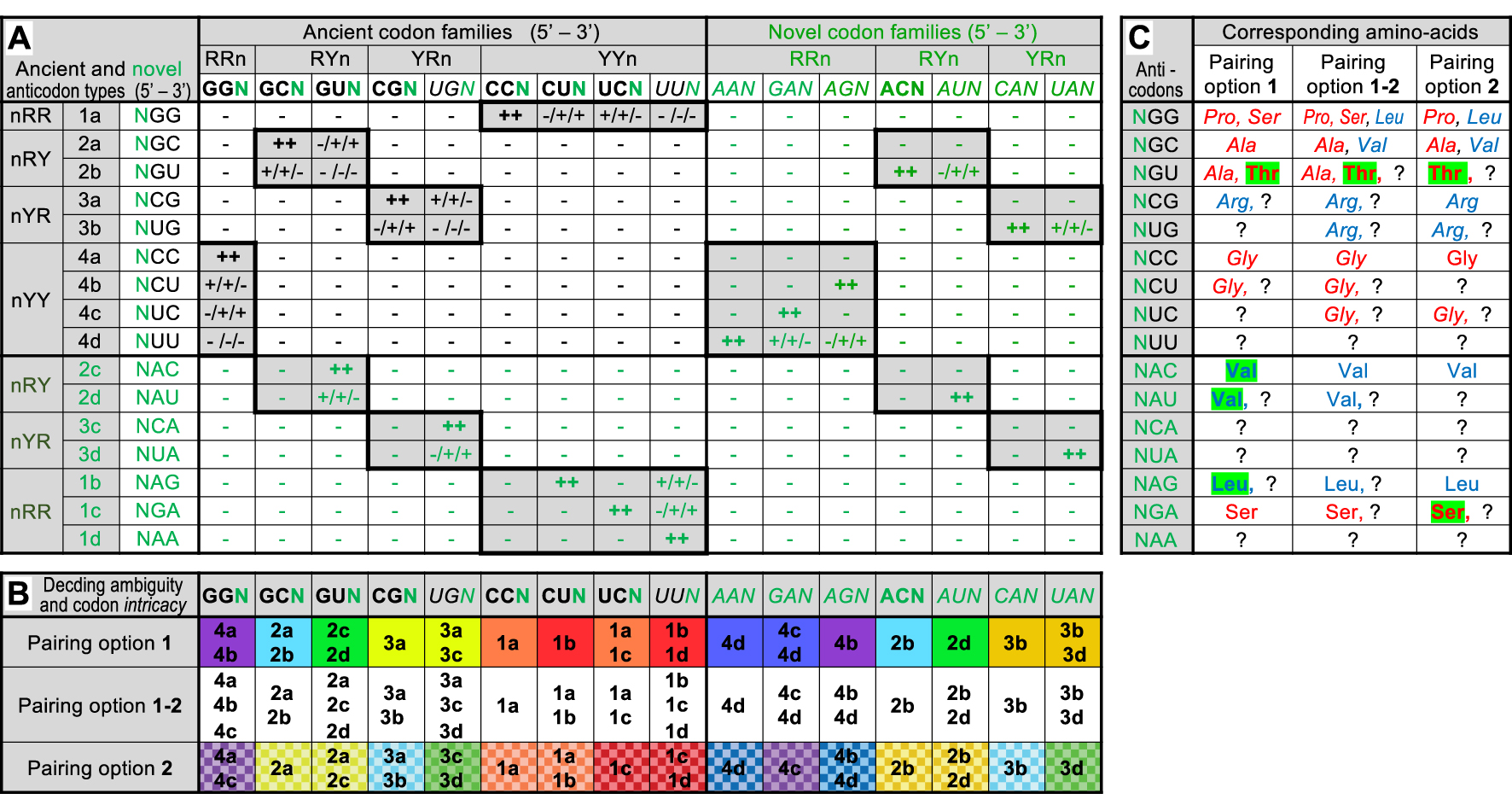
Codon–anticodon interaction matrix in the 64-codon G, C, U, A intermediate code and consequences. Part A: Same legend as Figure 1A except for symbols: ++ active pairing independent of chosen option (two G–C or C–G pairs, two A–U or U–A pairs, or one G–C or C–G and one A–U or U–A pairs at positions 1 and 2 of codons); + active pairing dependent of chosen option (one G–C, C–G, A–U or U–A pair plus one G ∙ U or U ∙ G pair at positions 1 and 2 of codons); − no pairing (any other combination). Novel codons, anticodons and their interactions are highlighted in green. Note that the upper left part of the matrix is identical to Figure 1A, except for N replacing B in ancient codon and anticodons families. Part B: Same legend as Figure 1B. Predicted codon family intricacy is visualized by color backgrounds (omitted for clarity under pairing option 1–2) with purple to green colors for purines and yellow to red colors for pyrimidines at the codon position responsible for the phenomenon. Part C: Same legend as Figure 1C. Novel a.a.s in the coding repertoire of each option (compare to Figure 1C) are highlighted over green backgrounds. Masquer
Codon–anticodon interaction matrix in the 64-codon G, C, U, A intermediate code and consequences. Part A: Same legend as Figure 1A except for symbols: ++ active pairing independent of chosen option (two G–C or C–G pairs, two A–U or U–A ... Lire la suite
The pairing matrix illustrates the phenomena of coding ambiguity and codon intricacy defined above. Figure 1B shows that 2 of the 6 readable codon families under pairing options 1 or 2 (not the same ones depending on the option) can be recognized by 2 distinct anticodon types each, suggesting a source of decoding ambiguity. The potentially ambiguous codon families are GGB and GCB for option 1 or GGB and CGB for option 2). Note that GGB, common to both options can be read by 3 distinct anticodon types under option 1–2. The prediction of a decoding ambiguity here is only tentative in absence of knowledge of the relationship between anticodons and a.a.s. More interestingly, the matrix also predicts that codons of distinct families are recognized by the same anticodon type, implying shared coding significance (intricacy) whichever a.a. may be concerned. Under options 1 or 2, the phenomenon affects 2 pairs of codon families, but it extends to 2 pairs plus 1 trio under option 1–2. The codon families predicted to share coding significance are CGB/UGB (sharing anticodon type BCG) and CCB/UCB (sharing anticodon type BGG) for pairing option 1 and GCB/GUB (sharing anticodon type BGC) and CCB/CUB (sharing anticodon type BGG) for pairing option 2. The phenomenon of codon intricacy plays a critical role in this work (see below).
Attempting to deduce which a.a.s could have been associated to this hypothetical precursor code is of course very difficult. Yet remarkable features emerge when one considers the significance of corresponding codons in unsplit families of the modern code (Figure 1C). If codon significance has been conserved, pairing option 1 suggests that Ala, Arg, Gly, Pro and Ser were present in the 27-codon precursor code. For reasons discussed later, it is unlikely that Arg, was an early a.a. (see Discussion). Ignoring it, it is notable that the four other a.a.s are activated by class II aaRS (and 3 of them by aaRS of the same subclass IIA, see below). Pairing option 2 predicts the presence of Ala, Arg, Gly, Leu, Pro and Val. The same remark holds for Arg but the replacement of Ser by Leu and Val eliminates the aaRS homogeneity (an argument in favor of option 1, see below). Pairing option 1–2 logically predicts the sum of all a.a.s. Note that the codon–anticodon interaction matrix built on hypothetical primitive RNA molecules made of G, A and U (Supplementary Figure S2) instead of G, C and U yields equivalent results in terms of coding capacity, potential decoding ambiguity and codon intricacy but its predictions in terms of coding repertoire are much poorer (see Discussion).
5. Extended codon–anticodon pairing matrix to 4-nucleobase RNA molecules
Building on the previous G, C, U precursor code, the arrival of a second purine within primitive RNA molecules extends it into a 64-codon structure if that second purine differs from G in its interactions with the two pyrimidines. A priori, two logical possibilities exist: either the novel purine (arbitrarily designated A′) pairs with both pyrimidines in an inverted order of free energies (A′–U higher than A′ ∙ C), hence creating a second low energy pair to consider in the pairing options, or it ignores the previously preferred pyrimidine (as does the actual A with C). To facilitate reading, only the second possibility is discussed here (Figure 2), the first one is illustrated by Supplementary Figure S3 and examined in the Discussion. For the same reasons as above, the theoretical matrix of 4096 interactions (64 × 64) can be reduced to 256 (16 × 16) combinations out of which only 64 correspond to R–Y facing at both the first and second position of codons (here again, two identical such matrices exist to accommodate R–Y facing at position 3 of codons).
Keeping the same pairing options as before, the G, C, U, A matrix exhibits two interesting features (Figure 2A). First, the novel codon families (those containing A) are immediately readable by the ancient set of anticodons (devoid of A). This is true for all 7 of them under the option 1–2 and for 6 of them under pairing options 1 (AUN ignored) or 2 (UAN ignored). This peculiarity was probably a critical factor for the successful evolution of the code as it allows the temporal continuity of peptide synthesis before novel anticodons (containing A) could be associated to a.a.s (see Discussion). With a total of 12 (options 1 or 2) or 15 (option 1–2) codon families immediately readable, the global coding capacity of the novel RNA molecules (containing A) is even slightly higher than that of the A-lacking ancient RNA molecules (75% instead of 67% under pairing options 1 or 2, and 94% instead of 89% under the option 1–2).
Second, only few novel anticodon types are required to extend the coding capacity of the intermediate code to 100% (all 16 codon families being readable). Of the 7 novel anticodon types (Figure 2A), only 3 are needed under pairing options 1 (NAC, NAU and NAG) or 2 (NCA, NUA and NGA), and only 1 is needed under pairing option 1–2 (either NAG, NGA or NAA). Such a parsimonious requirement of novel anticodons could also have been a critical issue for the successful evolution of the code if the association of novel anticodons with a.a.s were a rate-limiting process. Note that the full coding capacity of the intermediate code with a limited subset of anticodons is consistent with the always lower number of tRNA species compared to sense codons in modern organisms [38]. It also suggests that non-sense codons and release factors were not ancestral features of the code but represent evolved mechanisms (see Discussion).
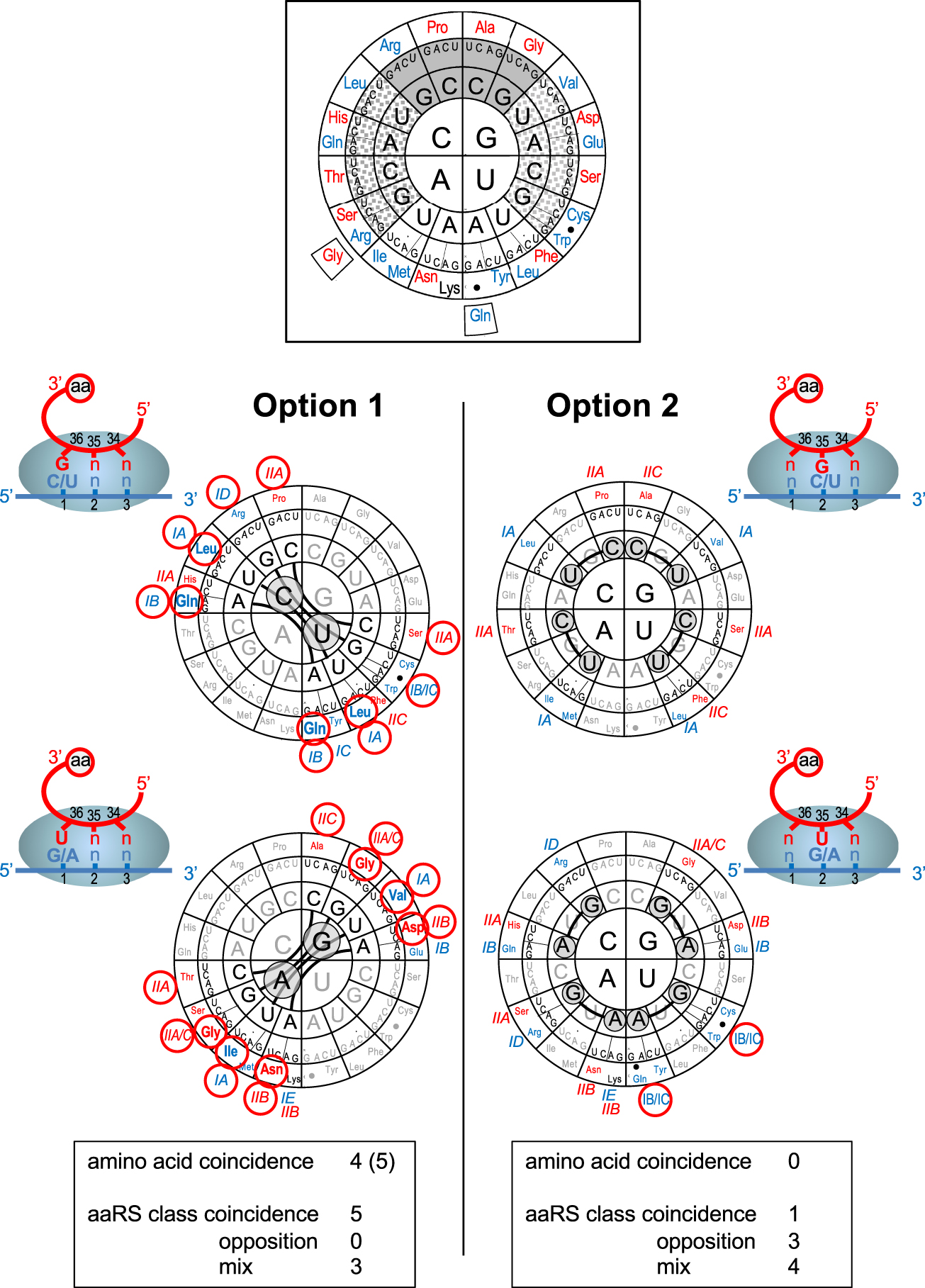
Predicted codon intricacy under pairing options 1 and 2. Top insert: The genetic code is represented according to [39], with nucleotides at positions 1, 2 and 3 of codons from the center to the periphery. Strong codon–anticodon helices (average −13.0 kJ/mole) are shown over plain grey background, weak codon–anticodon helices (average −4.2 kJ/mole) are shown over white background and intermediate ones (average −9.2 kJ/mole) over dotted grey background. Corresponding a.a.s are indicated in the outer circle (color code refers to aaRS class as in Table 1). Bold points indicate nonsense codons. Coding significance is shown for the standard code, the variant forms used in this work (see text) are shown on the periphery. Bottom part: Predicted codon intricacy under pairing options 1 and 2 (cartoons). Intricate pairs of codon families are visualized by thick lines. Nucleotides responsible for the phenomenon are circled over grey backgrounds (pyrimidines have been separated from purines for clarity). Amino acid and aaRS class coincidence between intricate pairs of codon families are highlighted by red circles (aaRS subclasses are according to [52]). Tables at bottom recapitulate results for each pairing option (see text and Supplementary Table S2). Masquer
Predicted codon intricacy under pairing options 1 and 2. Top insert: The genetic code is represented according to [39], with nucleotides at positions 1, 2 and 3 of codons from the center to the periphery. Strong codon–anticodon helices (average ... Lire la suite
The full coding capacity of the intermediate code is accompanied by a limited increase of the decoding ambiguity (Figure 2B). Half of the 16 codon families are recognized by 2 anticodon types under pairing options 1 or 2, the other 8 remain readable by a single anticodon type (under the option 1–2, only 4 codon families remain readable by a single anticodon type whereas 4 others are recognized by 3 anticodon types). But more importantly, the full coding capacity of the intermediate code is accompanied by a maximal increase of codon intricacy, each codon sharing an anticodon with at least one other codon of a distinct family (Figure 2B). Eight pairs of codon families are formed under pairing options 1 or 2 (not the same pairs between the two options) or 4 tetrads of codon families under the option 1–2. The 64-codon intermediate code is, therefore, fully intricate, a critical point in this work (see below). Note that in each pair (pairing options 1 or 2), one codon family is recognized by a single anticodon type, the other by the same anticodon type plus another one. In the tetrads (option 1–2), one codon family is recognized by a single anticodon type, two families by two anticodon types and the last family by three anticodon types.
