


 CC-BY 4.0
CC-BY 4.0
1. Introduction
As of mid-May 2021, the SARS-CoV-2 pandemic has caused more than 160 million cases with more than 3.4 million deaths reported to the World Health Organization (https://covid19.who.int/). At the same time, over 1.4 million vaccine doses have been administered throughout the world, and a race with the virus has been engaged in which, as more people are immunized through natural infection or because of vaccination, new virus variants emerge and take over the previously dominating strains [1]. These variants are selected because they are more apt to circulate in the presence of herd immunity. It remains to understand how far the virus can evolve in the face of the human immune response while maintaining its fitness, and how efficient the current vaccines will be in the long term to stop its dissemination and the economic tall imposed on society.
The development of novel and efficient vaccines has been made at an unprecedented pace, in part because the timing was ripe to test novel strategies such as mRNA vaccines, which had only been tried as anti-cancer strategies [2, 3]. Or such as the use of adenovirus vectors, which had been advanced during the 2014–2016 Ebola outbreak in West Africa, an epidemic that had receded before large clinical vaccine trials could be deployed [4]. What these successful anti-Covid-19 vaccines have in common is the immunogen that they carry, obtained by applying new concepts of structure-based vaccine design [5]. The virus spike protein was thus stabilized in an inactive state mimicking its otherwise metastable “pre-fusion” form present at the surface of infectious virus particles. Indeed, the spike protein is the sole target of virus neutralizing antibodies, but is intrinsically metastable as is must come apart to allow virus entry into cells by inducing the fusion of the viral envelope with the target cell membrane. Structural biology played a very important role in devising strategies to cope with its instability by engineering stable versions that preserve the relevant epitopes to be presented to the immune system to elicit strongly neutralizing antibodies. Previous studies on other enveloped viruses, such as HIV and the respiratory syncytial virus (RSV, responsible for severe bronchiolitis in infants and the elderly or immunocompromised individuals), which also display a highly unstable envelope protein at their surface, had paved the way for the coronavirus work [6, 7]. For both HIV and RSV, the situation is more complex as the epitopes targeted by the most potently and broadly neutralizing antibodies are only present in the intact ectodomain folded in its pre-fusion form. We now know, however, that the most potently neutralizing antibodies against the SARS-like viruses are directed to the receptor binding domain (RBD). This domain has the advantage that it can be produced independently of the rest of the spike, presenting by itself relevant epitopes to the immune system to elicit neutralizing antibodies [8]. Yet the formulation used in the current vaccines, using the full-length spike protein stabilized with two internal proline mutations, has proven remarkably efficient to stimulate the production of protecting antibodies, especially when administered in an mRNA form [9, 10].
I provide below a description of the ACE2 receptor and the spike protein together with an analysis of the conservation of the surfaces of interaction in both proteins across mammals and across SARS-like coronaviruses. I also summarize the knowledge gathered over the years on the mechanism of membrane fusion driven by enveloped viruses and how they apply to SARS-CoV-2. I end by illustrating that the mutations observed in the variants of concern display a clear clustering on three regions of the spike. In particular, in the receptor binding domain, they map to a rim all around the surface of interaction with ACE2.
2. The sarbecoviruses
The Coronaviridae family is divided into two subfamilies. The classic coronaviruses are grouped within the sub-family Orthocoronavirinae, which contains four genera: α-, β-, γ- and δ-coronaviruses. Only the α- and -β-Coronavirus genera include human pathogenic viruses. The SARS-like viruses form a subgenus, termed Sarbecovirus, within the β-Coronavirus genus. Of the four subgenera in this genus, only a subset of the sarbecoviruses were found to use ACE2 as receptor [11]. In this context, it is interesting to note that the human NL63 seasonal coronavirus, which belongs to the α-Coronavirus genus and is therefore quite distant to the sarbecoviruses, also uses ACE2 as receptor [12], although its receptor binding domain and its mode of recognition of ACE2 are unrelated to those of the sarbecoviruses [13], suggesting a convergence in evolution to use the same receptor. Another finding worth mentioning is that, for most coronaviruses the receptor is a membrane bound peptidase. For instance, amino-peptidase N (APN) is used by the human seasonal α-coronavirus 229E [14] as well as the porcine α-coronaviruses [15]. APN is a metal-dependent aminopeptidase playing multiple physiological functions, pain regulation, blood pressure homeostasis, angiogenesis of tumor cells, among others [16]. Similarly, the β-coronavirus Middle-East respiratory syndrome virus (MERS-CoV) uses dipeptidyl peptidase 4 (DPP4) as receptor [17]. DPP4 is a serine protease cleaving at the N terminus of physiological peptides involved in controlling apoptosis, immune regulation and signal transduction [18, 19, 20]. The catalytic activity is not required for virus recognition of any of these peptidase receptors, and the binding site recognized by the spike protein is away from the catalytic site. Yet it is intriguing that various coronaviruses from different genera have converged to use cell-surface peptidases for entry, and the potential relations to their pathogenicity remain an open field of investigation [21].
3. The ACE2 receptor
In 2003 Farzan and colleagues identified the angiotensin-converting enzyme 2 (ACE2) as the main receptor for the severe acquired respiratory syndrome coronavirus (SARS-CoV) [22]. The new coronavirus, SARS-CoV-2, was shown to use the same receptor [23]. ACE2 is a type I single-pass trans-membrane protein present at the cell surface of multiple tissues in various organisms. It is a component of the renin–angiotensin system (RAS) that controls blood volume and systemic vascular resistance, playing a crucial role in protection from high blood pressure and cardiovascular disease [24]. The other components of this hormone system are the soluble aspartic protease renin and the angiotensinogen (AGT) precursor, as well as the membrane-bound angiotensin converting enzyme (ACE) and different G-protein coupled receptors (GPRs) specific for the various angiotensin peptides derived from AGT cleavage. Cleavage of AGT by renin is the rate-limiting step [25] and is tightly regulated to maintain normal blood pressure, avoiding hypertension and other cardiovascular diseases. AGT is a 452 amino acid (aa) long circulating protein belonging to the serpin family of protease inhibitors [26, 27], albeit with no inhibitor function. Renin cleaves AGT at its N-terminus to generate angiotensin I (Ang-I) [28, 29], a decapeptide that is the substrate for ACE, which converts it to Ang-II (Figure 1). The Ang II octapeptide hormone is important for blood pressure homeostasis through its interaction with the GPR AP1, leading to increased blood pressure. The role of the carboxypeptidase ACE2 is to remove the C-terminal phenylalanine residue of Ang II to make Ang (1-7) [30, 31, 32], which binds to the GPR Mas [33, 34]. The Ang (1, 7)/GPR Mas interaction leads to vasodilation and protection from excessive blood pressure, contributing to vascular pressure homeostasis. Infection by a virus is known to cause downregulation of the expression if its entry receptor, as seen for instance with HIV and its CD4 receptor [35]. The reason is that low amounts of receptor in infected cells helps virus propagation away from the site in which the virus was replicated within an organism to more easily reach other tissues. SARS-CoV-2 downregulation of the ACE2 receptor may thus result in an imbalance in the Ang II/Ang(1/7) peptide ratio, potentially contributing to its pathogenicity.
The renin–angiotensin system and the ACE2 structural organization. (A) ACE2 processing of the angiotensin peptides. Shown is a partial amino acid sequence (truncated to leave out residues 51 to 481, with the break marked by a wiggly symbol) of angiotensinogen (ATG). The amino-terminus of AGT is generated co-translationally by signalase cleavage in the ER lumen (grey scissors). Upon environmental clues, secreted ATG exposes its N-terminal segment for cleavage by renin to generate the decapeptide angiotensin I (Ang I), highlighted in a yellow background [28, 29]. The renin cleavage site is labeled and marked by the black scissor to the right. The angiotensin converting enzyme (ACE) then cleaves Ang I at the site indicated by the left black scissor, to generate Ang II, a highly active peptide that stimulates vasoconstriction via interaction with its G-protein coupled receptor AP1 present at the plasma membrane of multiple cells. Unchecked Ang II leads to high blood pressure and vascular disease, and this effect is balanced by ACE2 (purple scissors below the sequence), which has a carboxypeptidase activity and cleaves the C-terminal residues of both, Ang I and Ang II, to generate Ang (1-9) and Ang (1-7), respectively.ACE2 cleaves the C-terminal residue of Ang II with about 400-fold the efficiency of Ang-1 cleavage to make Ang (1-9). Ang (1-7) induces vasodilation through its interaction with the GPR Mas. The activity of ACE-2 is therefore critical in the RAS system. Circled in green and red are, respectively, a glycosylated asparagine and a cysteine that forms a labile disulfide bond with another ATG cysteine located 120 aa downstream in the aa sequence (not shown). Both residues have been shown to play a role in recognition and cleavage by renin [29]. (B) Linear diagram of ACE, ACE2 and Collectrin, showing that ACE2 originated as a chimeric protein with its N-terminal domain derived from ACE and its C-terminus from collectrin. The domain boundaries are indicated, and vertical arrows point to different loops in the collectrin domain that are cleaved to release soluble ACE2 to the medium. ADAM17 cleaves in the loop 652–659, while TMPRSS2 cleaves in the loop 697–716 [36]. (C) Organization of ACE2 on membranes. The structure of ACE2 was determined by cryo-EM as a binary complex with the multi-spanning transmembrane amino acid transporter B0AT1 (left panel, PDB:6M18) and as a ternary complex with the SARS-CoV-2 spike’s receptor-binding domain (RBD) (right panels, PDB:6M17) [37]. The plasma membrane is indicated in the left panel, as a guide. ACE2 is shown in surface representation whereas B0AT1 and the RBD are shown as ribbons in blue and purple, respectively. In the left panel, ACE2 is colored green and yellow according to domains as in the linear diagram, with the two protomers shown in different shades of green and yellow. Similarly, both protomers of the B0AT1 transporter are shown in different blue tints. Note that the transmembrane helices of ACE2 form integral part of the transporter and contribute to its dimer interface. In the right panel, ACE2 is colored according to its amino acid sequence conservation from an alignment of 24 ACE2 sequences from various mammals. This alignment shows that the surface conservation is high within the cleft that carries the active site. The RBD binds away from this cleft at an exposed surface which is variable across mammalian species.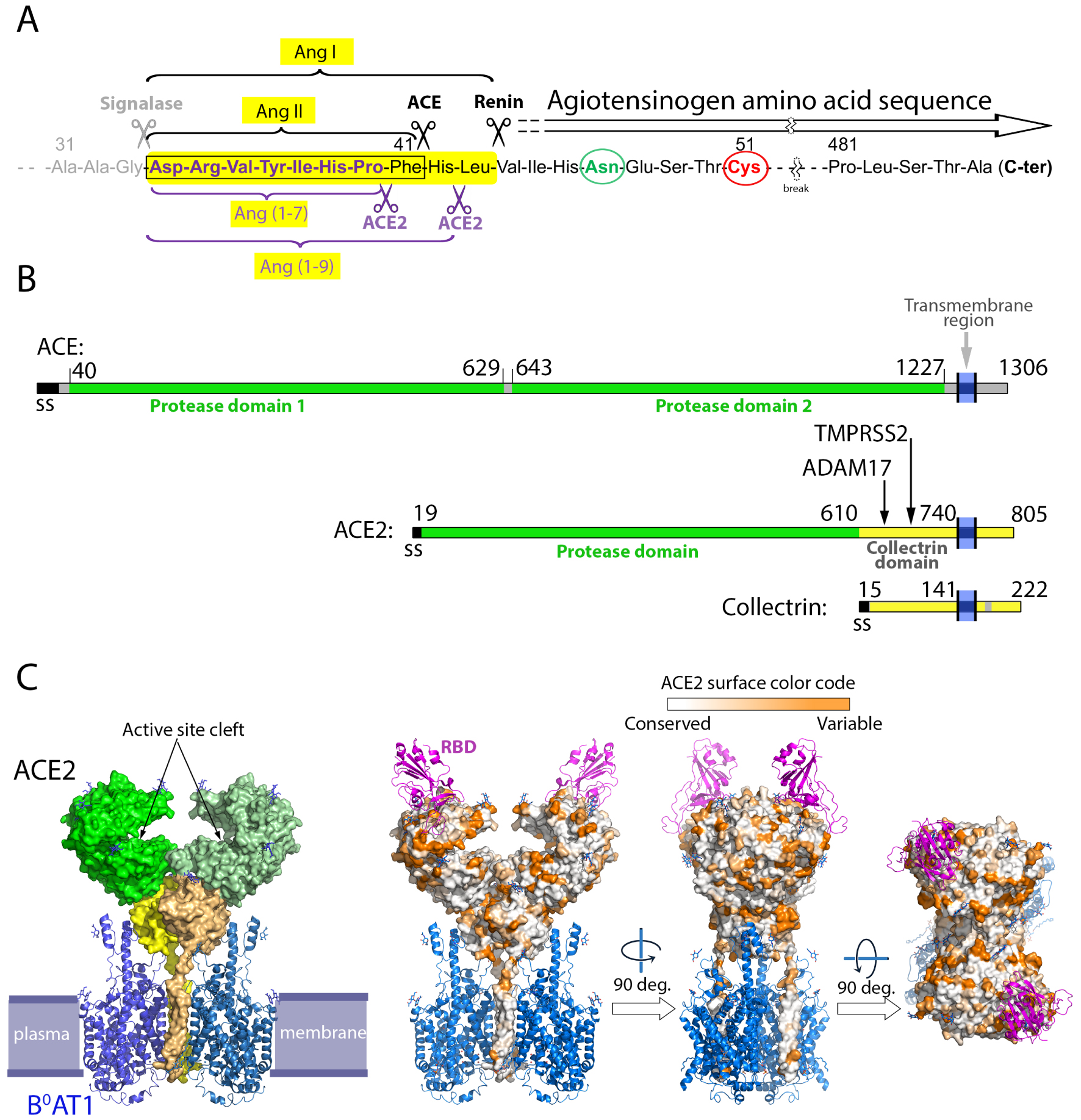
Human ACE2 was first identified in 2000 [30, 31] as a paralog of the angiotensin-converting enzyme (ACE), discovered in 1956 [38]. The corresponding gene, located at position p22.2 in the X chromosome, contains 18 exons, and gives rise to several ACE2 isoforms via alternative splicing. The longest isoform is 805 amino acids long and is expressed in a broad range of human tissues, including endothelial cells in blood vessels, enterocytes in the intestine, alveolar cells in the lung, nasal epithelial cells, all organs that are targeted by SARS-CoV-2. The organization of this ACE2 isoform is depicted in Figure 1B. Its ectodomain is composed of two structural domains [39]: an N-terminal catalytic domain with zinc metallopeptidase activity exhibiting 42% amino acid sequence identity with each the two catalytic domains of ACE [40] (Figure 2). The ACE2 protease domain is followed by a collectrin-like domain, which associates with the neutral amino acid transporter B0AT1 (Figure 1C), at least in intestinal epithelial cells. It has been shown that this association is required for expression of the transporter on the luminal surface of intestinal epithelial cells [41, 42]. The collectrin domain of human ACE2 shares 50% sequence identity with human collectrin, a type I single-pass trans-membrane glycoprotein that binds amino acid transporters of the B0 type, such as B0A1 and B0A3. These transporters, which belong to the solute carrier 6 (SLC6) family [43], are only active when collectrin is bound [44]. The activity of collectrin was also proposed to play a role in blood pressure homeostasis [45].