As before, one can attempt to associate each anticodon type with one or a few a.a.(s) using the significance of codons in the unsplit families of the modern code as guide (Figure 2C). Note that all pairing options yield the same final list of 8 a.a.s (a tautological result), but their order of appearance differs when comparing to the precursor code (Figure 1C). Under option 1, Leu, Thr and Val are added to the earlier list of a.a.s (Ala, Gly, Pro, Ser and Arg?). Under option 2, Ser and Thr are added to Ala, Gly, Leu, Pro, Val and Arg?). Thr is common to both options and it is the only novel a.a. under the option 1–2. Because Thr corresponds to an ancient anticodon type (NGU, formerly BGU), it is tempting to imagine that it was already present in the 27-codon precursor code (associated to a BGU anticodon). This idea changes nothing under pairing option 2 because the BGU anticodon type is not active. But if pairing options 1 or 1–2 applied, the association of Thr to the BGU anticodon type instead of Ala as imagined on Figure 1C would have extended the coding repertoire by yet another a.a. activated by a class II aaRS (actually Thr is activated by an aaRS of subclass IIA, like Gly, Pro and Ser, further extending this already notable homogeneity).
6. Codon intricacy distinguishes between the pairing options
As mentioned above, the three alternative pairing options generate different predictions of codon intricacy (see Figure 2B). Now, when intricated codons are reported to the modern code (Figure 3), a drastically distinct picture emerges between option 1 and option 2 (and consequently option 1–2, not shown). Under option 1, multiple cases of coincidence of a.a.s and aaRS are observed compared to none (for a.a.s) or only one (for aaRS) for option 2. It looks as if the modern codon families had conserved the traces of the ancestral codon intricacy predicted under pairing option 1. This conservation is detailed in Supplementary Table S2.
The first example is represented by the presence of Leu in both the CUN codon family (unsplit) and the UUN codon family split between the UUR codons (encoding Leu) and the UUY codons (encoding Phe) in the standard and variant forms of the code. This can be interpreted as a reminiscence of the fact that, in the 64-codon intermediate (see Figure 2A), CUN and UUN codons were read by the same anticodon type (NAG, attributed to Leu), before the emergence of the new anticodon type (NAA) allowed the late introduction of Phe for the UUY codons only (because NAA is unable to read the CUN codons).
A second striking example of coding coincidence appears if one considers the variant form of the code used by nuclear genes in many unicellular eukaryotes of distinct evolutionary lineages such as Ciliates [53, 54], Diplomonads [55], Chlorophytes [56], Kinetoplastids [57] and others (reviewed in [46, 47, 58]. In all those species, the UAR codons encode Gln (and sometimes Glu) instead of serving as stop codons. Beside the intrinsic interest of the phenomenon of context dependent translational termination related to it [59], this coding deviation is remarkable because of the predicted intricacy of the UAR codons with the CAR codons which also encode Gln (Figure 3). Here again, this striking coincidence may be the reminiscence of shared pairing of the ancestral UAN and CAN codons with the anticodon type NUG before the new anticodon type (NUA) allowed the late introduction of Tyr for the UAY codons only (NUA does not read the CAN codons) and late evolution transformed UAR into stop codons, but not in all lineages of unicellular eukaryotes.
The next two coding coincidences concern the GAY and AAY codons, encoding Asp and Asn respectively, and the GUN and AUH (or AUY) codons, encoding Val and Ile, respectively. In both cases, the a.a.s are not identical but closely related. Asn may have replaced Asp on the AAY codons after emergence of the transamidation pathway to synthesize the aminoacyl-tRNAs [60, 61]. Similarly, the chemical proximity between Val and Ile (both activated by aaRS of subclass IA) makes it possible that the two ancestral GUN and AUN codon families, that shared the NAU anticodon type, were ambiguous for the two a.a.s.
Altogether, of the eight pairs of codon families predicted to have been intricated under pairing option 1, four exhibit obvious coincidences in the modern code when a.a.s are considered (Figure 3). A possible fifth coincidence is only mentioned here for memory because it only exists in one of the numerous variations observed in mitochondrial codes (most of which concern the wobble position). The AGR codons encode Gly in urochordate mitochondria [62], like its intricated codon family GGN. This coincidence, however, is probably more relevant of late codon reassignments than of the ancestral intricacy because the AGG codon encodes a variety of other a.a.s in the mitochondrial code of green algae [63] and the AGR codons encode Ser or are used as stop codons in other mitochondria.
The three last pairs of intricate codon families predicted under pairing option 1 (CGN/UGN, CCN/UCN and GCN/ACN) do not show obvious a.a. coincidence in the modern code, but hide remarkable coincidence if one examines the aaRS used to activate the encoded a.a.s (Supplementary Table S2). The first two pairs are derived from the earliest intricate pairs (CGB/UGB and CCB/UCB) in the 27-codon precursor code (see Figure 1). The CGN/UGN pair is very difficult to interpret because it is unlikely that Arg was the original a.a. and the UGN family is the most mysterious one in the modern code (see Discussion). Yet, all 3 encoded a.a.s of the modern code (Arg, Cys and Trp) are activated by class I aaRS (not the same subclasses, however). No obvious a.a. coincidence is available for the CCN/UCN pair but the two encoded a.a.s (Pro and Ser) are activated by aaRS of the same subclass IIA. It is possible that these two a.a.s were ambiguously encoded by both codon families. This decoding ambiguity may have been very ancestral because, in the 27-codon precursor code, the CCB and UCB codon families interacted with the same anticodon type (BGG, see Figure 1). A similar argument can be made for the GCN and ACN families because, as mentioned above, the BGU anticodon type may have been associated very early with Thr, creating an Ala-Thr decoding ambiguity for the GCB family before the formation of the GCN and ACN codon families. Note that Ala and Thr are also activated by class II aaRS (although not the same subclass).
At this stage, it becomes possible to eliminate pairing option 2 (and consequently option 1–2 as well) and to conclude about the existence of pairing option 1 as the rule for the codon–anticodon interactions during the first phases of the code. In other words, the first active codon–anticodon duplexes must have been made of an obligatory Watson–Crick pair at their central position but tolerated weaker purine-pyrimidine pairs at the two flanking positions before pairing stringency increased at position 1 of codons and, probably latter, at position 3. The late increased stringency at position 3 has resulted from the chemical modifications of tRNA molecules (see [39]). At position 1, it has probably resulted from the evolution of the translational apparatus (see Discussion). In the 27-codon precursor code imagined, the central position was, therefore, only occupied by a G–C or C–G pair (no difference is considered in this work) and, in the 64-codon stage, this position also accepted A–U or U–A pairs, but not G ∙ U or U ∙ G pairs. By contrast, both positions 1 and 3 accepted G ∙ U or U ∙ G pairs in addition to the Watson–Crick pairs. The same conclusion would hold true for hypothetical precursors of the modern nucleobases so long as their relative pairing preferences remain qualitatively similar and confer quantitatively sufficient free energy (see Discussion).
7. Reconstruction of the temporal order of amino acid and aaRS recruitment during evolution of the genetic code
Combining the postulated evolution of the nucleobase composition of primitive RNA molecules with the evolution of the pairing stringency at position 1 of codons defines three successive periods in the evolution of the code (Figure 4). During the first period, the relaxed G ∙ U or U ∙ G pairing at position 1 permitted to initiate a first code with primitive RNA molecules having a yet incomplete nucleobase composition. The synthesized peptides must have been short (18 of the 27 codons were potentially coding, see Figure 1) and made of a limited repertoire of a.a.s. The conservation of the same relaxed pairing rule while the nucleobase composition of RNA molecules was reaching completion generated a second period during which slightly longer peptides could have been synthesized by an ultimately 64-codon code without the need for an immediate expansion of the coding repertoire (48 of the 64 codons can be read by ancient anticodons). The dissociation between coding capacity and repertoire was probably critical in the successful evolution of the code. It allowed the gradual formation of novel anticodons to eventually bring the coding capacity to 100% (64 coding codons out of 64). It is quite possible that this second period lasted a long time before the pairing stringency increased at the first codon position, probably under the influence of the evolution of the ribosomes [64, 65]. This event created a transition to the third period during which some previously unsplit codon families became split by the increasing differentiation at the third codon position as previously proposed [39], hence permitting the completion of the coding repertoire. Note that, on a purely logical basis, the increased stringency at codon position 1 (necessary to eliminate the ancestral codon intricacy) and position 3 (necessary to reduce the coding degeneracy) are not linked. But the interactions of anticodons with the ribosomal grip may have established such a linkage [39].
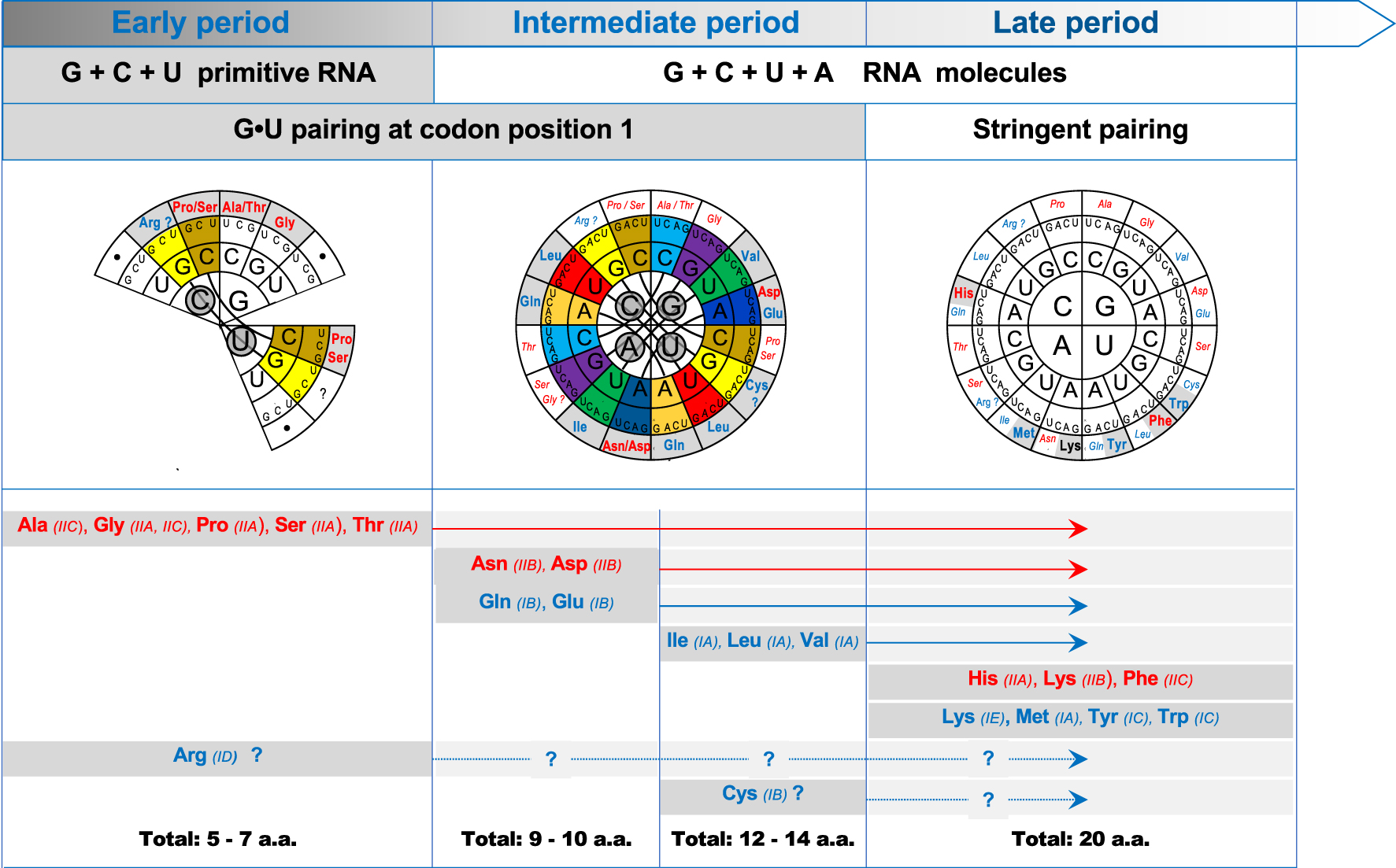
Reconstruction of the evolutionary steps of the genetic code. The figure illustrates the genetic code (drawn according to [39]) in the 3 successive periods predicted in this work on the basis of the nucleotide composition of primitive RNA molecules and G ∙ U/U ∙ G pairing tolerance at position 1 of codons (top). Codon family intricacy is visualized by colored sectors (see Figure 2) and thick lines with responsible nucleotides circled over grey backgrounds. Predicted significance of each codon family at each period is represented on the outer circle (color code of a.a.s as in Table 1, new a.a.s of the period are in bold type over grey backgrounds, black dots: non-coding codons). The evolution of the coding repertoire is recapitulated at the bottom part. The intermediate period has been subdivided based on the postulated usage of ancient anticodons before novel anticodons (see text and Table 2). A.a.s are listed alphabetically at their first period or subperiod of appearance in the coding repertoire (corresponding aaRS subclass are under brackets, color code as in Table 1). Note the uncertainty for Arg and Cys (see text) and the limited number of differences between the fully-coding code of the late period and the modern code. Masquer
Reconstruction of the evolutionary steps of the genetic code. The figure illustrates the genetic code (drawn according to [39]) in the 3 successive periods predicted in this work on the basis of the nucleotide composition of primitive RNA molecules and ... Lire la suite
Against this background, the evolution of the coding repertoire can be tentatively reconstructed a posteriori using the significance of codons in the modern code as guide. However, to do this, one first needs to determine the relative order of arrival of a.a.s in each split codon family during their hypothesized evolution from unsplit precursors. Two criteria can be used: (i) the remaining coding coincidence between intricate pairs of codon families (see above) and (ii) a priority of extant ancient anticodons before the formation of new ones to read the novel codons of the intermediate code. This priority postulate splits the intermediate period in two successive subperiods depending on the usage of ancient or novel anticodons (Figure 4). When both criteria are combined (Table 2), it can be concluded that Leu preceded Phe for the UUN family, Ile preceded Met for the AUN family, Gln preceded His and Tyr for the CAN and UAN families, respectively, Asp preceded Glu for the GAN family and Asn preceded Lys for the AAN family. Note that the biochemical relationship between Asp and Asn and the transamidation mechanism [60, 61] suggest a possible early ambiguity between these two amino acids for the AAN codon family. The same argument applies between Glu and Gln for the CAN and UAN codon families.
Sequential order of amino acid arrival within split codon families
| Ancient codons | Novel codons | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| UGN | UUN | AAN | GAN | AGN | AUN | CAN | UAN | ||
| Cys Trp | Phe Leu | Asn Lys | Asp Glu | Ser Arg | Ile Met | His Gln | Tyr (Gln) | ||
| Intricate family and coding significance | CGN Arg | CUNLeu | GAN Asp Glu | AAN Asn Lys | GGN Gly | GUNVal | UAN Tyr (Gln) | CANHis Gln | |
| Ancient active anticodon | NCU (Gly) | Ser Arg | |||||||
| Ancient inactive anticodon | NUU | Asn Lys | Asp Glu | ||||||
| NUC | Asp Glu | ||||||||
| NUG | His Gln | Tyr (Gln) | |||||||
| Novel anticodon | NAC | ||||||||
| NAU | Ile Met | ||||||||
| NCA | Cys Trp | ||||||||
| NUA | Tyr (Gln) | ||||||||
| NAG | Phe Leu | ||||||||
| NAA | Phe Leu | ||||||||
| Deduced order | First a.a. | ? | Leu | Asn/Asp | Asp | Ser/Gly ? | Ile | Gln/Glu | (Gln/Glu) |
| Last a.a. | ? | Phe | Lys | Glu | Arg ? | Met | His | Tyr | |
The 8 split (italics) codon families of the modern code sorted between ancient ones (lacking A, black) and novel ones (containing A, green) according to Figure 2 are listed in line 2 with their coding significance (left and right a.a.s are encoded by nnY and nnR codons, respectively; color code refers to aaRS class as in Table 1). Codon significance in the variant nuclear code used in this work is under brackets (see text). For each codon family, line 3 indicates its intricate partner predicted under pairing option 1 (same color code, bold type: unsplit family) with coding significance. Lines 4–7 indicate active pairing of ancient anticodon types (devoid of A) with novel codons after transition from the precursor code (see Figure 1) to the intermediate code (see Figure 2). Note that the NCU anticodon (formerly BCU) was tentatively attributed to Gly in the precursor code and that the 3 other anticodons were regarded as inactive for lack of matching codons. Lines 8–13 indicate active pairing of novel anticodon types (containing A) with ancient and novel codons, see Figure 2). Lines 14 and 15 recapitulate, for each split family, the order of appearance of a.a.s deduced from the combination of codon family intricacy and the postulate of ancient codon priority (see text).