Structural organization of the coronavirus spike protein. (A) Linear diagram of the spike protein indicating the position of the individual subunits S1 and S2, the transmembrane (TM) segments, the individual domains and the cleavage sites. The amino acid numbers correspond to the SARS-CoV-2 sequence, original Wuhan strain. The “head” and “stalk” regions indicated in B are also labeled. The fusion peptide is indicated by a white hashed region downstream the S2′ cleavage site. A thin purple box encompasses a 140 residues long region of S2 (aa 772-912 in SARS-CoV-2 S), encompassing the FP and the S2′ cleavage site, that was not resolved in the available cryo-EM structures of coronavirus S2 [46, 47] (see also Figure 3). (B) Ribbon representation of the intact spike protein as modeled by molecular dynamics (MD) [48] on the viral membrane and based on intermediate resolution cryo-electron tomography structures of the spike on inactivated intact virus particles [49, 50, 51]. The polypeptide chain of the protomer in the foreground is colored according to domains as in A. The lipids on the MD modeled membrane and the complete sugar chains of the glycans attached to the protein are shown as sticks color-coded by atom type (carbon, nitrogen, oxygen and sulfur in light grey, blue, red and yellow, respectively). The approximate dimensions are provided on the side, with the flexible “stalk” and the more globular dynamic “head” portions of the trimeric spike labeled. The C-terminus of the TM segment in the intraviral side of the membrane is labeled in pink. (C, D) Cryo-EM structures of the spike ectodomain, showing only the head portion, with the front protomer colored according to domains as in A. The trimer is shown in the closed form with three RBDs in the “down” conformation (C) and with one RBD in the “up” conformation (D) in a partially open spike. On virions, the spike head displays a dynamic equilibrium between the closed conformation and partially open forms. Virions also display inactive post-fusion S2 spikes, presumably resulting from spontaneous S1 shedding and premature spike activation [49] (see Figure 3). The models displayed correspond to PDB:7bnm (C) and 7bnn (D), determined at a resolution of about 3.5 Å [52].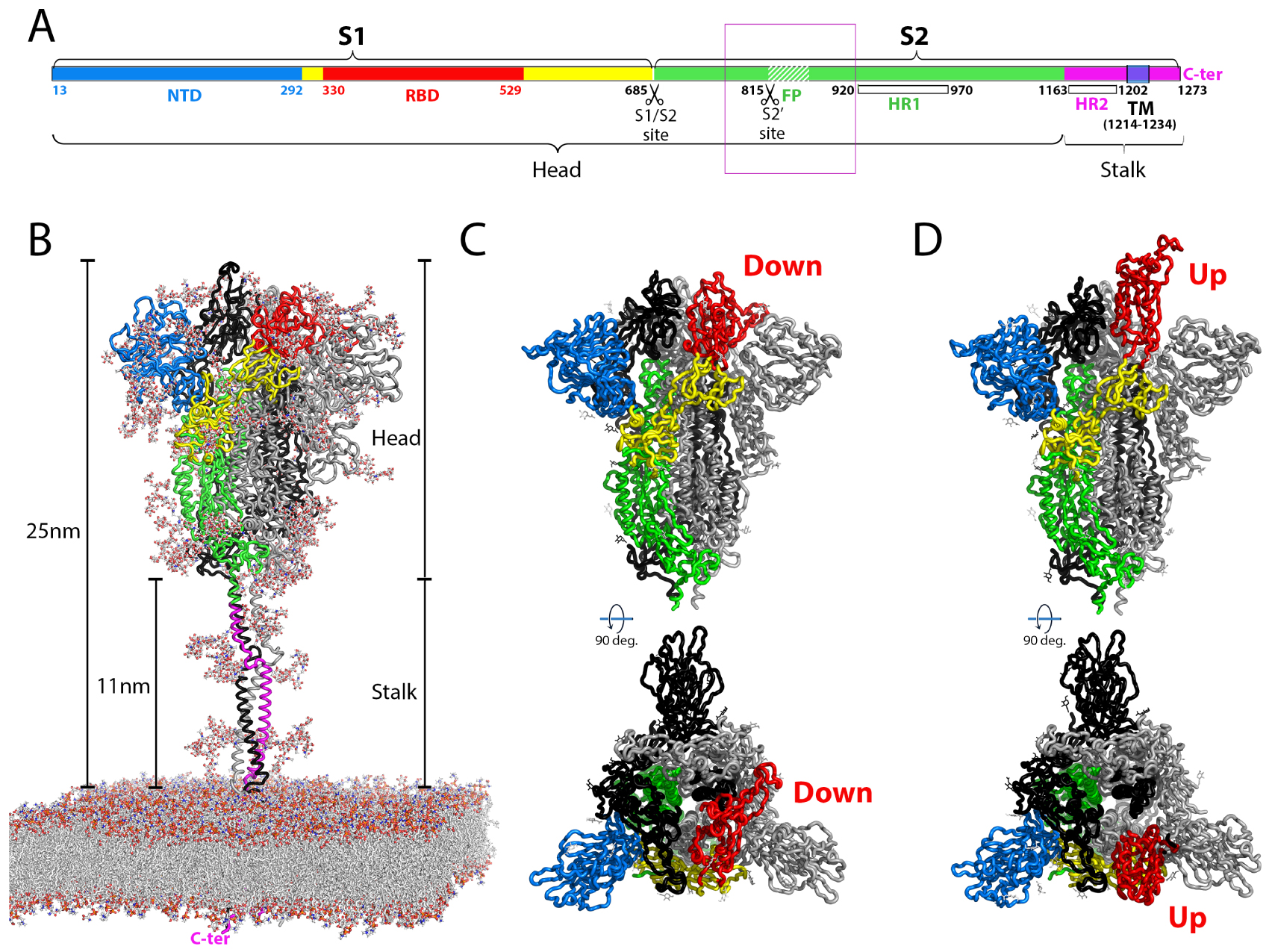
The spike protein binds at the membrane-distalmost surface of ACE2, away from the active site cleft (Figure 1C, right panels). Amino acid sequence comparisons show that the surface patch recognized by the RBD is variable across different mammalian species, contrary to the highly conserved cleft harboring the ACE2 catalytic site. The structure also shows that the two spike-interacting sites present in the (ACE2/B0A1) dimer are located such that two RBDs from the same spike cannot bind simultaneously, implying that two different spike trimers bind to one receptor dimer [37]. This situation has the potential of clustering the spikes on the virion side facing the cell membrane, where each spike trimer may bind up to three ACE2 dimers, each bound to a second spike and resulting in cluster formation. No study has so far addressed the stoichiometry of binding and whether the proposed clustering has a functional effect for virus entry, either in triggering fusion directly at the plasma membrane or by stimulating endocytosis with subsequent fusion in late endosomes.
3.1. The coronavirus spike glycoprotein
The sarbecovirus S polypeptide chain is roughly 1300 amino acids long with multiple asparagine-linked glycans in its ectodomain. It forms a trimer anchored to the viral membrane via a C-terminal transmembrane helix (Figure 2). S is activated by proteolytic cleavage to generate two subunits: S1 (roughly the N-terminal half of S), and S2, the membrane-anchored C-terminal half. SARS-CoV-2 features an insertion of a polybasic sequence at the S1/S2 cleavage site, which allows for S processing by furin, a trans-Golgi network resident protease in the virus producer cells. Cleavage by furin appears not to take place extensively, and most S trimers were reported to be only partially cleaved (i.e., cleavage occurring in only one or two of the three protomers of the trimer) [47]. Such partial cleavage may have functional consequences, as proposed in Figure 3B.
Spike protein activation to drive membrane fusion during virus entry. (A) Diagram summarizing the currently accepted main steps of the virus fusion process triggered by receptor binding. The fusion protein is anchored via its C-terminal end to the viral membrane, and is proteolytically activated by cleavage into an N-terminal subunit (NSU, in yellow), and a C-terminal subunit (CSU, in red and blue). The fusion peptide, indicated in green and labeled FP (i), is located immediately downstream the cleavage site. Upon proteolytic activation it becomes the N-terminus of the CSU. This activating cleavage renders the fusion protein metastable, with the CSU spring-loaded underneath an NSU crown (i). The CSU reacts to clues such as receptor binding or acidification of the milieu (i.e., in an endosome, depending on the virus) to trigger fusion via a major conformational rearrangement.In the case of the coronaviruses, the current model posits that receptor binding induces shedding of the NSU (i.e., the S1 subunit) from the trimer (ii), which allows the CSU to spring out and project the FP into the cell membrane, where it inserts firmly in the first step of the fusogenic rearrangement (iii). This “elongated intermediate” form is only transient, and folds back into a “hairpin” conformation in which the N-terminal half (in red) remains bound to the target membrane in the form of a central, trimeric, α-helical coiled coil, whereas the C-terminal half of the CSU runs in the opposite way along the grooves of the central coiled coil. Hairpin formation thus entails bringing into juxtaposition the N- and C-terminal ends of the CSU, thereby forcing the two membranes against each other (iii, iv and v). Fusion occurs first by merging of the outer leaflets, a process called “hemi-fusion” (v) and is followed by formation of a fusion pore (vi), which then enlarges to give rise to a single fused membrane. (adapted from [53]). (B) Current understanding of the SARS-CoV-2 activation process to drive membrane fusion. The Spike trimer is shown with the S1 subunit in surface representation and S2 as ribbons. One protomer is in colors and the others in grey and black. The S1 subunit of the front protomer in the trimer is shown in yellow to match the NSU represented in A. The front S2 subunit is ramp colored blue–green–yellow–orange–red from its N- to C-terminus (see bar under panel C), highlighting that its N-terminal moiety is spring-loaded underneath the S1 crown, whereas its C-terminal portion enters the stalk, as indicated in Figures 2A and B. The ACE2 receptor in complex with B0AT1 in the cell membrane is shown in surface representation with the two protomers of the ACE2 dimer differentiated in dark and pale red, while B0AT1 is in light grey. The