The last two families, AGN and UGN, are more problematic. The significance of the AGR codons is highly variable in the mitochondrial genomes, varying between Ser, Gly and stop (see above). The AGG codon alone is even more variable, varying between Ala, Lys, and Met [63]. Together with the question of Arg (see Discussion), it seems more likely that the AGN family initially coded for Ser or, more probably, was ambiguous between Gly and Ser given its intricacy with the GGN family (note that Ser and Gly are activated by aaRS of the same subclass IIA). For the UGN family, the situation is even worse. Besides being the only codon family split in 3 parts in the standard code, the significance of its codons is also highly variable in the variant forms of the code. The non-sense codon UGA often encodes Trp and sometimes Cys in several nuclear as well as organellar codes. It can also encode Gly in bacteria and Selenocysteine (Sel) in specific cases. Although it is tempting to consider that this family initially encoded Cys, this cannot be formally deduced from the present work (see Discussion). Finally, application of the above postulate of ancient anticodon priority in the intermediate code leads to the conclusion that, in the intermediate period, Asn/Asp and Gln/Glu that use ancient anticodons arrived before Ile, Leu and Val that use novel anticodons (Table 2).
From above considerations, the chronology of events during the evolution of the code can be summarized as illustrated by Figure 4. Starting from a simple precursor code of 27 codons, 18 of which were potentially coding, the early period was compatible with the synthesis of short peptides composed of 5 a.a.s, Ala, Gly, Pro, Ser and Thr, plus at least one other a.a. corresponding to the CGB and UGB codons (the arrival of Arg and Cys in unclear, see Discussion). The 5 early a.a.s identified are all activated by aaRS of class II. This homogeneity is even more striking when one remembers that Pro, Ser and Thr are all activated by enzymes of subclass IIA, Ala is activated by an enzyme of subclass IIC and Gly is activated by two enzymes of, respectively, subclasses IIA and IIC [60, 52]. Enzymes of these 2 subclasses have subsequently been used for the activation of the late a.a.s. His and Phe, respectively.
At the transition to the intermediate period, the majority (30 of 37) of novel codons (containing A), had the immediate possibility of interacting with the ancient anticodons (lacking A). Since 3 ancient anticodon types (NUU, NUC and NUG) were potentially inactive (for lack of corresponding codon in the precursor code, see Figure 1), it is possible that they have contributed to the immediate expansion of the coding repertoire by the arrival of Asn and Asp, activated by a subclass IIB aaRS, and Gln and Glu the first a.a.s to be activated by class I aaRS (subclass IB). It is attractive to think that class I aaRS appeared during this period from the complementary strand of the same nucleic acid as the earlier class II aaRS as proposed by Rodin and Ohno [66]. Whatever the origin, it is remarkable that all 3 immediately subsequent a.a.s, Ile, Leu and Val (and possibly Cys as well, see Discussion) are all activated by class I aaRS. At this stage, the 64-codon intermediate code had reached its full coding capacity and its repertoire totalized at least 12 a.a.s (possibly 14 depending upon which a.a.s were encoded by the CGN and UGN intricated families, see Discussion). The full coding capacity permitted the synthesis of longer peptides. All codon families except perhaps GAN were unsplit, their intricacy was complete (8 pairs) and several families were probably ambiguous (GAN could have been ambiguous between Asp and Glu instead of being split).
At the transition to the late period, the increase of pairing stringency at position 1 of codons eliminated their intricacy and reduced the decoding ambiguity but a further expansion of the coding repertoire was not possible before the splitting of some codon families. With their coding significance as deduced above (see Table 2), 6 late a.a.s could be added (His, Lys, Met, Phe, Tyr and Trp) to the repertoire. As opposed to the remarkable homogeneity of the early periods, these a.a.s are activated by aaRS of either class I or class II in equal numbers (note that Lys is activated by two aaRS, one of each class).
The transition from the late period to the modern genetic code involved only minor changes in some (but not all) versions of the code such as installing UAR and UGA as stop codons instead of Gln and Trp, respectively, the assignment of Ile instead of Met to the AUA codon and of Arg (instead of Ser or Gly ?) to the AGR codons. The frequent variations observed for these codons, including the coding of pyrrolysine (Pyl) and selenocysteine (Sel), is consistent with this late evolution.
The evolution of the code presented here is, of course, schematic as it only relies on the internal logic of triplet pairing matrices in hypothetical primitive RNA molecules without concern for the actual molecular mechanisms involved in the decoding process. Yet, the predicted stepwise development of the coding repertoire is in excellent agreement with previous conclusions based on independent data such as the prebiotic abundance of a.a.s [16, 67], the complexity of their biosynthetic pathways [17] or their role in protein function [68, 69]. In particular, the chronological order obtained here matches remarkably well the suspected role of atmospheric oxygen in the selective recruitment of the late a.a.s, as deduced from their chemical reactivity [70]. It looks as if, the early and intermediate periods defined here correspond to the evolution of living cells in the reductive environment preceding the first accumulation of atmospheric oxygen while the late period started after this oxidative transition (see below).
8. Discussion
During more than five decades, the genetic code has been contemplated in multiple manners, in search of its logic and possible origin. The present work only adds a very modest contribution to an impressive list of previous investigations. Its main interest relies on the observation of numerous actual coincidences between pairs of codons whose ancestors were predicted to have been entangled (codon intricacy) under the pairing option 1 (see Figure 3). This pairing option highlights the special role played by the central position of the codon–anticodon duplexes compared to its two flanking positions. This idea is not novel, the specificity of the central position relative to the first one has been previously recognized with regard to codon assignment and mutational robustness of the code [28, 39, 42, 44, 71]. Its functional importance is further illustrated by the fact that, within the modern ribosomal decoding center, the extent of degeneracy tolerated at the third codon position is determined by the level of stability of the base pair at the central position [72].
The idea of a code starting with an obligatory high energetic pair at the central position of the codon–anticodon duplexes and less stringent requirements at the two flanking positions, followed by a subsequent increase of pairing stringency at the first and then the third positions of codons, has been briefly mentioned before as the 2.1.3 hypothesis [42, 73], but without a detailed analysis of its consequences. Here, I show that the tolerance of low energetic pairs at the first position of codons generated an initial intricacy between codons that has left so many traces in the modern code that it cannot have been otherwise. When, why and how the pairing stringency at position 1 has subsequently increased remains an open question. The answers are probably hidden in the three-dimensional interactions between the codon–anticodon duplex and the components of the modern ribosomes [39]. But it is interesting to remember that many years ago Weissenbach and colleagues [74] have discovered that a single yeast tRNALeu harboring the UAG anticodon was able to read all six leucine codons (CUN and UUR) in extracts of interferon-treated mouse cells, confirming the persistent functionality of a G ∙ U pair at the first position of codons in the modern code, at least under these conditions.
Similarly, the idea of a sequential order of appearance of the nucleobases into primitive RNA molecules is not novel. It is even central to investigations on the RNA world and the prebiotic formation of purines and pyrimidines [75, 76, 77]. But the late arrival of A relative to the three other nucleobases, as proposed here, has not been previously considered. A stepwise construction of the 4-nucleobase code from a GC-only precursor has been proposed with a G–C–A intermediate [77]. Unfortunately, this composition does not allow the formation of two distinct base pairs in primitive RNA duplexes, a fundamental aspect to initiate an active code, as shown in this work. This formation is possible in the hypothetical G, A and U composition of the primitive RNA world recently examined [78]. The complete codon–anticodon matrix constructed with hypothetical primitive RNA molecules composed of G, A and U (Supplementary Figure S2) yields equivalent numerical predictions in terms of coding capacity, ambiguity and codon intricacy as the precursor code proposed in Figure 1. But its coding repertoire is extremely difficult to predict because most of the codon families predicted to be ancient are split in the modern code, suggesting that split families preceded unsplit ones instead of the opposite. This leaves the hypothetical G, C and U composition of primitive RNA molecules proposed here as the best possibility. Furthermore, the fact that A (or its deaminated derivative I) is rarely found at the wobble position of modern anticodons [11, 38, 39] is consistent with the idea that active anticodons existed before the arrival of A.
The major difficulty with 3-nucleobase molecules is that replication cannot proceed by base complementarity, as previously discussed for primitive RNA of G, A and U composition [78]. The same difficulty exists for the G, C and U composition. One possibility would be that the early RNA molecules on which the genetic code emerged (that may have been very short) were not replicating by classical base complementarity but simply synthesized more or less randomly or with the a.a.s themselves serving as chemical guides as imagined in the stereochemical theory. However, if this possibility is relatively easy to imagine for the anticodons, it is obviously more difficult for the codons as some degree of conservative replication is needed to start a hereditary process. Therefore, one can further speculate that the actual starting point of the code was not a 27-codon precursor as proposed (Figure 1) but the encountering between two distinct sources of primitive RNA molecules of different compositions. The first one, that eventually would led to anticodons and tRNA molecules, was initially composed of G, C, U nucleotides and a.a.s, and synthesized chemically. The second one was composed of the four nucleobases and able to replicate by base complementarity and eventually led to viruses and primitive mRNA molecules (and subsequently genes). If so, the precursor code could have been made of 27 anticodons, as proposed, but 64 codons instead of 27. This possibility does not significantly alter the predicted coding capacity of the precursor code because, as discussed before for the intermediate code, most codon families can be read by the limited set of ancient anticodon types (Figure 2).
Beside RNA replication, the hypothetical absence of A in the early phase of the code looks also difficult to imagine considering its critical role in the modern molecular mechanisms of decoding. For example, A is present in the NCCA extension of the tRNA acceptor stem without which a.a.s. could not be activated (in addition to the need of ATP for the reaction). Similarly, A is present in the universally conserved C ∙ A pair within the peptidyl transferase center of the ribosomal RNA and is also present in the ribosomal grip [39, 79]. But the part of the genetic code examined here only concerns the codon–anticodon interactions (where A is not needed), not the catalytic machineries of a.a. activation and peptide bond formation (where A is needed). It is unclear how these different parts joined one another at the origin but, as mentioned above, it cannot be excluded that they initially emerged from distinct pools of primitive RNA molecules. The divergent prebiotic formation of the purines and pyrimidines [76] and the complex prebiotic chemistry at the origin of primitive RNA molecules [80], may have generated differences in their initial composition.
From the purely logical point of view, the early nucleobases of the primitive RNA molecules that initiated the precursor code did not necessarily need to be identical to the nucleobases of modern RNA molecules. The only requirement is that two purine-pyrimidine pairs of different free energies can be formed in triplet duplexes. However, beside hypoxanthine (I), the choice appears limited [81]. Inosine (I) also comes naturally to mind as the common precursor of both adenosine and guanosine. But its lower energetic differentiation (compared to G) for the two pyrimidines would reduce the distinction between positions 1 and 2 of codons which appears so important here (the codon–anticodon pairing matrix with I instead of G would be equivalent to the pairing option 1–2 but with the two weak pairs simultaneously tolerated). Similarly, the theoretical possibility that two hypothetical purines existed simultaneously with opposite pairing preferences for the two pyrimidines cannot be disregarded. This possibility has been examined here (Supplementary Figure S3). Its predictions are equivalent in terms of codon intricacy to those of the proposed 64-codon intermediate with the four modern nucleobases (only the coding degeneracy is increased). But it is, of course, impossible to predict the coding repertoire unless each purine is associated to G or A, respectively (as shown in Supplementary Figure S3 to allow direct comparison with Figure 2).
An important aspect of the present proposal directly contradicts common ideas on the origin of the code. The 64-codon intermediate was entirely coding, at least at some point in time, leaving no space for stop codons. This idea is opposite to the hypothesis according to which the extension of the coding repertoire relied on the gradual takeover of earlier stop codons under a selective pressure to form longer peptides [82]. However, it seems unlikely that termination factors were already existing in the earliest periods of the code when some codons remained non-coding for the mere lack of corresponding anticodons. Instead, it can be argued that it is the late arrival of these factors that helped the formation of novel stop codons from previously coding ones, by analogy to what is observed in the multiple variant forms of the modern code [83]. Another important aspect of the present proposal differs from the common view of a generalized primordial ambiguity that would have been gradually reduced as the translational machinery evolved [84, 85] or as novel codons gradually emerged [86]. Here, the predicted decoding ambiguity always remains precisely circumscribed to specific codons at every stage of the evolution of the code. Yet, if precisely defined, the overall decoding ambiguity of the code cannot be quantitatively estimated in absence of any knowledge about the relative concentrations of the distinct codons and anticodons in the pools of primitive RNA molecules.
Overall, the best argument for the rationale used in this work is the remarkable consistency of its predictions on the evolution of the coding repertoire with previous conclusions drawn from considerations of prebiotic chemistry or the biosynthesis and chemical reactivity of a.a.s [16, 17, 67, 70, 87]. All a.a.s predicted here to have appeared in the late period of a theoretical evolutionary scheme that is solely defined by the codon–anticodon interactions under a precise pairing option (see Figure 4) correspond to those predicted to have been selected into proteins after the major oxidative shift generated by the first accumulation of atmospheric oxygen [70]. Among the 14 other a.a.s, whose first occurrence is predicted here in the early or intermediate periods, 12 correspond to the early set according to the same criterion. The last two, Arg and Cys, raise questions. This problem is not new. Back in 1989, Taylor and Coates [42] noted that Arg does not fit the hydrophobicity pattern of other a.a.s with respect to the codon table. Furthermore, Arg remains a rare a.a. in proteins (on average) despite having 6 codons. And those codons are the major contributor to the variation of codon usage that exists across the domains of life [88]. In this work, Arg is predicted to have appeared in the early period but its class I aaRS put it in opposition to all other a.a.s of the same period. Furthermore, the multistep biosynthetic pathway of Arg is more consistent with a relatively late a.a. than an early one [17]. The fact that, in aptamer experiments, Arg largely dominates all other a.a.s in RNA interactions [26] probably explains its unique place in the evolution of the code. Arg could have replaced another a.a., now disappeared from proteins, as soon as the evolution of biochemical pathways allowed its synthesis, perhaps in multiple occasions. Its biosynthetic precursor ornithine (Orn) has been proposed in place of Arg in the ancestral code [77]. The case of Cys is also problematic. In the modern code, it is encoded by the UGN codon family that, according to this work, was previously intricated with the CGN codon family (encoding Arg or Orn?). The UGN family also encodes Trp and contains a stop codon, UGA, in the standard code. However, UGA often encodes Cys or Trp, and sometimes Gly, in the variant codes. And it encodes Sel in specific circumstances. According to its chemical reactivity [70], Cys should be a late a.a. that is considered to have played a major role in the adaptation of proteins to the biospheric oxygenation [89]. But the presence of sulfur may also have played a key role in the early phases of life [90]. In this work, Cys has been arbitrarily attributed to the intermediate period (see Figure 4) but without clear-cut argument (see Table 2). It is also possible that it is one of the late a.a. or, to the contrary, was already present in the early period since the UGB family remained non assigned.
The consistency of the coding repertoire predicted here also indicates that, besides the few exceptions mentioned above, codon assignment has only undergone limited changes during the long evolution of the code, as imagined from the very beginning [3]. This idea is, of course, counterbalanced by the flexibility demonstrated in many examples [47, 58, 62]. Over 30 different codes have now been listed1, 2 and it looks likely that this ever-growing list is far from complete. Codon reassignment can occur by several mechanisms and the results may be selected for various reasons. But globally, the frozen code melted with such elegance and parsimony that not all its historical traces were irreversibly erased.
Acknowledgments
I thank A. Danchin, M. Delarue, C. Fairhead, G. Fischer, C. Gaillardin, H. Grosjean, R. Koszul, B. Llorente, C. Marck, G. Pelletier, G-F. Richard, J-L. Souciet, J. Weissenbach and E. Westhof for insightful discussions and critical reading of the manuscript. I am indebted to E. Westhof for sharing unpublished material.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crbiol.47 or from the author.