central step (labeled “(0)”) shows the spike trimer with a closed head, which is in equilibrium with partially open forms (as the one shown in Figure 2C). ACE2 binding shifts the equilibrium towards open forms (step labeled (1)). Structural studies have shown that in the open head the S1/S2 interactions are weaker, especially when all three RBDs are in the up conformation [54]. It was proposed that fully furin processed spikes are unstable and spontaneously shed S1 after displaying the open-head conformation (step (−1), resulting in premature conformational change and insertion of the post-fusion S2 into the viral membrane (step (−2) [47]). In this panel, the post-fusion form is indicated with a hand-drawn connection between residues 772 and 912, which are disordered in the cryo-EM structures [46, 47]. Note that the S2 conformation of the left panel corresponds to step (vi) in Figure 3A, and not to that of step (ii), which depicts the CSU still in the spring-loaded pre-fusion conformation. Step (1) in B corresponds to a step in between (i) and (ii) in A, where the spike protein has already bound the ACE2 receptor. Pulling forces arising from the flow or other fluctuations in between the bound particle and the cell then result in S1 shedding, a step that requires completion of the cleavage between S1 and S2 on the spike. This cleavage is carried out by TMPRRSS2 at the surface of some cells, or by cathepsin upon endocytosis of the receptor-bound virions [55]. S1 shedding in this context leads to insertion of the fusion loop into the target cell membrane upon springing out of the HR1 helix to reach the target membrane, as indicated in step (iii) in A. The HR2 portion then completes the hairpin conformation by binding to the grooves of the central, trimeric coiled coil formed by the HR1 helix, which is the structure represented in step (3). (C) HR1 spring-out. The four panels represented illustrate the transition between steps (1) and (3) of Figure 2B, with only S2 represented in ribbons ramp-colored from N- to C-termini according to the color-coded bar underneath. For clarity, only one of the three protomers of the S2 trimer is shown. The left panel shows the HR1 moiety together with segments upstream the S2′ cleavage site tucked underneath the S1 crown in the pre-fusion form. The S1 subunit and the ACE2 receptor are schematically diagrammed and colored according to Figure 3A in steps (i) and (ii). Dissociation of the receptor-bound S1 crown induces HR1 to spring out and reach the target membrane (second panel). Simultaneously, the S2′ cleavage site, which is buried and inaccessible in the spring-loaded form of S2, becomes exposed, to be cleaved by either TMPRSS2 at the cell surface, or the cathepsins in endosomes. The curved black arrows indicate the subsequent refolding of S2 into the stable post-fusion hairpin, bringing the two membranes together as indicated in Figure 3A, steps (iv) to (vi). It is not known whether cleavage at the S2′ site occurs before or after hairpin formation, and the two options are represented (third and fourth panel). The fusion peptide (FP) has been shown to be required at the S2′ N-terminus for membrane fusion to take place, but the cleavage site becomes exposed only after the conformational change. Its location in the uncleaved form is too far to reach the target membrane (as labeled). We postulate that the region immediately upstream of HR1 is also involved in target membrane insertion. Amino acid sequence analyses indicate that this region, termed “internal fusion peptide” in the Figure, has a strong propensity to form an amphipathic helix. Its binding to the outer leaflet of the target membrane would allow the right leverage to bring the two membranes into apposition even if the S2′ occurs after the conformational change. Otherwise the S2 trimer would to flip and end inserting into the viral membrane, as observed on particles and as indicated in Figure 3B, left panel. The effect of the IFP segment has not yet been experimentally tested, however.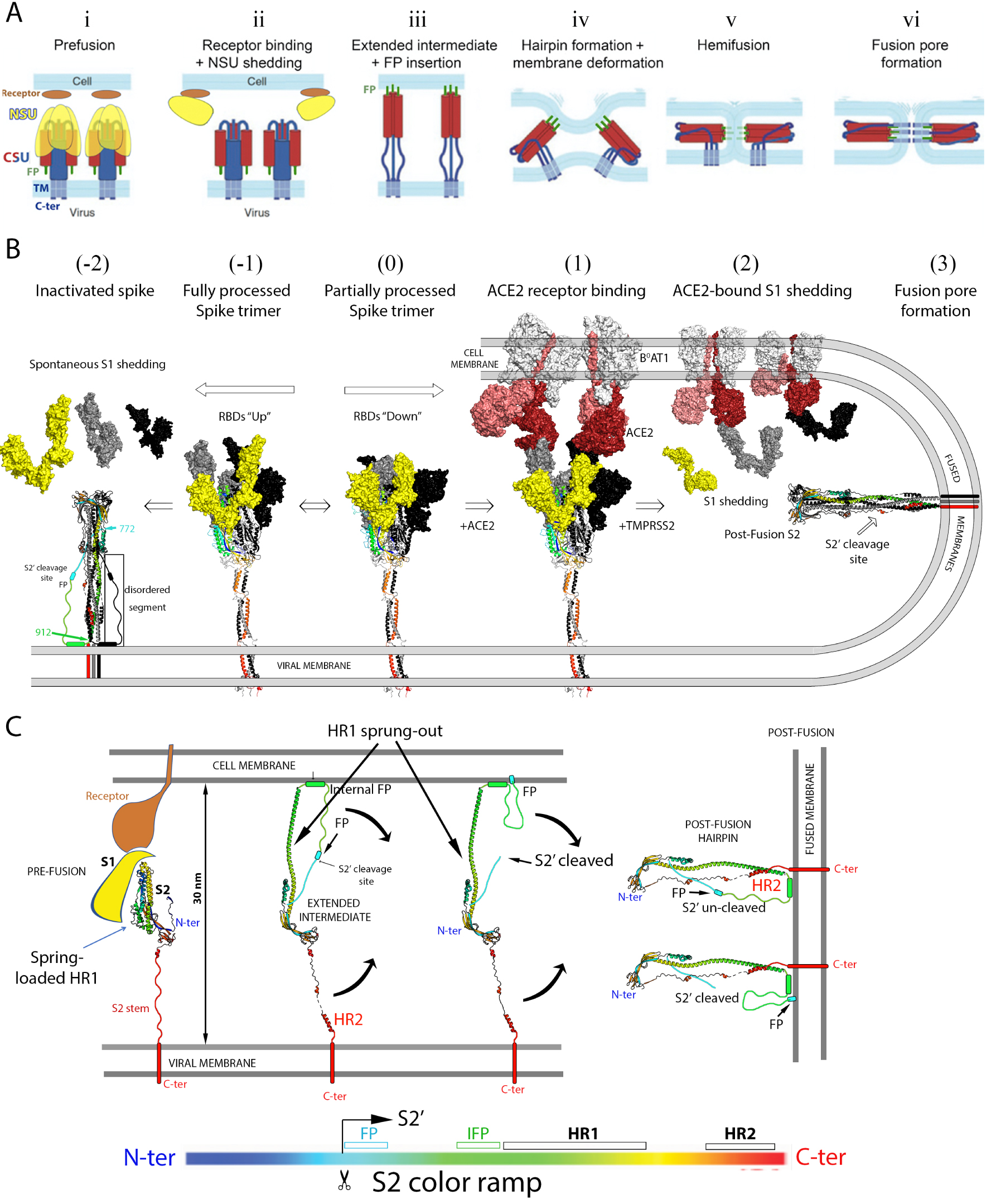
4. The furin cleavage site insertion
This spike polybasic site has been shown to be involved in interactions with neuropilin 1 [56, 57]. The neuropilins bind vascular endothelial growth factors (VEGFs) at the carboxyl-terminal sequence generated by furin cleavage of the VEGF precursor [58]. Neuropilins are present in the olfactory epithelium, and could play a role in the observed anosmia in Covid-19 patients [59] and potentially in the virus invasion of the central nervous system [60]. The same polybasic furin site has also been proposed to interact with the nicotinic acetylcholine receptor (nAChR) [61], by analogy to a similar motif present in the glycoprotein of rabies virus and in several snake neurotoxins [62]. No evidence has been provided so far, however, for a direct interaction between the nAChR and the polybasic site. Indirect evidence lies in the fact that ivermectin (IVM), an antiparasitic compound that is also a positive allosteric effector of the α7 neuronal nACHR [63] was shown to attenuate the neurological symptoms in the hamster model for Covid 19 [64]. IVM did not reduce the viral load in the infected hamsters, only the Covid 19 pathological symptoms, contradicting a previous report [57] suggesting that IVM had a direct antiviral effect. In any case, whatever the role of the polybasic sequence, independent studies have demonstrated that SARS-CoV-2 mutants in which it is deleted are less pathogenic than the parent virus [65, 66].
5. The spike protein on the viral membrane
The S trimer projects about 25 nm from the viral membrane, displaying a prominent, globular yet dynamic “head” and a flexible “stalk” region (Figure 2A). The three-dimensional (3D) structure of the spike head has been determined by cryogenic electron microscopy (cryo-EM) and single-particle averaging for several coronaviruses, the first of which were reported back in 2016 [67, 68] (reviewed in [69]) to resolutions of about 3 Å or better, including that of the SARS-CoV-2 spike [47, 70, 71]. The full spike has been visualized on SARS-CoV-2 virions by cryo-electron tomography analyses of inactivated virus particles using sub-tomogram averaging to reach a resolution beyond 1 nm [49, 50, 51]. S1, which remains non-covalently attached to S2 in the cleaved trimer, contains two structural domains, an N-terminal domain (NTD) and the RBD. The fold of both of these domains is independent of the rest of the chain (Figure 2). The structural studies have shown that the RBD displays a dynamic equilibrium between two conformations, termed “down” and “up”. The spike head trimer has thus been captured in four different forms, a completely closed form with all three RBDs in the down conformation, an open conformation with all three RBDs up, and intermediately open forms with one or two RBDs up [54, 52]. Only in the up conformation the RBD can engage ACE2, which shifts the equilibrium towards the open form of the spike.
5.1. The S2 subunit: a “class I” fusion protein
S2 is responsible for catalyzing the membrane fusion reaction required for entry. This reaction follows S1 dissociation from the spike trimer upon ACE2 binding, which releases S2 from important constrains derived by folding within the context of the S precursor. Structural studies have characterized three structural classes of viral membrane fusion proteins (reviewed by [72]). S2 belongs to class I, together with the fusion proteins found in multiple enveloped viruses, such as paramyxoviruses, influenza viruses, retroviruses and other viral families. All of them undergo a functional conformational change from a metastable pre-fusion to a highly stable post-fusion form to drive fusion. The energy stored in the metastable form is coupled to target-membrane binding to catalyze lipid merger. In particular, the proteins of the three classes were shown to adopt a highly stable “hairpin” conformation in the post-fusion form. In the prototypic class I proteins, such as influenza virus HA2 or HIV-1 gp41, the N-terminal half forms a long “N-helix” that makes a central, parallel trimeric coiled coil, while the C-terminal half runs antiparallel to the N helix (in some cases as a “C-helix, like in gp41) along the grooves of the coiled coil. The resulting “hairpin” is such that its N- and C-terminal ends are at the same end of a long trimeric rod. A hydrophobic segment termed “fusion peptide” (FP) is located immediately upstream the N helix. This segment is responsible for insertion into the target membrane during the first steps of the fusogenic conformational rearrangement, a step absolutely required to drive the fusion process, as outlined in Figure 3A. The prototypical class I fusion proteins, such as those in influenza virus or in retroviruses, have the fusion peptide at the N-terminal end, and the hairpin is formed exclusively by the region in between the FP and the C-terminal TM anchor. In the coronavirus spike protein, similar to the paramyxovirus counterpart, this is not the case as the fusion peptide is at the N-terminus of S2′, which is internal to S2 (see Figure 2A), and regions upstream the S2′ cleavage site are also present as part of the hairpin. The structural similarities between coronavirus S2 and the paramyxovirus fusion (F) protein suggest that they had a common origin [46], forming a distinct subclass compared to the prototypical class I proteins. There are two heptad-repeat regions in the S2′ moiety, termed HR1 and HR2 (labeled in Figure 2A), which have a strong potential to form alpha helices. In the prefusion S trimer, the HR1 region is spring-loaded in the form a bundle of short helical segments under a “crown” formed by the S1 moiety (Figure 3), while HR2 is part of the flexible stalk (Figure 2B). To stabilize the prefusion spike trimer used in the current vaccines was engineered to introduce proline residues at the connections between the short helices of the spring-loaded HR1 [73, 74, 75]. Because prolines are helix-breaking residues, the mutated HR1 cannot spring out to forming a long helix as shown in Figure 3C, resulting in an overall stabilized from that is non-fusogenic.
5.2. Membrane fusion mechanism
The S trimer becomes metastable upon proteolytic maturation into S1 and S2. Subsequent binding to ACE2 on cells shifts the equilibrium of the spike toward the open form, within which the S1/S2 interactions were shown to be weaker [54]. Since each spike is bound to more than one receptor, mechanical constraints have the potential of exerting forces pulling the S1 subunits in opposite directions, ripping off the spike head and releasing the receptor-bound S1 moiety from the spike surface (Figure 2B). Loss of the S1 crown on the activated spike in turn causes the HR1 moiety to spring out and form a long helix and projecting the FP a distance of about 15 nm to reach and stably insert into the target membrane (Figure 3C). This inserted form of S2, bridging the two membranes at a distance of about 30 nm, is a transient long extended intermediate trimer. The HR2 moiety, which is initially distantly located, near the viral membrane, eventually binds to the grooves of the trimeric HR1 coiled coil to make the trimer of hairpins observed in the post-fusion form (shown in Figure 3C, right panel, where only one protomer of the trimer is shown for clarity). In its final conformation, S2 thus forces viral and target membrane against each other, overcoming the repulsion between the two outer leaflets, as in steps (iv) and (v) in Figure 3A. Repulsion arises when two membranes are brought under 1 nm distance due to the required dehydration of the lipid heads to allow direct contact [76, 77]. The hairpin conformation of the fusion protein thus catalyzes the membrane fusion reaction by forcing the close apposition of the outer lipid bilayers, lowering the kinetic barrier for fusion, which is an overall exothermic reaction. Membrane fusion ensues via a hemifusion (step (v) in Figure 3A) in which only the outer leaflets merge, followed by formation of a fusion pore (step vi), which then broadens to make a single, fused membrane.