French version
1. Introduction
Plus d’un demi-siècle après l’élucidation du tableau des codons [1], l’origine du code génétique reste la question la plus fascinante de tous les processus biologiques dans lesquels des produits sont nécessaires pour synthétiser les éléments de leur propre synthèse. Cette difficulté conceptuelle est ici accentuée par l’interaction entre deux classes distinctes de molécules, l’ARN et les protéines, dont les origines et les interactions élargissent le spectre des possibilités. Depuis les premiers examens de la table des codons [2, 3, 4] jusqu’aux résultats les plus récents sur les détails chimiques de la machinerie traductionnelle [5, 6] et les phylogénies de ses éléments fonctionnels [7, 8, 9, 10, 11], l’origine possible du code génétique a été explorée de différents points de vue (voir [12]). Trois grandes théories ont émergé. Dans la théorie de la coévolution [13, 14, 15], la principale force motrice de l’évolution du code génétique est recherchée dans l’émergence séquentielle de nouveaux acides aminés (a.a.s, ci-après) au sein des systèmes biochimiques primordiaux. On soutient que les a.a.s ayant les chaînes latérales les plus simples ou nécessitant le plus petit nombre d’étapes biochimiques pour leur biosynthèse, ainsi que ceux existant dans les environnements prébiotiques [16], sont probablement les premiers à être entrés dans le répertoire de codage. La corrélation entre les voies de biosynthèse des a.a.s et la classe d’ARNt synthétases (aaRS) les activant [17] apporte un soutien à cette hypothèse. Par ailleurs, la théorie stéréochimique postulée il y a longtemps [18, 19], qui met l’accent sur l’existence d’une affinité chimique entre les a.a. et les molécules d’ARN pour expliquer le tableau des codons, a également reçu un soutien ultérieur [20, 21, 22, 23, 24] et peut maintenant être testée par l’évolution expérimentale des aptamères [25, 26]. Enfin, la théorie de la minimisation des erreurs postule que la table des codons a été sélectionnée au cours de l’évolution de manière à minimiser les effets des erreurs de mutation et de traduction [27, 28, 29, 30]. Cette idée, qui s’oppose à la vision originale d’un accident gelé [3], a été soumise à des évaluations théoriques récentes [31, 32, 33, 34, 35], en tenant compte également des variantes des tables de codons dans divers organismes [36].
Au cours des dernières décennies, la table de codons standard a fait l’objet de nombreuses recherches visant à déterminer l’origine du code dans sa logique interne. L’hypothèse wobble formulée il y a longtemps [37] pour expliquer la dégénérescence générale du codage dans les boîtes de deux ou quatre codons (exceptionnellement trois) a évolué vers des listes exhaustives d’espèces d’ARNt déduites des séquences complètes des génomes et des descriptions détaillées de leurs modifications chimiques et de leurs interactions avec les ribosomes pour expliquer la spécificité de la position du troisième codon par rapport aux deux premiers [38, 39, 40]. Les règles d’attribution des codons qui ont été recherchées dans les corrélations entre les positions des a.a.s et celles des nucléotides dans les codons à la même période précoce [41], ont permis de mettre en évidence la deuxième position des codons comme un déterminant majeur dans la différenciation entre les a.a. hydrophobes et hydrophiles [42]. L’observation que tous les codons avec C en deuxième position codent des a.a.s activés par des aaRS de classe II [43], alors que presque tous ceux avec U codent des a.a.s activés par des aaRS de classe I (ceux avec G ou A sont mélangés à parts égales) a suggéré un modèle de différenciation par des choix binaires asymétriques successifs comme origine du code [44]. Une autre distinction de la position centrale du codon par rapport à la première (et bien sûr à la troisième) est également suggérée par le fait que tous les codons avec C à cette position appartiennent à des boites de 4 codons complètement dégénérées (familles non scindées de codons) alors que tous les codons avec A à cette position appartiennent à des boites à 2 codons (familles scindées de codons). Les deux types de familles sont en nombre égal pour les codons avec G ou U à la position centrale. Aucune des asymétries ci-dessus n’existe pour la première position du codon.
Récemment, un progrès majeur dans notre vision du code génétique et du processus de décodage a été réalisé avec le réexamen complet de la thermodynamique des interactions codon–anticodon en tenant compte des modifications chimiques connues des molécules d’ARNt et des interactions structurelles des duplex codon–anticodon avec l’appareil de traduction [39]. Trois classes de familles de codons ont été définies en fonction de l’énergie libre calculée correspondant à l’appariement des deux premières positions des codons avec les anticodons. Les valeurs moyennes vont de −13 kJ/mole pour deux paires G–C à −4,2 kJ/mole pour deux paires A–U avec un chiffre intermédiaire de −9,2 kJ/mole pour une paire G–C et une paire A–U. De plus, en considérant les caractéristiques structurelles de l’épingle à cheveux des ARNt portant l’anticodon dans ses interactions avec le site de décodage ribosomique [45], ce travail a montré comment des modifications spécifiques de l’ARNt déterminent des structures non canoniques en position wobble pour équilibrer la stabilité thermodynamique entre les paires synonymes codon–anticodon. Les auteurs ont conclu que le code a commencé à un stade précoce riche en GC, limité aux interactions codon–anticodon de plus haute énergie libre, et a évolué vers sa forme moderne par l’incorporation progressive d’interactions codon–anticodon de plus en plus faibles stabilisées par les modifications chimiques des molécules d’ARNt en parallèle avec l’évolution de l’appareil de traduction. La corrélation entre l’énergie libre moyenne des hélices codon–anticodon (telle que définie par les nucléotides aux deux premières positions du codon) et la dégénérescence des familles de codons (définies par les deux mêmes positions) suggère que les familles non scindées de codons (boites de 4) ont précédé les familles scindées (boites de 2, exceptionnellement 3) au cours de l’évolution du code. Le fait que les variations connues du code [46, 47] ne se retrouvent qu’au sein des familles de codons de niveaux d’énergie plus faible ou intermédiaire, et presque toujours dans les familles scindées corrobore ce point de vue.
Dans le présent travail, j’ai délibérément ignoré les contraintes structurelles et fonctionnelles du mécanisme de synthèse des protéines malgré leur importance évidente (voir Discussion) pour me concentrer sur la logique interne des interactions entre triplets en supposant: (i) des options d’appariement distinctes à chacune des trois positions des duplex et (ii) l’existence de molécules d’ARN primitives hypothétiques composées de divers ensembles de nucléobases. Les résultats suggèrent l’émergence possible d’un code précurseur construit sur des molécules d’ARN primitives composées d’une seule purine (G ou précurseur) et de deux pyrimidines (C et U ou précurseurs) si les paires G ∙ U étaient tolérées aux positions 1 ou 2 des codons. Avec un maximum de 27 codons, ce code aurait pu permettre l’incorporation de 5 à 7 a.a.s. distincts dans des peptides courts, selon l’option d’appariement choisie. La conservation des mêmes options d’appariement pendant l’expansion du code précurseur à l’arrivée de A (ou son précurseur) a produit un intermédiaire hypothétique entièrement codant dans lequel les codons étaient interconnectés les uns aux autres dans un réseau défini en raison de leurs interactions communes avec certains anticodons. Les traces de ce phénomène, appelé ici « intrication des codons » (pour le distinguer de la dégénérescence de codage) sont visibles dans les formes standard et variantes du code moderne. Elles démontrent que les premiers duplex codon–anticodon étaient obligatoirement constitués d’une paire purine-pyrimidine de type Watson–Crick en position centrale (ce qui explique son caractère unique) mais pas en première ni en troisième position des codons où des interactions purine-pyrimidine plus faibles étaient initialement tolérées. Un ordre chronologique d’apparition des a.a.s et des aaRS peut être déduit de ce schéma évolutif, qui est remarquablement cohérent et conforme aux conclusions indépendantes.
2. La déconstruction du code génétique moderne révèle des traces d’un possible précurseur de 27 codons
Outre les spécificités de la deuxième position des codons brièvement mentionnées ci-dessus et résumées dans le tableau supplémentaire S1, le code génétique moderne révèle une asymétrie supplémentaire si l’on examine les 27 codons restants (9 sous-familles de 3 codons chacune) après avoir supposé une absence de l’une ou l’autre des quatre bases azotées dans les molécules d’ARN (Tableau 1). En l’absence de C (un code construit sur d’hypothétiques molécules d’ARN primitives composées uniquement de G, A et U), 2 familles de codons sont non scindées (GGD pour Gly et GUD pour Val) et 7 sont scindées entre deux a.a.s et/ou stop. L’absence de G (ARN primitif A, C et U) ou de U (ARN primitif G, A et C) donne des résultats plus équilibrés avec, dans chaque cas, 5 familles non scindées pour 4 scindées (à noter que la sous-famille AUH est non scindée contrairement à la famille AUN car tous les codons correspondent à Ile dans le code standard). En revanche, un code construit sur des molécules d’ARN primitives hypothétiques composées uniquement de G, C et U (absence de A) présente 7 familles non scindées pour seulement 2 familles scindées (et une absence totale de tout codon stop). En gardant l’hypothèse que les familles de codons non scindées dans le code moderne sont plus représentatives de sa forme ancestrale que les familles scindées [39], cette asymétrie favorise l’idée d’un code précurseur à 27 codons construit sur des molécules d’ARN primitives composées de G, C et U seulement. Notons que 7 des 8 familles non scindées du code moderne sont présentes dans un tel code (seule la famille ACN codant pour Thr est manquante, ce qui n’exclut pas nécessairement Thr des premiers a.a.s, voir ci-dessous).
Déconstruction du code génétique moderne
| Composition of primitive RNA molecules | Missing nucleotide | Significance of remaining codons in the modern code | |
|---|---|---|---|
| Unsplit families | Split families | ||
| G + A + U | C | GAD > Asp + Glu | |
| AGD > Ser + Arg (Gly) | |||
| GGD > Gly | AAD > Asn + Lys | ||
| GUD > Val | AUD > Ile + Met | ||
| UGD > Cys + Trp + stop | |||
| UAD > Tyr + stop (Gln) | |||
| UUD > Phe + Leu | |||
| A + C + U | G | ACH > Thr | AAH > Asn + Lys |
| AUH$ > Ile | CAH > His + Gln | ||
| CCH > Pro | UAH > Tyr + stop (Gln) | ||
| CUH > Leu | UUH > Phe + Leu | ||
| UCH > Ser | |||
| G + A + C | U | GGV > Gly | GAV > Asp + Glu |
| GCV > Ala | AGV > Ser + Arg (Gly) | ||
| ACV > Thr | AAV > Asn + Lys | ||
| CGV > Arg | CAV > His+ Gln | ||
| CCV > Pro | |||
| G + C + U | A | GGB > Gly | |
| GCB > Ala | |||
| GUB > Val | UGB > Cys + Trp | ||
| CGB > Arg | UUB > Phe + Leu | ||
| CCB > Pro | |||
| CUB > Leu | |||
| UCB > Ser | |||
En partant du code moderne, le tableau indique les sous-familles de codons restantes si l’un des quatre nucléotides était manquant dans d’hypothétiques molécules d’ARN primitif. Dans tous les cas, 9 sous-familles de 3 codons restent avec soit D (A, G ou U), H (A, C ou U), V (A, G ou C) ou B (G, C ou U) en position 3 des codons. Compte tenu de la signification de chaque codon dans le code moderne, les familles sont soit non scindées (les trois codons ont une signification identique), soit scindées (signification distincte). Les a.a.s correspondants sont indiqués en bleu s’ils sont activés par une aaRS de classe I ou en rouge s’ils sont activés par une aaRS de classe II (notez que Lys peut être activé par une aaRS de l’une ou l’autre classe selon les organismes, [48]). Les parenthèses indiquent les a.a.s codés dans les variantes du code utilisé dans ce travail (Gln remplace les codons stop dans la famille UAN, et Gly remplace Arg dans la famille AGN, voir le texte). $ Contrairement à la famille AUN qui code à la fois Ile et Met dans le code standard et dans de nombreuses variantes du code, la sous-famille AUH n’est pas scindée si l’on adopte sa signification dans le code standard (ne code que Ile) mais reste scindée entre Ile et Met dans de nombreuses variantes du code.
La composition G, C et U des molécules d’ARN primitives (absence de A) semble également la plus favorable des quatre possibilités en termes de formation de structures secondaires d’ARN car elle permet la formation d’une paire à haute énergie (G–C) et d’une paire à faible énergie (G ∙ U). La possibilité de former deux paires d’énergies libres distinctes existe également pour la composition G, A et U (absence de C) mais avec une différenciation énergétique plus faible (A–U et G ∙ U). Cette possibilité n’existe pas pour les deux autres compositions hypothétiques (absence de G ou C). Une déconstruction plus poussée du code génétique moderne en supposant des molécules d’ARN encore plus primitives, composées de seulement deux nucléobases, ne donne pas de résultats plus concluants, sauf que G et C pourraient avoir été les premières nucléobases, comme cela a déjà été proposé [39, 49, 50, 51].
3. Justification de la présente enquête
L’observation ci-dessus m’a incité à examiner plus avant la possibilité que le code génétique ait pu évoluer à partir d’un précurseur de 27 codons qui a commencé dès que des molécules d’ARN primitif sont devenues capables de former deux paires purine-pyrimidine distinctes, différenciées par leurs énergies libres, c’est-à-dire contenant trois nucléobases distinctes. Cela aurait pu être réalisé avec une seule purine capable de s’associer avec deux pyrimidines (comme dans l’hypothèse G, C et U ci-dessus) ou avec une seule pyrimidine capable de s’associer avec deux purines (comme dans l’hypothèse G, A et U ci-dessus). Les deux possibilités ont été examinées mais, pour faciliter la lecture, la seconde sera réservée à la discussion. Pour la même raison, la possibilité que certaines bases azotées anciennes dans les molécules d’ARN primitives hypothétiques ne soient pas identiques aux G, A, C et U modernes (par exemple, la présence d’hypoxanthine (I) offre une possibilité intéressante) ne sera examinée que dans la Discussion.
Le point de départ de ce travail est l’examen exhaustif de toutes les interactions par paires entre tous les codons et anticodons possibles formés dans des molécules d’ARN hypothétiques composées d’ensembles sélectionnés de purines (R) et de pyrimidines (Y), en supposant des triplets de nucléotides et des options d’appariement indépendantes à chaque position des duplex des triplets. Cette stratégie a d’abord été appliquée à des molécules d’ARN primitives hypothétiques à 3 nucléobases (composées de 1R/2Y ou 2R/1Y), formant 27 triplets générant 729 interactions possibles deux à deux. Elle a ensuite été étendue aux molécules d’ARN à 4 nucléobases (composées de 2R/2Y), formant 64 triplets générant 4096 interactions possibles deux à deux. Dans chaque cas, une interaction R–Y est exigée à chaque position des triplets, réduisant le nombre d’interactions possibles à examiner à seulement 64 (4 × 4 × 4) ou 512 (8 × 8 × 8) pour 3 ou 4 nucléobases, respectivement. Pour chacune de ces interactions, 3 options d’appariement, illustrées dans la figure supplémentaire S1A, ont été envisagées. Dans l’option 1, on suppose qu’une paire R–Y à haute énergie (type Watson–Crick) est obligatoire en position 2 des codons alors qu’une paire plus faible (type G ∙ U) est également tolérée en position 1. Dans l’option 2, la paire à haute énergie est obligatoire en position 1 des codons, mais une paire plus faible est également tolérée en position 2. Enfin, dans l’option 1–2, on suppose qu’une paire plus faible est tolérée en position 1 ou 2, mais pas aux deux simultanément (il convient de noter que les résultats de l’option 1–2 sont équivalents à la somme des résultats de l’option 1 et de l’option 2). Dans toutes les options, toutes les paires R–Y sont tolérées à la position 3 des codons.
Les matrices d’appariement codon–anticodon qui en résultent présentent deux propriétés intrinsèques et fondamentales : une ambiguïté de décodage, c’est-à-dire qu’un même codon peut être reconnu par plus d’un anticodon et une intrication des codons, c’est-à-dire que des codons distincts peuvent être reconnus par le même anticodon (Figure supplémentaire S1B). Les codons et anticodons affectés par ces propriétés diffèrent selon l’option d’appariement choisie, mais restent toujours définis avec précision. Cette distinction est essentielle pour comparer les prédictions théoriques avec le code moderne (voir ci-dessous).