Spike mutations appearing in recently emerged SARS-CoV-2 variants of concern. (A) Linear diagram of the mature spike protein colored to match the diagram of Figure 3A. The grey bar underneath represents the complete polypeptide, including its signal sequence. On this bar are plotted the mutations appearing on the variants of concern annotated immediately below, as labeled.Note the clustering of mutations in the NTD, the RBD and near the furin cleavage site. (B) The spike head shown in surface representation with the protomers colored differently according to the key labeled “chain”. The mutations appearing in the variants are plotted on the surface colored according to the variant in which they appeared. The trimer in the top row is in the closed conformation, in which the ACE2 binding surface is mostly buried. The bottom panel shows an open spike head with the protease domain of ACE2 bound (yellow ribbons). Note that some of the mutations map directly to the surface of interaction with ACE2. The right panels show an “open book” representation, with the front subunit (labeled A) rotated 180° and showing the B and C subunits of the back, to highlight the buried surfaces in the trimer (colored differently as indicated in the key). Note that some of the mutations map to these internal surfaces, which are likely to affect the dynamics of the S trimer and may play a role in allosteric changes.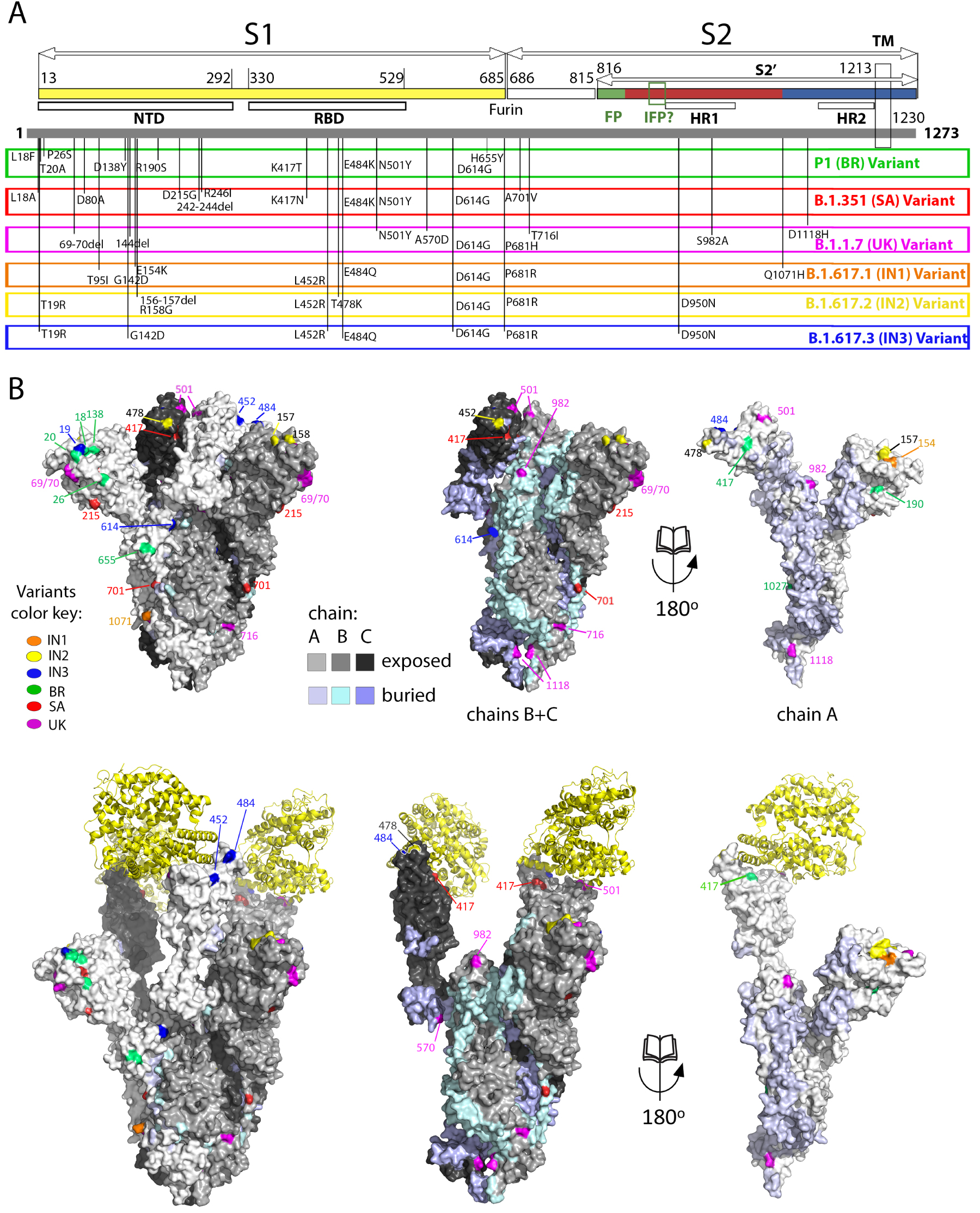
6. The spike protein in the SARS-CoV-2 variants of concern
New SARS-CoV-2 variants with mutations increasing inter-individual transmission or allowing immune evasion have emerged recently and are displacing the initial circulating viral strains [78, 79, 80, 81]. Genomic changes in these VOCs have been associated with increased person-to-person transmission and/or escape from post-infection or post-vaccination immunity. Although the mutations in VOCs occur throughout the viral genome, there are key mutations in the spike protein that control virus transmission and/or are targeted by neutralizing antibodies. The first variant identified presented the D614G mutation in the spike [82], a mutation has now become fixed in the circulating virus and was shown to increase virus transmissibility [83, 78, 84, 85, 86, 87]. As demonstrated recently for the B.1.1.7 variant [88], the variants may also cause more severe disease. The new variants are often referred to by country or region where they were first detected: B.1.1.7 is called the UK variant, B.1.351 the South Africa (SA) variant, P.1 the Brazil (BR) variant, B.1.429 the California variant. Novel variants have also emerged in India, which is undergoing a large surge of the Covid 19 epidemic. Although the predominant strain circulating there has been B.1.1.7, a new lineage, B.1.617, is taking over and is diversifying into several sub-lineages [89, 90, 91, 92, 93, 94, 95]. This variant has now reached other countries, notably the UK where about half of the new cases belong to variant 1.617.2.
Figure 4 lists the positions on the spike of the mutations that have appeared in the main VOCs, including the three new Indian sub-lineages. These mutations cluster in in several regions of the spike head, in particular the N-terminal domain. There is also clustering in the RBD and near the S1/S2 cleavage sites. Some mutations map to the HR1 helix and the chain reversal region in the post-fusion hairpin (between the red and blue halves of S2′ colored as in the diagram of Figure 3A). Although the majority of the mutations map to the trimer surface, a number of them affect contact residues buried at the trimer interface (see Figure 4B, right panels). This is the case for the mutations in the S2 moiety and those that are in S1 near the S1/S2 site (from residue 570 on). An analysis of the amino acid conservation of the spike protein among sarbecoviruses predicted to use ACE2 and infecting different mammals (Figure 5) showed that the location of the VOC mutations at the spike surface lies in variable regions, as expected. Importantly, although the most variable surface maps to the NTD, the ACE2 binding surface of the RBD is is also highly variable (compare the 4th and 5th columns in Figure 5A)—in line with the observation that the ACE2 patch that is recognized by the RBD is also variable across mammals (Figure 1C). The open spike exposes a conserved region of S2 at the spike top that corresponds to the spring-loaded S2 (Figure 5A, right panels). This region does not appear to be immunogenic, most likely because the completely open spike is exposed only transiently, immediately before its irreversible fusogenic conformational change, as indicated by arrows in Figure 5B. The location and relative orientation of the ACE2 binding surface of the RBD (yellow patch in Figure 5B, especially in the 5th panel) is also compatible with a “ripping off” effect wen more than one ACE2 is bound, as they can pull the RBD in opposite directions, thereby disrupting the spike and inducing the conformational change.
Analysis of the amino acid conservation of the spike protein from an alignment of 28 different sarbecoviruses infecting different mammal species predicted to bind ACE2. The selection was based in the conservation of cysteines 480 and 488, which form a disulfide bond that conforms the ACE2 binding surface of the RBD. The Uniprot accession codes of the sequences used are: P0DTC2, A0A023PTS3, A0A0P0INJ4, A0A0U2IWM2, A0A2D1PX29, A0A2D1PXA9, A0A3G5BJ32, A0A3Q8AKM0, A0A4Y6GL47, A0A6B9WHD3, A0A6G6A2R8, A0A6G8I328, A0A6G9KF73, A0A6G9ZWX7, A0A6G9ZWY9, A0A6G9ZX63, A0A6G9ZXI6, A0A6G9ZXM6, A0A6G9ZY67, A0A6G9ZYB3, A0A6G9ZYE7, A0A6H0C589, A0A6H0C6F4, A0A6H0C6Q6, A0A6H0C790, A0A6H0C7A7, A0A6H0C7Y4, A0A6H0JGD1. (A) The spike is shown in the closed (left panels) and open (right) conformations, all in exactly the same orientation, a side view in the first raw and a top view in the second row. The very left and right panels are shown, as a guide, colored as domains as in Figure 2. The central two panels have the surface of ACE2 recognition in blue (labeled “ACE2 patch”), showing that this surface is not accessible in the closed spike. The conservation is plotted as indicated in the horizontal bar at the top. Comparison between the second third columns, and between fourth and fifth, shows that the ACE2 patch is part of a variable surface, in line with the observation that the ACE2 receptor surface that it recognizes also varies across species (see Figure 1C, right panels). The top views also show that in the open spike, a conserved surface of S2 (in green in the sixth column), becomes exposed. This surface, which corresponds to the spring-loaded HR1 helix, is non-immunogenic as is essentially not exposed on infectious particles. This Figure was prepared using the atomic models PDB:7BNM (closed spike) AND 6A98 (open spike). (B) Equilibrium between closed, partially open and open conformations of the spike. The lateral trimers are shown as a guide to define the color code used, with the RBDs in three shades of blue, with pale, middle and dark blue matching the light, middle and dark grey of the remainder of the corresponding protomer. To facilitate the description, the protomers have been labeled A, B and C. The four middle trimers display, in addition, the ACE2 patch in yellow, and the mutations in the variants listed in Figure 4 indicated in the same color code as in Figure 4. Note that these mutations map to regions that are variable across sarbecoviruses (compare with rows 2 and 5 above). The second row in B has all three RBDs down, the third panel has the RBD of protomer A up and the others down, , the fourth panel has RBDs A and C up, and the fourth panel all three RBDs up to make an open spike. Double arrows between panels indicate reversibility between the various conformations. Note that the three RBDs up conformation is unstable and appears to lead to irreversible S1 shedding, as indicated in Figure 3. This is represented by the single arrow pointing down in the fifth panel. The atomic models used in this panel are: PDB 7BNM (closed spike), 7BNN (one RBD up), 7BNO (2 RBDs up), 6A98 (open spike, three RBDs up).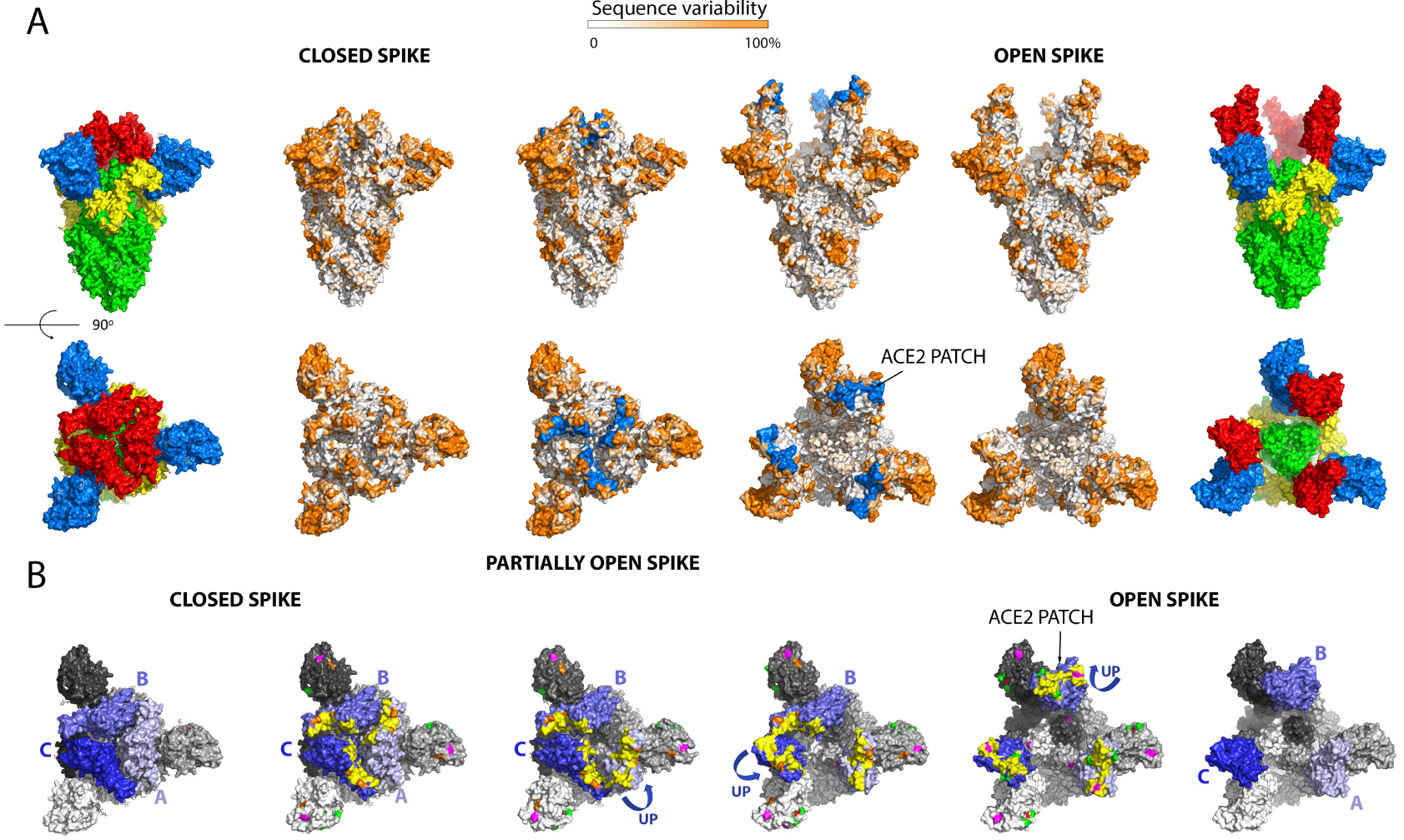
Surface analysis of the RBD, location of main epitopes and mutations in VOCs. (A,B) The RBD surface colored according to its solvent accessibility in the closed conformation of the spike (A) and according to the amino acid conservation (B) as indicated in the color-coded bar. (C) The ACE2 binding surface was superposed in yellow on the same view as in A. The glycan chains interacting with the RBD on the closed spike are shown as sticks colored according to atom type: nitrogen blue, oxygen red, and carbon atoms light grey for the N343 glycan attached to the represented protomer, and yellow for those emanating from adjacent protomers (N149 in a neighboring NTD and N343 in the adjacent RBD), showing that they mask part of the surface not buried by protein atoms as displayed in panel A. Of note, molecular dynamics simulations suggest a gating role of the N343 glycan to allow swinging of the RBD to the Up conformation [96]. The lower panel shows a top view without highlighting the ACE2 patch. (D) The mutations appearing in the VOCs are marked on the same view as in C (without the glycan chains, for clarity). The location of the epitope of the major classes of neutralizing antibodies are indicated by ovals, color coded by the antibody class. The most potently neutralizing antibodies reported belong to classes 1 and 2, which interfere with ACE2 binding. Those in class 1 require the RBD in the Up conformation to bind, as their epitopes include a buried patch in the closed spike (compare with A, left panel). In contrast, those in class 2 can bind the spike in both, open or closed conformations. Note that the mutations on the variants make a rim at the periphery of the ACE2 binding patch, seen more clearly in the top vie (lower panel).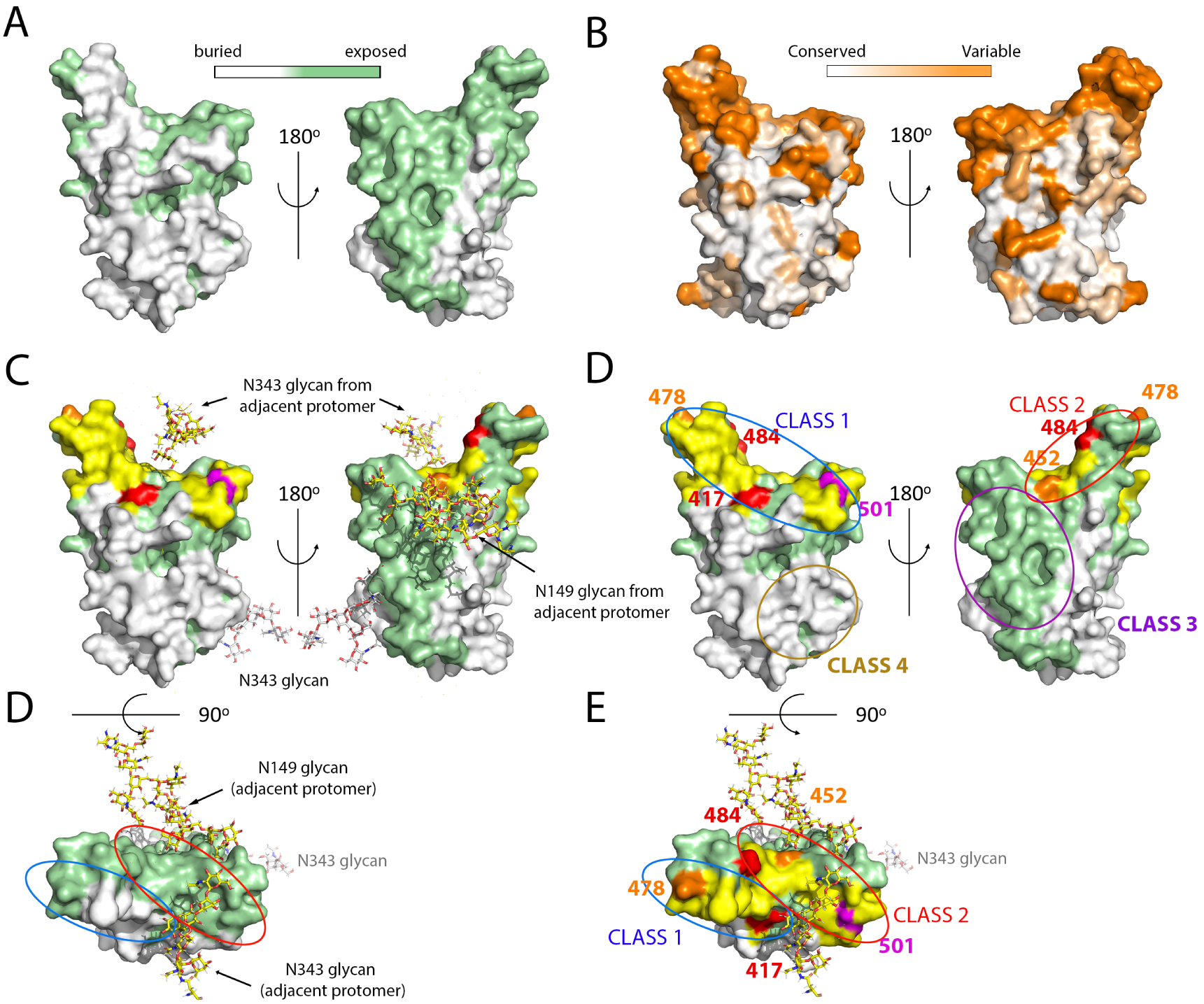
7. VOC mutations in the RBD and escape to neutralizing antibodies
Although the immune response against the spike is directed to multiple sites [97], the RBD has been shown to be the target of the vast majority of the most potent human neutralizing antibodies against SARS-CoV-2. Multiple reports describing the cloning and isolation of monoclonal antibodies (mAbs) from convalescent individuals and vaccinees, together with structural studies by X-ray crystallography and/or cryo-EM have been reported [98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 8, 108]. These studies attest to the potency of the RBD-specific mAbs. Overall, they have been categorized into four broad classes, depending on the location of their epitope at the RBD surface [98]. The most strongly neutralizing mAbs, acting with near picomolar efficiently, target the ACE2 binding patch (yellow surface in Figure 6) [103]. These antibodies are divided into two classes: those in class 1 require the RBD in the up conformation to bind, whereas those in class 2 bind irrespective of having the RBD up or down. The location of the corresponding epitopes is outlined in Figure 6D, which shows that the epitope lo class 1 mAbs includes a region that is buried in the closed conformation of the spike, whereas the class 2 does not (compare panel D, top with panel A, as well as the bottom panels of C and D). Two additional categories of antibodies have been identified in which the epitope is away from the ACE2 patch: class 3 mAbs target an exposed surface, whereas the epitope of class 4 is buried and require at least two RBDs in the up conformation for binding [109, 110, 111, 112]. Class 4 mAbs are among those with the broadest reactivity across sarbecoviruses, [101], as can be seen by comparing panels D and B of Figure 6. The mutations in the VOCs all result in reduced binding of the mAbs of classes 1 and 2, suggesting that these are the dominant form controlling virus circulation in the population in general.