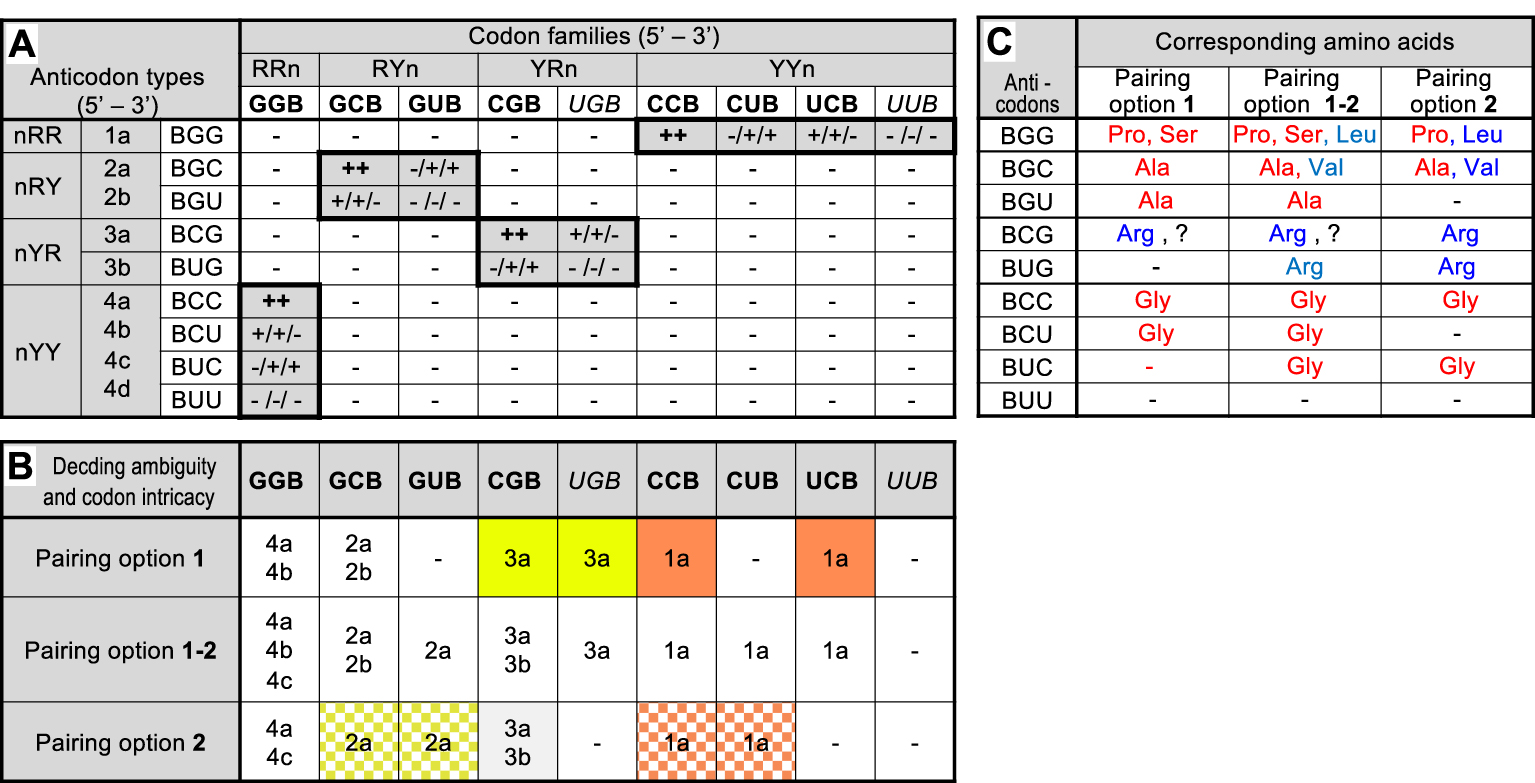
Matrice d’interaction codon–anticodon dans le code précurseur hypothétique G, C, U à 27 codons et conséquences. Partie A : matrice d’interaction entre les 9 familles de codons (troisième ligne) et les 9 types d’anticodons (troisième colonne) qui peuvent se former dans les molécules d’ARN primitives composées des 3 nucléotides G, C et U si l’on ignore la troisième position du codon/première position de l’anticodon (B = non A). Les codons et les anticodons ont été classés selon leurs séquences 5′ à 3′ (R : purine, Y : pyrimidine, n : tout nucléotide) et les types d’anticodons ont été numérotés arbitrairement (deuxième colonne). Les codons en caractères gras ou italiques correspondent respectivement aux familles non scindées ou scindées du code moderne. Les interactions dans lesquelles les 3 positions des duplex codon–anticodon impliquent une paire purine-pyrimidine sont mises en évidence par des cases ombrées entourées de lignes épaisses. Notez qu’une paire purine-pyrimidine est toujours supposée à la troisième position du codon/première position de l’anticodon, c’est-à-dire que la matrice présentée est en fait la somme de deux matrices indépendantes mais identiques respectant cette condition. Les résultats prévus de chaque interaction deux à deux en termes de formation d’un duplex codon–anticodon actif sont symbolisés par ++ appariement actif indépendant de l’option choisie (deux paires G–C ou C–G aux positions 1 et 2 des codons) : + appariement actif dépendant de l’option choisie (une paire G–C ou C–G et une paire G ∙ U ou U ∙ G aux positions 1 et 2 des codons) ; les résultats sont présentés à gauche, à droite ou au centre pour l’option 1, l’option 2 ou l’option 1–2, respectivement ; − aucun appariement (toute autre combinaison). Partie B : Résumé de l’ambiguité de décodage et de l’intrication des familles de codons correspondant à chaque option d’appariement (voir la figure complémentaire S1 pour un exemple). Le tableau indique tous les types d’anticodons (indiqués par leur numéro) dont on prédit la lecture pour chaque famille de codons dans le cadre de chaque option d’appariement. Les familles correspondant à plus d’un type d’anticodon sont potentiellement ambiguës. Les familles sans type d’anticodon (−) sont potentiellement non codantes. Les familles partageant le même type d’anticodon sont mises en évidence par des fonds de couleur similaires (ignorés pour des raisons de clarté pour l’option d’appariement 1–2). Partie C: Association attendue des a.a.s aux types d’anticodon comme déduit de la signification des codons des familles non scindées dans la forme standard du code moderne (? : signification des codons dans une famille scindée). La couleur des acides aminés est liée à la classe de leur aaRS respectif (bleu : classe I, rouge : classe II). – : type d’anticodon inactif (absence de codon apparenté sous l’option d’appariement choisie). Masquer
Matrice d’interaction codon–anticodon dans le code précurseur hypothétique G, C, U à 27 codons et conséquences. Partie A : matrice d’interaction entre les 9 familles de codons (troisième ligne) et les 9 types d’anticodons (troisième colonne) qui peuvent se former ... Lire la suite
4. Propriétés remarquables de la matrice d’appariement codon–anticodon du code précurseur à 27 codons
Les 27 triplets possibles issus de l’assemblage aléatoire de nucléotides dans des molécules d’ARN primitives composées de G, C et U génèrent 729 interactions deux à deux qui peuvent être simplifiées en 81 combinaisons si l’on ignore la position 3 des codons (les 27 codons peuvent être classés en 9 familles de 3 codons chacune) et la première position des anticodons (dans l’orientation 5′–3′). La matrice d’interaction codon–anticodon qui en résulte (Figure 1A) présente 16 combinaisons où G et Y se font face aux deux positions 1 et 2 des codons (en fait, il existe deux matrices de ce type si l’on considère également que G–Y est requis à la position 3 des codons, mais les deux matrices ont des structures identiques et n’ont pas besoin d’être détaillées ici). Selon l’option d’appariement choisie pour les positions 1 et 2 des codons (ici, la paire forte est G–C et la paire faible optionnelle est G ∙ U), 6 (options 1 ou 2) ou 8 (option 1–2) des 9 familles de codons sont lisibles par l’ensemble des anticodons (UUB est toujours exclu, B = non A). Les 6 familles sont GGB, GCB, CGB, UGB, CCB et UCB selon l’option 1 ou GGB, GCB, GUB, CGB, CCB et CUB selon l’option 2. Dans les deux cas, les trois familles de codons restantes (celles avec U en deuxième ou première position, respectivement) ne peuvent pas former de duplex avec un anticodon et sont donc prédites comme étant non codantes. Le code précurseur imaginé ici devrait donc être potentiellement codant à environ 67% si les options 1 ou 2 sont retenues et à environ 89% si l’option 1–2 est retenue, ce qui correspond à la synthèse de peptides courts. En supposant que les 3 nucléobases soient en quantités équimolaires, ces molécules d’ARN primitives auraient eu une continuité de lecture de 6 à 8 codons en moyenne (selon l’option d’appariement réelle), c’est-à-dire qu’elles auraient pu être suffisantes pour la synthèse des domaines peptidiques les plus simples. De même, la matrice d’appariement montre que certains des 9 types d’anticodons possibles restent incapables de former un duplex avec aucun des codons. Il s’agit de BUG, BUC et BUU dans l’option d’appariement 1, de BGU, BCU et BUU dans l’option d’appariement 2 et de BUU seul dans l’option 1–2. Ces anticodons inutiles dans le code précurseur à 27 codons peuvent avoir formé un réservoir utile lors de l’expansion ultérieure du code (voir ci-dessous).
La matrice d’appariement illustre les phénomènes d’ambiguïté du codage et d’intrication des codons définis ci-dessus. La Figure 1B montre que 2 des 6 familles de codons lisibles selon les options d’appariement 1 ou 2 (pas les mêmes selon l’option) peuvent être reconnues par 2 types d’anticodons distincts chacun, ce qui suggère une source d’ambiguïté de décodage. Les familles de codons potentiellement ambiguës sont GGB et GCB pour l’option 1 ou GGB et CGB pour l’option 2). Notez que GGB, commun aux deux options, peut être lu par 3 types d’anticodons distincts sous l’option 1–2. La prédiction d’une ambiguïté de décodage ici n’est que provisoire en l’absence de connaissance de la relation entre les anticodons et les a.a.s. Plus intéressant encore, la matrice prédit également que les codons de familles distinctes sont reconnus par le même type d’anticodon, ce qui implique une signification partagée (intrication) quel que soit l’a.a. concerné. Selon les options 1 ou 2, le phénomène touche 2 paires de familles de codons, mais il s’étend à 2 paires plus 1 trio selon l’option 1–2. Les familles de codons dont on prévoit qu’elles partageront la signification de codage sont CGB/UGB (partageant le type d’anticodon BCG) et CCB/UCB (partageant le type d’anticodon BGG) pour l’option 1 et GCB/GUB (partageant le type d’anticodon BGC) et CCB/CUB (partageant le type d’anticodon BGG) pour l’option 2. Le phénomène d’intrication des codons joue un rôle essentiel dans ce travail (voir ci-dessous).
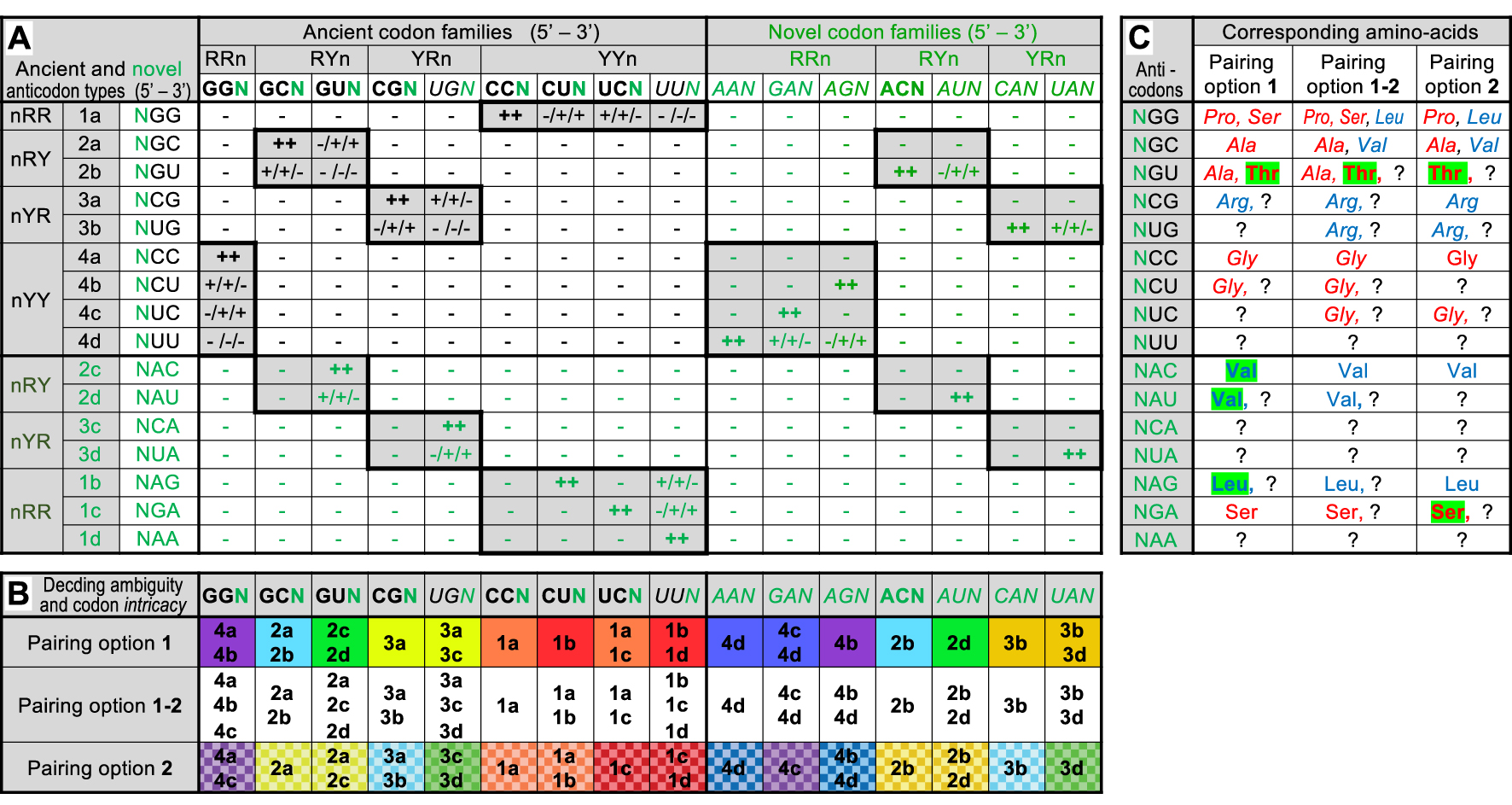
Matrice d’interaction codon–anticodon dans le code intermédiaire à 64-codons G, C, U, A et conséquences. Partie A: Même légende que la Figure 1A, sauf pour les symboles : ++ appariement actif indépendant de l’option choisie (deux paires G–C ou C–G, deux paires A–U ou U–A, ou une paire G–C ou C–G et une paire A–U ou U–A aux positions 1 et 2 des codons) ; + appariement actif dépendant de l’option choisie (une paire G–C, C–G, A–U ou U–A plus une paire G ∙ U ou U ∙ G aux positions 1 et 2 des codons) ; – aucun appariement (toute autre combinaison). Les nouveaux codons, anticodons et nouvelles interactions sont mis en évidence en vert. Notez que la partie supérieure gauche de la matrice est identique à la Figure 1A, sauf que N remplace B dans les anciennes familles de codons et d’anticodons. Partie B : Même légende que la Figure 1B. L’intrication prévue des familles de codons est visualisée par des fonds de couleur (omis pour des raisons de clarté dans l’option d’appariement 1–2) avec des couleurs violettes à vertes pour les purines et des couleurs jaunes à rouges pour les pyrimidines à la position du codon responsable du phénomène. Partie C: même légende que la Figure 1C. Les nouveaux a.a.s. du répertoire de codage correspondant à chaque option (par rapport à la Figure 1C) sont mis en évidence sur fond vert. Masquer
Matrice d’interaction codon–anticodon dans le code intermédiaire à 64-codons G, C, U, A et conséquences. Partie A: Même légende que la Figure 1A, sauf pour les symboles : ++ appariement actif indépendant de l’option choisie (deux paires G–C ou C–G, ... Lire la suite
Il est bien sûr très difficile de déduire quelles sont les a.a.s qui auraient pu être associés à ce code précurseur hypothétique. Cependant, des caractéristiques remarquables émergent lorsque l’on considère la signification des codons correspondants dans les familles non scindées du code moderne (Figure 1C). Si la signification des codons a été conservée, l’option d’appariement 1 suggère que Ala, Arg, Gly, Pro et Ser étaient présents dans le code précurseur à 27 codons. Pour les raisons évoquées plus loin, il est peu probable que Arg, soit un a.a. précoce (voir Discussion). Si l’on ne tient pas compte de ce fait, il est à noter que les quatre autres a.a.s sont activés par des aaRS de classe II (et 3 d’entre eux par des aaRS de la même sous-classe IIA, voir ci-dessous). L’option d’appariement 2 prédit la présence d’Ala, Arg, Gly, Leu, Pro et Val. La même remarque vaut pour Arg, mais le remplacement de Ser par Leu et Val élimine l’homogénéité des aaRS (un argument en faveur de l’option 1, voir ci-dessous). L’appariement de l’option 1–2 prédit logiquement la somme de tous les a.a.s. Notez que la matrice d’interaction codon–anticodon construite sur d’hypothétiques molécules d’ARN primitif constituées de G, A et U (Figure supplémentaire S2) au lieu de G, C et U donne des résultats équivalents en termes de capacité de codage, d’ambiguïté potentielle de décodage et d’intrication des codons mais ses prédictions en termes de répertoire de codage sont beaucoup plus pauvres (voir Discussion).
5. Matrice d’appariement codon–anticodon étendue aux molécules d’ARN à 4 nucléobases.
En s’appuyant sur le code précurseur G, C, U précédent, l’arrivée d’une seconde purine au sein des molécules d’ARN primitives l’étend à une structure à 64 codons si cette seconde purine diffère de G dans ses interactions avec les deux pyrimidines. A priori, deux possibilités logiques existent : soit la nouvelle purine (arbitrairement désignée A′) s’apparie avec les deux pyrimidines dans un ordre inversé d’énergies libres (A′–U supérieur à A′–C), créant ainsi une deuxième paire de faible énergie à prendre en compte dans les options d’appariement, soit elle ignore la pyrimidine précédemment préférée (comme le fait A avec C). Pour faciliter la lecture, seule la deuxième possibilité est examinée ici (Figure 2), la première étant illustrée par la figure supplémentaire S3 et examinée dans la discussion. Pour les mêmes raisons que ci-dessus, la matrice théorique de 4096 interactions (64 × 64) peut être réduite à 256 (16 × 16) combinaisons, dont seulement 64 correspondent à un appariement R–Y à la fois à la première et à la deuxième position des codons (ici encore, deux matrices identiques existent pour tenir compte de l’appariement R–Y à la position 3 des codons).
En conservant les mêmes options d’appariement qu’auparavant, la matrice G, C, U, A présente deux caractéristiques intéressantes (Figure 2A). Premièrement, les nouvelles familles de codons (celles contenant A) sont immédiatement lisibles par l’ancien ensemble d’anticodons (dépourvus de A). Cela est vrai pour les 7 familles sous l’option 1–2 et pour 6 d’entre elles sous les options d’appariement 1 (AUN ignoré) ou 2 (UAN ignoré). Cette particularité a probablement été un facteur critique pour la réussite de l’évolution du code car elle permet la continuité temporelle de la synthèse des peptides avant que les nouveaux anticodons (contenant A) puissent être associés aux a.a.s (voir Discussion). Avec un total de 12 (options 1 ou 2) ou 15 (option 1–2) familles de codons immédiatement lisibles, la capacité globale de codage des nouvelles molécules d’ARN (contenant A) est même légèrement supérieure à celle des anciennes molécules d’ARN dépourvues de A (75% au lieu de 67% avec les options d’appariement 1 ou 2, et 94% au lieu de 89% avec l’option 1–2).
Deuxièmement, seuls quelques nouveaux types d’anticodons sont nécessaires pour étendre la capacité de codage du code intermédiaire à 100% (les 16 familles de codons devenant toutes lisibles). Sur les 7 nouveaux types d’anticodons (Figure 2A), seuls 3 sont nécessaires selon l’option d’appariement 1 (NAC, NAU et NAG) ou 2 (NCA, NUA et NGA), et seul 1 est nécessaire selon l’option d’appariement 1–2 (soit NAG, NGA ou NAA). Cette exigence parcimonieuse de nouveaux anticodons aurait également pu être un élément crucial pour la réussite de l’évolution du code si l’association de nouveaux anticodons avec les a.a.s était l’étape limitante. Il faut noter que la capacité de codage complète du code intermédiaire avec un sous-ensemble limité d’anticodons est cohérente avec le nombre toujours plus faible d’espèces d’ARNt par rapport aux codons sens dans les organismes modernes [38]. Cela suggère également que les codons non sens et les facteurs de libération n’étaient pas des caractéristiques ancestrales du code mais représentent des mécanismes évolués (voir Discussion).
La pleine capacité de codage du code intermédiaire s’accompagne d’une augmentation limitée de l’ambiguïté du décodage (Figure 2B). La moitié des 16 familles de codons sont reconnues par 2 types d’anticodons selon l’option d’appariement 1 ou 2, les 8 autres restent lisibles par un seul type d’anticodon (selon l’option 1–2, seules 4 familles de codons restent lisibles par un seul type d’anticodon alors que 4 autres sont reconnues par 3 types d’anticodon). Mais plus important encore, la pleine capacité de codage du code intermédiaire s’accompagne d’une augmentation maximale de l’intrication des codons, chaque codon partageant un anticodon avec au moins un autre codon d’une famille distincte (Figure 2B). Huit paires de familles de codons sont formées selon les options d’appariement 1 ou 2 (pas les mêmes paires entre les deux options) ou 4 tétrades de familles de codons selon l’option 1–2. Le code intermédiaire à 64 codons est donc totalement intriqué, un point critique dans ce travail (voir ci-dessous). Notez que dans chaque paire (options 1 ou 2), une famille de codons est reconnue par un seul type d’anticodon, l’autre par le même type d’anticodon plus un autre. Dans les tétrades (option 1–2), une famille de codons est reconnue par un seul type d’anticodon, deux familles par deux types d’anticodon et la dernière famille par trois types d’anticodon.
Comme auparavant, on peut tenter d’associer chaque type d’anticodon à un ou quelques a.a.s en utilisant comme guide la signification des codons des familles non scindées du code moderne (Figure 2C). Notez que toutes les options d’appariement donnent la même liste finale de 8 a.a.s (un résultat tautologique), mais leur ordre d’apparition diffère lorsque l’on compare avec le code précurseur (Figure 1C). Avec l’option 1, Leu, Thr et Val sont ajoutés à la liste précédente des a.a.s (Ala, Gly, Pro, Ser et Arg ?). Avec l’option 2, Ser et Thr sont ajoutés à Ala, Gly, Leu, Pro, Val et Arg ?). Thr est commun aux deux options et c’est le seul nouvel a.a. de la liste avec l’option 1–2. Comme Thr correspond à un ancien type d’anticodon (NGU, anciennement BGU), il est tentant d’imaginer qu’il était déjà présent dans le code précurseur à 27 codons (associé à un anticodon BGU). Cette idée ne change rien sous l’option d’appariement 2, car le type d’anticodon BGU n’est pas actif. Mais si les options d’appariement 1 ou 1–2 s’appliquaient, l’association de Thr au type d’anticodon BGU au lieu de Ala comme imaginé sur la Figure 1C aurait étendu le répertoire de codage par un autre a.a. activé par un aaRS de classe II (en fait Thr est activé par un aaRS de sous-classe IIA, comme Gly, Pro et Ser, étendant encore cette homogénéité déjà significative).
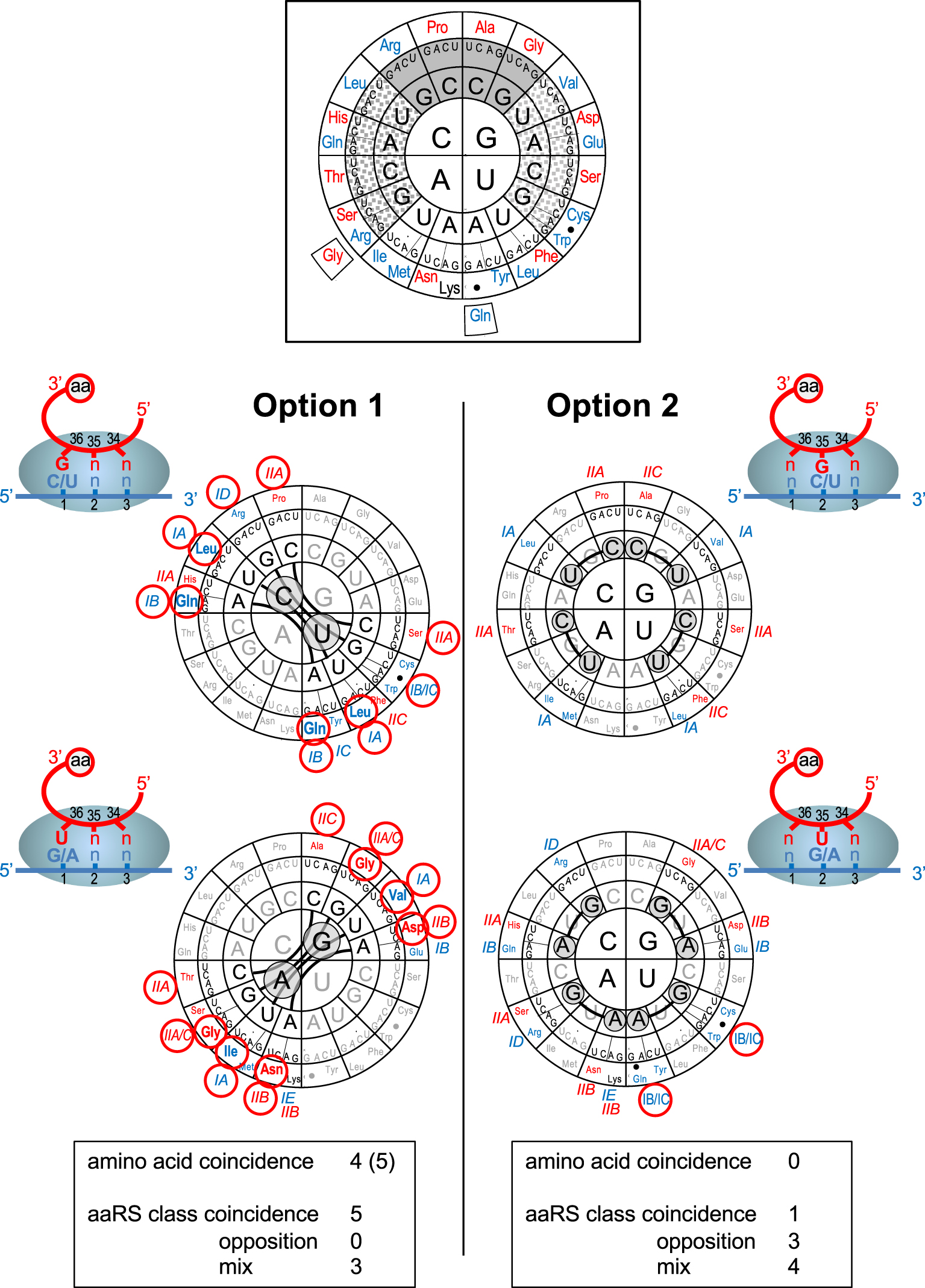
Prédiction de l’intrication des codons dans le cadre des options d’appariement 1 et 2. Encadré haut : Le code génétique est représenté selon [39], avec les nucléotides aux positions 1, 2 et 3 des codons du centre vers la périphérie. Les hélices codon–anticodon fortes (moyenne −13,0 kJ /mole) sont représentées sur fond gris clair, les hélices codon–anticodon faibles (moyenne −4,2 kJ /mole) sont représentées sur fond blanc et les hélices intermédiaires (moyenne −9,2 kJ /mole) sur fond gris pointillé. Les a.a.s correspondants sont indiqués dans le cercle extérieur (le code couleur se réfère à la classe d’aaRS comme dans le Tableau 1). Les points en gras indiquent les codons stop. La signification des codons est indiquée pour le code standard, les formes variantes utilisées dans ce travail (voir texte) sont indiquées en périphérie. Partie inférieure : Intrication prédite des codons selon les options d’appariement 1 ou 2 (schémas). Les paires intriquées de familles de codons sont visualisées par les lignes épaisses. Les nucléotides responsables du phénomène sont encerclés sur fond gris (les pyrimidines ont été séparées des purines pour plus de clarté). La coïncidence des classes d’acides aminés et d’aaRS entre les paires intriquées de familles de codons est mise en évidence par des cercles rouges (les sous-classes d’aaRS sont conformes à [52]). Les tableaux en bas récapitulent les résultats pour chaque option d’appariement (voir le texte et le Tableau supplémentaire S2). Masquer
Prédiction de l’intrication des codons dans le cadre des options d’appariement 1 et 2. Encadré haut : Le code génétique est représenté selon [39], avec les nucléotides aux positions 1, 2 et 3 des codons du centre vers la périphérie. ... Lire la suite
6. L’intrication des codons permet de distinguer entre les options d’appariement
Comme mentionné ci-dessus, les trois options d’appariement alternatives génèrent des prédictions différentes de l’intrication des codons (voir Figure 2B). Or, lorsque les codons intriqués sont rapportés au code moderne (Figure 3), une image radicalement différente apparaît entre l’option 1 et l’option 2 (et par conséquent l’option 1–2, non représentée sur la figure). Dans le cadre de l’option 1, on observe de multiples cas de coïncidence entre a.a.s et aaRS, alors qu’il n’y en a aucun (pour les a.a.s) ou un seul (pour les aaRS) pour l’option 2. Il semble que les familles de codons modernes aient conservé les traces de l’intrication ancestrale des codons prédite par l’option d’appariement 1. Cette conservation est détaillée dans le tableau supplémentaire S2.
Le premier exemple est représenté par la présence de Leu dans la famille de codons CUN (non scindée) et la famille de codons UUN scindée entre les codons UUR (codant Leu) et les codons UUY (codant Phe) dans les formes standard et variantes du code. Cela peut être interprété comme une réminiscence du fait que, dans l’intermédiaire à 64 codons (voir Figure 2A), les codons CUN et UUN étaient lus par le même type d’anticodon (NAG, attribué à Leu), avant que l’émergence du nouveau type d’anticodon (NAA) ne permette l’introduction tardive de Phe pour les codons UUY uniquement (car NAA est incapable de lire les codons CUN).
Un deuxième exemple frappant de coïncidence de codage apparaît si l’on considère la forme variante du code utilisé par les gènes nucléaires dans de nombreuses lignées évolutives distinctes d’eucaryotes unicellulaires comme les Ciliés [53, 54], les Diplomonades [55], les Chlorophytes [56], les Kinétoplastides [57] et d’autres (voir [46, 47, 58]). Dans toutes ces espèces, les codons UAR codent Gln (et parfois Glu) au lieu de servir de codons stop. Outre l’intérêt intrinsèque du phénomène de terminaison traductionnelle dépendante du contexte qui lui est lié [59], cette déviation de codage est remarquable en raison de l’intrication prédite des codons UAR avec les codons CAR qui codent également Gln (Figure 3). Là encore, cette coïncidence frappante peut être la réminiscence de l’appariement partagé des codons ancestraux UAN et CAN avec le type d’anticodon NUG avant que le nouveau type d’anticodon (NUA) n’autorise l’introduction tardive de Tyr pour les codons UAY uniquement (NUA ne lit pas les codons CAN) et que l’évolution tardive ne transforme les UAR en codons stop, mais pas dans toutes les lignées d’eucaryotes unicellulaires.
Les deux coïncidences de codage suivantes concernent les codons GAY et AAY, codant respectivement Asp et Asn, et les codons GUN et AUH (ou AUY), codant respectivement Val et Ile. Dans les deux cas, les a.a.s ne sont pas identiques mais étroitement apparentés. Asn peut avoir remplacé Asp sur les codons AAY après l’émergence de la voie de transamidation pour synthétiser les ARNt aminoacylés [60, 61]. De même, la proximité chimique entre Val et Ile (tous deux activés par les aaRS de la sous-classe IA) rend possible que les deux familles ancestrales de codons GUN et AUN, qui partageaient le type d’anticodon NAU, aient été ambiguës pour les deux a.a.s.
Au total, sur les huit paires de familles de codons dont on a prédit l’intrication dans le cadre de l’option d’appariement 1, quatre présentent des coïncidences évidentes dans le code moderne lorsque l’on considère les a.a.s (Figure 3). Une cinquième coïncidence possible n’est mentionnée ici que pour mémoire car elle n’existe que dans l’une des nombreuses variations observées dans les codes mitochondriaux (dont la plupart concernent la position wobble). Les codons AGR codent Gly dans les mitochondries des Urochordés [62], tout comme sa famille intriquée de codons GGN. Cette coïncidence, cependant, est probablement plus significative de réassignations tardives de codons que de l’intrication ancestrale car le codon AGG code une variété d’autres a.a.s dans le code mitochondrial des algues vertes [63] et les codons AGR codent Ser ou sont utilisés comme codons stop dans d’autres mitochondries.
Les trois dernières paires de familles intriquées de codons prévues dans le cadre de l’option d’appariement 1 (CGN/UGN, CCN/UCN et GCN/ACN) ne présentent pas de coïncidence évidente d’a.a.s dans le code moderne, mais cachent une coïncidence remarquable si l’on examine les aaRS utilisées pour activer les a.a.s codés (Tableau supplémentaire S2). Les deux premières paires sont dérivées des familles intriquées les plus anciennes (CGB/UGB et CCB/UCB) du précurseur à 27 codons (voir Figure 1). Le couple CGN/UGN est très difficile à interpréter car il est peu probable que Arg soit l’a.a. originel et la famille UGN est la plus mystérieuse du code moderne (voir Discussion). Pourtant, les 3 a.a.s du code moderne (Arg, Cys et Trp) sont tous activés par des aaRS de classe I (pas les mêmes sous-classes, cependant). Aucune coïncidence évidente entre a.a.s n’est disponible pour la paire CCN/UCN, mais les deux a.a.s codés (Pro et Ser) sont activés par des aaRS de la même sous-classe IIA. Il est possible que ces deux a.a.s aient été codés de manière ambiguë par les deux familles de codons. Cette ambiguïté de décodage peut avoir été très ancestrale car, dans le code précurseur à 27 codons, les familles de codons CCB et UCB interagissaient avec le même type d’anticodon (BGG, voir Figure 1). Un argument similaire peut être avancé pour les familles GCN et ACN car, comme mentionné ci-dessus, le type d’anticodon BGU peut avoir été associé très tôt à Thr, créant une ambiguïté de décodage Ala-Thr pour la famille GCB avant la formation des familles de codons GCN et ACN. Notez que Ala et Thr sont également activés par deux aaRS de même classe II (bien que ce ne soit pas la même sous-classe).
À ce stade, il devient possible d’éliminer l’option d’appariement 2 (et par conséquent l’option 1–2 également) et de conclure à l’existence de l’option d’appariement 1 comme règle pour les interactions codon–anticodon pendant les premières phases du code. En d’autres termes, les premiers duplex codon–anticodon actifs devaient être constitués d’une paire Watson–Crick obligatoire en position centrale, mais toléraient des paires purine-pyrimidine plus faibles aux deux positions latérales avant que la rigueur d’appariement n’augmente en position 1 des codons et, probablement plus tard en position 3. L’augmentation tardive de la rigueur en position 3 est due aux modifications chimiques des molécules d’ARNt (voir [39]). En position 1, elle a probablement résulté de l’évolution de l’appareil de traduction (voir Discussion). Dans le code précurseur à 27 codons imaginé, la position centrale n’était donc occupée que par une paire G–C ou C–G (aucune différence n’est prise en compte dans ce travail) et, au stade 64 codons, cette position acceptait également les paires A–U ou U–A, mais pas les paires G ∙ U ou U ∙ G. En revanche, les positions 1 et 3 acceptaient les paires G ∙ U ou U ∙ G en plus des paires Watson–Crick. La même conclusion s’appliquerait aux précurseurs hypothétiques des bases azotées modernes tant que leurs préférences d’appariement relatives restent qualitativement similaires et confèrent une énergie libre quantitativement suffisante (voir Discussion).

Reconstruction des étapes de l’évolution du code génétique. La figure illustre le code génétique (dessiné selon [39]) au cours des 3 périodes successives prédites par ce travail sur la base de la composition nucléotidique des molécules d’ARN primitives et de la tolérance d’appariement G ∙ U/U ∙ G à la position 1 des codons (barres du haut). L’intrication des familles des codons est visualisée par des secteurs colorés (voir Figure 2) et des lignes épaisses avec les nucléotides responsables encerclés sur des fonds gris. La signification prévue de chaque famille de codons à chaque période est représentée sur le cercle extérieur (code couleur des a.a.s comme dans le Tableau 1, les nouveaux a.a.s de la période sont en caractères gras sur fond gris, points noirs: codons non codants). L’évolution du répertoire de codage est récapitulée dans la partie inférieure. La période intermédiaire a été subdivisée sur la base de l’utilisation supposée des anciens anticodons avant les nouveaux anticodons (voir le texte et le Tableau 2). Les a.a.s. sont énumérés par ordre alphabétique à leur première période ou sous-période d’apparition dans le répertoire de codage (la sous-classe d’aaRS correspondante se trouve entre parenthèses, code couleur comme dans le Tableau 1). Notez l’incertitude pour Arg et Cys (voir le texte) et le nombre limité de différences entre le code entièrement codant de la période tardive et le code moderne. Masquer
Reconstruction des étapes de l’évolution du code génétique. La figure illustre le code génétique (dessiné selon [39]) au cours des 3 périodes successives prédites par ce travail sur la base de la composition nucléotidique des molécules d’ARN primitives et de ... Lire la suite
7. Reconstruction de l’ordre temporel du recrutement des acides aminés et des aaRS au cours de l’évolution du code génétique
La combinaison entre les évolutions postulées de la composition en nucléobases des molécules d’ARN primitives et de la rigueur d’appariement en position 1 des codons définit trois périodes successives dans l’évolution du code (Figure 4). Pendant la première période, l’appariement G ∙ U ou U ∙ G en position 1 des codons a permis d’initier un premier code avec des molécules d’ARN primitives ayant une composition en bases azotées encore incomplète. Les peptides synthétisés devaient être courts (18 des 27 codons étaient potentiellement codants, voir Figure 1) et constitués d’un répertoire limité d’acides aminés. La conservation de la même règle d’appariement relaché alors que la composition en bases azotées des molécules d’ARN arrivait à son terme a généré une deuxième période au cours de laquelle des peptides légèrement plus longs ont pu être synthétisés par un code qui allait vers 64 codons sans qu’il soit nécessaire d’étendre immédiatement le répertoire de codage (48 des 64 codons peuvent être lus par d’anciens anticodons). Cette dissociation entre capacité de codage et répertoire a probablement été déterminante pour la réussite de l’évolution du code. Elle a permis la formation progressive de nouveaux anticodons pour finalement aboutir à une capacité de codage à 100% (64 codons sur 64). Il est fort possible que cette deuxième période ait duré longtemps avant que la rigueur des appariements n’augmente à la première position des codons, probablement sous l’influence de l’évolution des ribosomes [64, 65]. Cet événement a créé une transition vers la troisième période au cours de laquelle certaines familles de codons auparavant non scindées ont été scindées par une différenciation croissante à la troisième position des codons, comme proposé précédemment [39], permettant ainsi de compléter le répertoire de codage. Il convient de noter que, sur une base purement logique, l’augmentation des rigueurs d’appariement à la position 1 des codons (nécessaire pour éliminer l’intrication ancestrale des codons) et à la position 3 (nécessaire pour réduire la dégénérescence du codage) ne sont pas liées. Mais les interactions des anticodons avec la poignée ribosomique (ribosomal grip) peuvent avoir établi un tel lien [39].
Dans ce contexte, l’évolution du répertoire de codage peut être provisoirement reconstituée a posteriori en utilisant comme guide la signification des codons dans le code moderne. Cependant, pour ce faire, il faut d’abord déterminer l’ordre relatif d’arrivée des a.a.s dans chaque famille scindée de codons au cours de son évolution hypothétique à partir du précurseur non scindé. Deux critères peuvent être utilisés: (i) la coïncidence de codage restante entre les paires de codons anciennement intriqués (voir ci-dessus) et (ii) une priorité donnée aux anciens anticodons sur les nouveaux pour lire les nouveaux codons du code intermédiaire. Ce postulat de priorité divise la période intermédiaire en deux sous-périodes successives selon l’utilisation des anciens ou des nouveaux anticodons (Figure 4). Lorsque les deux critères sont combinés (Tableau 2), on peut conclure que Leu a précédé Phe pour la famille UUN, Ile a précédé Met pour la famille AUN, Gln a précédé His et Tyr pour les familles CAN et UAN, respectivement, Asp a précédé Glu pour la famille GAN et Asn a précédé Lys pour la famille AAN. Notez que la relation biochimique entre Asp et Asn et le mécanisme de transamidation [60, 61] suggèrent une possible ambiguïté précoce entre ces deux acides aminés pour la famille des codons AAN. Le même argument s’applique entre Glu et Gln pour les familles de codons CAN et UAN.
Ordre séquentiel d’arrivée des acides aminés dans les familles scindées de codons
| Ancient codons | Novel codons | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| UGN | UUN | AAN | GAN | AGN | AUN | CAN | UAN | ||
| Cys Trp | Phe Leu | Asn Lys | Asp Glu | Ser Arg | Ile Met | His Gln | Tyr (Gln) | ||
| Intricate family and coding significance | CGN Arg | CUNLeu | GAN Asp Glu | AAN Asn Lys | GGN Gly | GUNVal | UAN Tyr (Gln) | CANHis Gln | |
| Ancient active anticodon | NCU (Gly) | Ser Arg | |||||||
| Ancient inactive anticodon | NUU | Asn Lys | Asp Glu | ||||||
| NUC | Asp Glu | ||||||||
| NUG | His Gln | Tyr (Gln) | |||||||
| Novel anticodon | NAC | ||||||||
| NAU | Ile Met | ||||||||
| NCA | Cys Trp | ||||||||
| NUA | Tyr (Gln) | ||||||||
| NAG | Phe Leu | ||||||||
| NAA | Phe Leu | ||||||||
| Deduced order | First a.a. | ? | Leu | Asn/Asp | Asp | Ser/Gly ? | Ile | Gln/Glu | (Gln/Glu) |
| Last a.a. | ? | Phe | Lys | Glu | Arg ? | Met | His | Tyr | |
Les 8 familles scindées de codons (en italique) du code moderne, triées entre anciennes (sans A, noir) et nouvelles (contenant A, vert) selon la Figure 2, sont énumérées à la ligne 2 avec leur signification de codage (les a.a.s de gauche et de droite sont codés par les codons nnY et nnR, respectivement ; le code de couleur se réfère à la classe d’aaRS comme dans le Tableau 1). La signification des codons dans la variante du code nucléaire utilisée dans ce travail est indiquée entre parenthèses (voir le texte). Pour chaque famille de codons, la ligne 3 indique son partenaire intriqué prédit dans l’option d’appariement 1 (même code couleur, en caractères gras : famille non scindée) avec sa signification de codage. Les lignes 4 à 7 indiquent les appariements actifs des anciens types d’anticodons (sans A) avec les nouveaux codons après la transition du code précurseur (voir Figure 1) au code intermédiaire (voir Figure 2). Notez que l’anticodon NCU (anciennement BCU) a été provisoirement attribué à Gly dans le code précurseur et que les 3 autres anticodons ont été considérés comme inactifs par manque de codons correspondants. Les lignes 8 à 13 indiquent les appariements actifs des nouveaux types d’anticodons (contenant A) avec des codons anciens et nouveaux, voir Figure 2). Les lignes 14 et 15 récapitulent, pour chaque famille scindée, l’ordre d’apparition des a.a.s déduit de la combinaison de l’intrication des familles de codons et du postulat de priorité des codons anciens (voir le texte).
Les deux dernières familles, AGN et UGN, sont plus problématiques. La signification des codons AGR est très variable dans les génomes mitochondriaux, variant entre Ser, Gly et stop (voir ci-dessus). Le codon AGG seul est encore plus variable, variant entre Ala, Lys et Met [63]. Avec la question de Arg (voir Discussion), il semble plus probable que la famille AGN ait initialement codé pour Ser ou, plus probablement, ait été ambiguë entre Gly et Ser étant donné son intrication avec la famille GGN (notez que Ser et Gly sont activés par des aaRS de la même sous-classe IIA). Pour la famille UGN, la situation est encore pire. En plus d’être la seule famille de codons scindée en 3 parties dans le code standard, la signification de ses codons est également très variable dans les formes variantes du code. Le codon non-sens UGA code souvent Trp et parfois Cys dans plusieurs codes nucléaires et organellaires. Il peut également coder Gly dans certaines bactéries et la Sélénocystéine (Sel) dans des cas spécifiques. Bien qu’il soit tentant de considérer que cette famille a initialement encodé Cys, cela ne peut pas être formellement déduit du présent travail (voir Discussion). Enfin, l’application du postulat de priorité des anticodons anciens dans le code intermédiaire conduit à la conclusion que, dans la période intermédiaire, Asn/Asp et Gln/Glu qui utilisent des anticodons anciens sont arrivés avant Ile, Leu et Val qui utilisent des anticodons nouveaux (Tableau 2).
À partir des considérations ci-dessus, la chronologie des événements au cours de l’évolution du code peut être résumée comme illustré par la Figure 4. Partant d’un simple code précurseur de 27 codons, dont 18 potentiellement codants, la première période était compatible avec la synthèse de peptides courts composés de 5 a.a.s, Ala, Gly, Pro, Ser et Thr, plus au moins un autre a.a. correspondant aux codons CGB et UGB (l’arrivée de Arg et Cys n’est pas claire, voir Discussion). Les 5 premiers a.a.s identifiés sont tous activés par des aaRS de classe II. Cette homogénéité est encore plus frappante quand on se rappelle que Pro, Ser et Thr sont tous activés par des enzymes de la sous-classe IIA, Ala est activé par une enzyme de la sous-classe IIC et Gly est activé par deux enzymes des sous-classes IIA et IIC respectivement [60, 52]. Les enzymes de ces deux sous-classes ont ensuite été utilisés pour l’activation des a.a.s tardifs, His et Phe, respectivement.
Lors de la transition vers la période intermédiaire, la majorité (30 sur 37) des nouveaux codons (contenant A), avaient la possibilité immédiate d’interagir avec les anciens anticodons (manquant de A). Comme 3 types d’anticodons anciens (NUU, NUC et NUG) étaient potentiellement inactifs (en raison de l’absence de codon correspondant dans le code précurseur, voir Figure 1), il est possible qu’ils aient contribué à l’expansion immédiate du répertoire de codage par l’arrivée de Asn et Asp, activés par des aaRS de sous-classe IIB, et Gln et Glu qui sont les premiers a.a.s à être activés par des aaRS de classe I (sous-classe IB). Il est intéressant de penser que les aaRS de classe I sont apparus pendant cette période à partir du brin complémentaire du même acide nucléique que les aaRS de classe II, comme le proposent Rodin et Ohno [66]. Quelle que soit cette origine, les trois a.a.s immédiatement suivants, Ile, Leu et Val (et peut-être aussi Cys, voir la discussion) sont tous activés par des aaRS de classe I. À ce stade, le code intermédiaire à 64 codons avait atteint sa pleine capacité de codage et son répertoire totalisait au moins 12 a.a.s (peut-être 14 selon les a.a.s codés par les familles intriquées CGN et UGN, voir Discussion). La capacité de codage complète a permis la synthèse de peptides plus longs. Toutes les familles de codons, sauf peut-être GAN, étaient non-scindées, leur intrication était complète (8 paires) et plusieurs familles étaient probablement ambiguës (GAN aurait pu être ambiguë entre Asp et Glu au lieu d’être scindée).
Lors de la transition vers la période tardive, l’augmentation de la rigueur d’appariement en position 1 des codons a éliminé leur intrication et réduit l’ambiguïté du décodage, mais une nouvelle expansion du répertoire de codage n’était pas possible avant la scission de certaines familles de codons. En se basant sur leur signification de codage telle que déduite ci-dessus (voir Tableau 2), 6 a.a.s tardifs ont pu être ajoutés au répertoire (His, Lys, Met, Phe, Tyr et Trp). Contrairement à la remarquable homogénéité des périodes précoces, ces a.a.s sont activés par des aaRS de classe I ou de classe II en nombre égal (notez que Lys est activée par deux aaRS, une de chaque classe).
Le passage de la période tardive au code génétique moderne n’a impliqué que des changements mineurs dans certaines (mais pas toutes) versions du code, comme l’installation de UAR et UGA comme codons stop au lieu de Gln et Trp, respectivement, l’affectation de Ile au lieu de Met au codon AUA et de Arg (au lieu de Ser ou Gly ?) aux codons AGR. Les variations fréquentes observées pour ces codons, y compris le codage de la pyrrolysine (Pyl) et de la sélénocystéine (Sel), sont cohérentes avec cette évolution tardive.
L’évolution du code présenté ici est, bien sûr, schématique car elle ne repose que sur la logique interne des matrices d’appariement de triplets dans des molécules d’ARN primitives hypothétiques sans se soucier des mécanismes moléculaires réels impliqués dans le processus de décodage. Cependant, le développement prédit du répertoire de codage est en excellent accord avec des conclusions préalables basées sur des données indépendantes telles que l’abondance des a.a.s prébiotiques [16, 67], la complexité de leurs voies de biosynthèse [17] ou leur rôle fonctionnel dans les protéines [68, 69]. En particulier, l’ordre chronologique obtenu ici correspond remarquablement bien au rôle présumé de l’oxygène atmosphérique dans le recrutement sélectif des derniers a.a.s, tel que déduit de leur réactivité chimique [70]. Il semble que les périodes précoces et intermédiaires définies ici correspondent à l’évolution des cellules vivantes dans le milieu réducteur précédant la première accumulation d’oxygène atmosphérique, alors que la période tardive a commencé après cette transition oxydative (voir ci-dessous).
8. Discussion
Pendant plus de cinq décennies, le code génétique a été envisagé de multiples façons, à la recherche de sa logique et de son origine possible. Le présent travail n’ajoute qu’une contribution très modeste à une liste impressionnante de recherches antérieures. Son intérêt principal repose sur l’observation de nombreuses coïncidences réelles entre paires de codons dont on prédit ici que les ancêtres étaient interconnectés (intrication des codons) en suivant l’option d’appariement 1 (voir Figure 3). Cette option met en évidence le rôle particulier joué par la position centrale des duplex codon–anticodon par rapport à ses deux positions latérales. Cette idée n’est pas nouvelle, la spécificité de la position centrale par rapport à la première a déjà été reconnue en ce qui concerne la signification des codons et la robustesse du code en termes de mutation [28, 39, 42, 44, 71]. Son importance fonctionnelle est encore illustrée par le fait que, dans le centre décodage des ribosomes modernes, l’étendue de la dégénérescence tolérée à la troisième position du codon est déterminée par le niveau de stabilité de la paire de base à la position centrale [72].
L’idée d’un code commençant par une paire obligatoire à haute énergie en position centrale des duplex codon–anticodon et des exigences moins strictes aux deux positions adjacentes, suivie d’une augmentation ultérieure de la rigueur d’appariement à la première puis à la troisième position des codons, a été brièvement évoquée précédemment comme l’hypothèse 2.1.3 [42, 73], mais sans analyse détaillée de ses conséquences. Ici, je montre que la tolérance des paires à faible énergie à la première position des codons a généré une intrication initiale entre les codons qui a laissé tant de traces dans le code moderne qu’il ne peut en être autrement. La question de savoir quand, pourquoi et comment la rigueur de l’appariement en position 1 a ensuite augmenté reste ouverte. Les réponses sont probablement cachées dans les interactions tridimensionnelles entre le duplex codon–anticodon et les composants des ribosomes modernes [39]. Mais il est intéressant de se rappeler qu’il y a de nombreuses années, Weissenbach et ses collègues [74] ont découvert qu’un seul ARNtLeu de levure portant l’anticodon UAG était capable de lire les six codons de la leucine (CUN et UUR) dans des extraits de cellules de souris traitées à l’interféron, confirmant la fonctionnalité persistante d’une paire G ∙ U à la première position des codons dans le code moderne, du moins dans ces conditions.
De même, l’idée d’un ordre séquentiel d’apparition des nucléobases dans les molécules d’ARN primitives n’est pas nouvelle. Elle est même au centre des recherches sur le monde à ARN et la formation prébiotique des purines et des pyrimidines [75, 76, 77]. Mais l’arrivée tardive de A par rapport aux trois autres nucléobases, comme proposé ici, n’a pas été prise en compte auparavant. Une construction progressive du code à 4 nucléobases à partir d’un précurseur uniquement GC a été proposée avec un intermédiaire G–C–A [77]. Malheureusement, cette composition ne permet pas la formation de deux paires de bases distinctes dans les duplex d’ARN primitifs, un aspect fondamental pour initier un code actif, comme le montre ce travail. Cette formation est possible dans l’hypothèse d’une composition G, A et U de l’ARN primitif récemment examinée [78]. La matrice complète d’appariements codon–anticodon construite avec des molécules d’ARN hypothétiques composées de G, A et U (Figure supplémentaire S2) donne des prédictions numériques équivalentes à celles du code précurseur proposé dans la Figure 1 en termes de capacité de codage, d’ambiguïté et d’intrication des codons. Mais son répertoire de codage est extrêmement difficile à prédire car la plupart des familles de codons dont on prédit l’ancienneté sont scindées dans le code moderne, ce qui suggère que les familles scindées auraient précédé les familles non scindées au lieu du contraire. Il reste donc l’hypothétique composition G, C et U des molécules d’ARN primitives proposée ici comme la meilleure possibilité. De plus, le fait que A (ou son dérivé désaminé I) soit rarement trouvé en position wobble des anticodons modernes [11, 38, 39] est cohérent avec l’idée que les anticodons actifs existaient avant l’arrivée de A.
La difficulté majeure des molécules à 3 nucléobases est que la réplication ne peut se faire par complémentarité des bases, comme précédemment discuté pour un ARN primitif de composition G, A et U [78]. La même difficulté existe pour la composition de G, C et U. Une possibilité serait que les premières molécules d’ARN grâce auxquelles le code génétique a émergé (qui peuvent avoir été très courtes) ne se répliquaient pas par complémentarité de base classique, mais étaient simplement synthétisées plus ou moins au hasard ou avec les a.a.s eux-mêmes servant de guides chimiques comme l’imagine la théorie stéréochimique. Cependant, si cette possibilité est relativement facile à imaginer pour les anticodons, elle est évidemment plus difficile pour les codons car un certain degré de réplication conservatrice est nécessaire pour lancer un processus héréditaire. Par conséquent, on peut également supposer que le point de départ réel du code n’était pas un précurseur de 27 codons comme proposé (Figure 1) mais la rencontre entre deux sources distinctes de molécules d’ARN primitives de composition différente. La première, qui aurait finalement donné naissance aux anticodons et aux molécules d’ARNt, était initialement composée de nucléotides G, C, U et d’a.a.s, et synthétisée chimiquement. La seconde, composée des quatre nucléobases et capable de se répliquer par complémentarité des bases, a finalement donné naissance à des virus et à des molécules d’ARNm primitives (puis à des gènes). Si c’est le cas, le code précurseur aurait pu être constitué de 27 anticodons, comme proposé, mais de 64 codons au lieu de 27. Cette possibilité ne modifie pas de manière significative la capacité de codage prévue du code précurseur car, comme nous l’avons vu précédemment avec le code intermédiaire, la plupart des familles de codons peuvent être lues par l’ensemble limité d’anciens types d’anticodons (Figure 2).
Outre la réplication de l’ARN, l’absence hypothétique de A dans la phase initiale du code semble également difficile à imaginer compte tenu de son rôle critique dans les mécanismes moléculaires modernes de décodage. Par exemple, A est présent dans l’extension NCCA de la tige accepteuse des ARNt sans laquelle les a.a.s ne pourraient pas être activés (en plus du besoin d’ATP pour la réaction). De même, A est présent dans la paire C ∙ A universellement conservée dans le centre peptidyl-transférase de l’ARN ribosomique et est également présent dans la poignée ribosomique [39, 79]. Mais la partie du code génétique examinée ici ne concerne que les interactions codon–anticodon (où A n’est pas nécessaire), et non les mécanismes catalytiques de l’activation des a.a.s et de la formation des liaisons peptidiques (où A est nécessaire). On ne sait pas comment ces différentes parties se sont liées entre elles à l’origine mais, comme mentionné ci-dessus, on ne peut exclure qu’elles aient initialement émergé de pools distincts de molécules d’ARN primitives. La formation prébiotique divergente des purines et des pyrimidines [76] et la chimie prébiotique complexe à l’origine des molécules d’ARN primitives [80], peuvent avoir généré des différences dans leur composition initiale.
D’un point de vue purement logique, les premières bases azotées des molécules d’ARN primitives qui ont initié le code précurseur n’avaient pas nécessairement besoin d’être identiques aux bases azotées des molécules d’ARN modernes. La seule exigence est que deux paires purine-pyrimidine d’énergies libres différentes puissent être formées. Cependant, à part l’hypoxanthine (I), le choix semble limité [81]. L’inosine (I) vient aussi naturellement à l’esprit en tant que précurseur commun de l’adénosine et de la guanosine. Mais sa différenciation énergétique plus faible (par rapport à G) pour les deux pyrimidines réduirait la distinction entre les positions 1 et 2 des codons qui semble si importante ici (la matrice d’appariement codon–anticodon avec I au lieu de G serait équivalente à l’option d’appariement 1–2 mais avec les deux paires faibles simultanément tolérées). De même, la possibilité théorique que deux purines hypothétiques aient existé simultanément avec des préférences d’appariement opposées pour les deux pyrimidines ne peut être écartée. Cette possibilité a été examinée ici (Figure supplémentaire S3). Ses prédictions sont équivalentes, en termes d’intrication des codons, à celles de l’intermédiaire à 64 codons proposé avec les quatre nucléobases modernes (seule la dégénérescence du codage est augmentée). Mais il est, bien sûr, impossible de prédire le répertoire à moins que chaque purine soit associée à G ou A, respectivement (comme le montre la figure supplémentaire S3 pour permettre une comparaison directe avec la Figure 2).
Un aspect important de la présente proposition est en contradiction directe avec les idées courantes sur l’origine du code. L’intermédiaire à 64 codons était entièrement codant, du moins à un moment donné, ne laissant aucune place aux codons stop. Cette idée est contraire à l’hypothèse selon laquelle l’extension du répertoire de codage reposait sur le remplacement progressif des codons stop antérieurs sous une pression sélective pour former des peptides plus longs [82]. Toutefois, il semble peu probable que les facteurs de relâchement existaient déjà dans les premières périodes du code lorsque certains codons restaient non codants par simple absence d’anticodons correspondants. Au contraire, on peut argumenter que c’est l’arrivée tardive de ces facteurs qui a favorisé la formation de nouveaux codons stop à partir de codons sens précédents, par analogie avec ce qui est observé dans les multiples variantes de codes modernes [83]. Un autre aspect important de la présente proposition diffère de l’opinion commune selon laquelle une ambiguïté primordiale généralisée aurait été progressivement réduite au fur et à mesure de l’évolution de la machinerie traductionnelle [84, 85] ou de l’émergence progressive de nouveaux codons [86]. Ici, l’ambiguïté de décodage prédite reste toujours précisément circonscrite à des codons spécifiques à chaque étape de l’évolution du code. Cependant, si elle est définie avec précision, l’ambiguité globale de décodage du code ne peut être estimée quantitativement en l’absence de toute connaissance sur les concentrations relatives des codons et anticodons distincts dans les pools de molécules d’ARN primitives.
Dans l’ensemble, le meilleur argument pour justifier le raisonnement utilisé dans ce travail est la remarquable cohérence de ses prédictions sur l’évolution du répertoire avec des conclusions préalables tirées de considérations sur la chimie prébiotique ou la biosynthèse et la réactivité chimique des a.a.s [16, 17, 67, 70, 87]. Tous les a.a.s prédits ici comme étant apparus dans la dernière période d’un schéma évolutif théorique uniquement défini par les interactions codon–anticodon sous une option d’appariement précise (voir Figure 4) correspondent à ceux prédits comme ayant été sélectionnés dans les protéines après le changement oxydatif majeur généré par la première accumulation d’oxygène atmosphérique [70]. Parmi les 14 autres a.a.s, dont la première apparition est prédite ici dans les périodes précoces ou intermédiaires, 12 correspondent à l’ensemble précoce selon le même critère. Les deux derniers, Arg et Cys, soulèvent des questions. Ce problème n’est pas nouveau. En 1989, Taylor et Coates [42] ont noté que Arg ne correspondait pas au modèle d’hydrophobie des autres a.a.s par rapport à la table des codons. De plus, l’Arg reste un a.a. rare dans les protéines (en moyenne) malgré ses 6 codons. Et ces codons sont les principaux responsables de la variation de l’utilisation des codons qui existe entre les différents domaines de la vie [88]. Dans ce travail, on prévoit que Arg est apparu dès la première période mais son aaRS de classe I le place en opposition avec tous les autres a.a.s de la même période. De plus, la voie de biosynthèse multi-étapes de Arg est mieux compatible avec un a.a. relativement tardif qu’avec un a.a. précoce [17]. Le fait que, dans les expériences d’aptamère, Arg domine largement tous les autres a.a.s dans les interactions avec l’ARN [26] explique probablement sa place unique dans l’évolution du code. Arg pourrait avoir remplacé un autre a.a., aujourd’hui disparu des protéines, dès que l’évolution des voies biochimiques a permis sa synthèse, peut-être à plusieurs reprises. Son précurseur biosynthétique, l’ornithine (Orn), a été proposé à la place de Arg dans le code ancestral [77]. Le cas de Cys est également problématique. Dans le code moderne, il est codé par la famille de codons UGN qui, selon ce travail, était auparavant intriquée avec la famille de codons CGN (codant Arg ou Orn ?). La famille UGN encode également Trp et contient un codon stop, UGA, dans le code standard. Cependant, UGA code souvent Cys ou Trp, et parfois Gly, dans les codes variants. Et il encode Sel dans des circonstances spécifiques. Selon sa réactivité chimique [70], Cys devrait être un a.a. tardif qui est considéré comme ayant joué un rôle majeur dans l’adaptation des protéines à l’oxygénation de la biosphère [89]. Mais la présence de soufre peut également avoir joué un rôle clé dans les premières phases de la vie [90]. Dans ce travail, Cys a été arbitrairement attribué à la période intermédiaire (voir Figure 4) mais sans argument précis (voir Tableau 2). Il est également possible qu’il soit l’un des derniers a.a.s ou, au contraire, qu’il ait déjà été présent dès la première période puisque la famille UGB est restée non assignée.
La cohérence du répertoire prédit ici indique également que, outre les quelques exceptions mentionnées ci-dessus, l’attribution des codons n’a subi que des changements limités au cours de la longue évolution du code, comme cela a été imaginé dès le début [3]. Cette idée est, bien sûr, contrebalancée par la flexibilité démontrée dans de nombreux exemples [47, 58, 62]. Plus de 30 codes différents1, 2 ont maintenant été répertoriés et il semble probable que cette liste sans cesse croissante soit loin d’être complète. La réaffectation des codons peut se faire par plusieurs mécanismes et les résultats peuvent être sélectionnés pour diverses raisons. Mais globalement, le code gelé a fondu avec une telle élégance et parcimonie que toutes ses traces historiques n’ont pas été irréversiblement effacées.
Remerciements
Je remercie A. Danchin, M. Delarue, C. Fairhead, G. Fischer, C. Gaillardin, H. Grosjean, R. Koszul, B. Llorente, C. Marck, G. Pelletier, G-F. Richard, J-L. Souciet, J. Weissenbach et E. Westhof pour des discussions approfondies et une lecture critique du manuscrit. Je suis redevable à E. Westhof d’avoir partagé des documents non publiés.
Données supplémentaires
Un complément d’informations est disponible sur le site internet de la revue en suivant le lien https://doi.org/10.5802/crbiol.47 ou auprès de l’auteur.