The RBD mutations that have appeared in the VOCs are not randomly distributed: they make a rim all around the periphery of the ACE2 binding surface (Figure 6D, bottom panel). This is in contrast to the variability observed when comparing across sarbecoviruses, in which the whole surface is variable. The notion emerging from this analysis is that the virus samples all mutations that are still compatible with binding to the human ACE2 yet interfering with antibody binding. It remains to be seen how far the virus can continue this trend and escape the human immune response while still maintaining its fitness.
8. Concluding remarks
This review covers most of the structural information available on the coronavirus spike protein, its interaction with ACE2, and the mechanistic information that can be extracted from the structural studies. The location of the mutations in the new SARS-CoV-2 variants, distributed throughout the spike surface, further suggest that there is epistasis between them and also that a likely allosteric effect may expose differently the three cleavage sites to furin, a feature that appears to be functionally important. The mutations may also affect in multiple ways the conformational change triggered by receptor binding. The biology of the coronavirus spike is far from having revealed its secrets, and there remains a great deal to learn. Still, the amount of information gathered on this protein in 18 months of the pandemic has been unprecedented, as is the pandemic itself. Only by understanding the spike function in mechanistic detail—as well that of the other viral proteins involved in other steps of the viral cycle—can we expect to be better prepared to face future outbreaks of emerging zoonotic viruses such as novel coronaviruses.
Acknowledgments
I thank my colleagues in the structural virology unit for discussions and the members of the Pasteur Coronavirus Task Force for support. All the structural illustrations used were prepared with The PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC. The plotting of amino acid conservation to the protein surface was done using EsPript [113]. I thank P. Guardado-Calvo for tips about using this software.
French version
1. Introduction
À la mi-mai 2021, la pandémie de SRAS-CoV-2 a provoqué plus de 160 millions de cas et plus de 3,4 millions de décès ont été signalés à l’Organisation mondiale de la santé (https://covid19.who.int/). Dans le même temps, plus de 1,4 million de doses de vaccin ont été administrées dans le monde et une course au virus s’est engagée, dans laquelle, à mesure que davantage de personnes sont immunisées par une infection naturelle ou grâce à la vaccination, de nouveaux variants du virus émergent et prennent le dessus sur les souches précédemment dominantes [1]. Ces variants sont sélectionnés parce qu’ils sont plus aptes à circuler en présence d’une immunité collective. Il reste à comprendre jusqu’à quel point le virus peut évoluer face à la réponse immunitaire humaine tout en conservant son aptitude, et quelle sera l’efficacité des vaccins actuels à long terme pour arrêter sa dissémination et le poids économique imposé à la société.
Le développement de vaccins nouveaux et efficaces s’est fait à un rythme sans précédent, en partie parce que le moment était propice pour tester de nouvelles stratégies telles que les vaccins à ARNm, qui n’avaient été essayés que comme stratégies anticancéreuses [2, 3]. Ou comme l’utilisation de vecteurs adénoviraux, qui avait été avancée lors de l’épidémie d’Ebola de 2014–2016 en Afrique de l’Ouest, une épidémie qui avait reculé avant que de grands essais cliniques de vaccins puissent être déployés [4]. Le point commun de ces vaccins anti-Covid-19 réussis est l’immunogène qu’ils portent, obtenu en appliquant de nouveaux concepts basés sur la structure de la protéine de spicule (S) [5]. Cette protéine a ainsi été stabilisée dans un état inactif imitant sa forme métastable « pré-fusion » présente à la surface des particules virales infectieuses. En effet, la protéine S est la seule cible des anticorps neutralisant le virus, mais elle est intrinsèquement métastable car elle doit se désagréger pour permettre l’entrée du virus dans les cellules en induisant la fusion de l’enveloppe virale avec la membrane de la cellule cible. La biologie structurale a joué un rôle très important dans la conception de stratégies permettant de faire face à son instabilité en élaborant des versions stables qui préservent les épitopes pertinents à présenter au système immunitaire pour susciter des anticorps fortement neutralisants. Des études antérieures sur d’autres virus enveloppés, tels que le VIH et le virus respiratoire syncytial (VRS, responsable de bronchiolites graves chez les nourrissons, les personnes âgées ou immunodéprimées), qui présentent également une protéine d’enveloppe très instable à leur surface, ont ouvert la voie aux travaux sur les coronavirus [6, 7]. Pour le VIH et le VRS, la situation est plus complexe car les épitopes ciblés par les anticorps les plus puissants et les plus largement neutralisants ne sont présents que dans l’ectodomaine intact plié sous sa forme de pré-fusion. Cependant, nous savons maintenant que les anticorps les plus puissants et les plus neutralisants contre les virus semblables au SRAS sont dirigés vers le domaine de liaison au récepteur (RBD). Ce domaine présente l’avantage de pouvoir être produit indépendamment du reste de la protéine, présentant par lui-même des épitopes pertinents pour le système immunitaire afin de susciter des anticorps neutralisants [8]. Cependant, la formulation utilisée dans les vaccins actuels, qui utilise la protéine S complète stabilisée par deux mutations internes en proline, s’est avérée remarquablement efficace pour stimuler la production d’anticorps protecteurs, en particulier lorsqu’elle est administrée sous forme d’ARNm [9, 10].
Je présente ci-dessous une description du récepteur ACE2 et de la protéine S ainsi qu’une analyse de la conservation des surfaces d’interaction des deux protéines chez les mammifères et chez les coronavirus du type SRAS. Je résume également les connaissances acquises au fil des ans sur le mécanisme de fusion membranaire piloté par les virus enveloppés et la manière dont elles s’appliquent au SRAS-CoV-2. Je termine en illustrant le fait que les mutations observées dans les variantes en cause présentent un regroupement clair sur trois régions de la pointe. En particulier, dans le domaine de liaison aux récepteurs, elles sont cartographiées sur un pourtour tout autour de la surface d’interaction avec l’ACE2.
2. Les Sarbecovirus
La famille des Coronaviridae est divisée en deux sous-familles. Les coronavirus classiques sont regroupés dans la sous-famille des Orthocoronavirinae, qui contient quatre genres : α-, β-, γ- et δ-coronavirus. Seuls les genres α- et -β-Coronavirus comprennent des virus pathogènes pour l’homme. Les virus semblables au SRAS forment un sous-genre, appelé Sarbecovirus, au sein du genre β-Coronavirus. Parmi les quatre sous-genres de ce genre, seul un sous-ensemble de sarbecovirus utilise l’ACE2 comme récepteur [11]. Dans ce contexte, il est intéressant de noter que le coronavirus saisonnier humain NL63, qui appartient au genre α-Coronavirus et qui est donc assez éloigné des sarbecovirus, utilise également l’ACE2 comme récepteur [12], bien que son domaine de liaison au récepteur et son mode de reconnaissance de l’ACE2 soient sans rapport avec ceux des sarbecovirus [13], ce qui suggère une convergence dans l’évolution pour utiliser le même récepteur. Un autre résultat qui mérite d’être mentionné est que, pour la plupart des coronavirus, le récepteur est une peptidase liée à la membrane. Par exemple, l’amino-peptidase N (APN) est utilisée par le α-coronavirus saisonnier humain 229E [14] ainsi que par les α-coronavirus porcins [15]. L’APN est une aminopeptidase métal-dépendante jouant de multiples fonctions physiologiques, régulation de la douleur, homéostasie de la pression sanguine, angiogenèse des cellules tumorales, entre autres [16]. De même, le β-coronavirus du syndrome respiratoire du Moyen-Orient (MERS-CoV) utilise la dipeptidyl peptidase 4 (DPP4) comme récepteur [17]. La DPP4 est une sérine protéase qui clive l’extrémité N-terminale des peptides physiologiques impliqués dans le contrôle de l’apoptose, la régulation immunitaire et la transduction des signaux [18, 19, 20]. L’activité catalytique n’est pas requise pour la reconnaissance par le virus de l’un de ces récepteurs peptidases, et le site de liaison reconnu par la protéine S est éloigné du site catalytique. Pourtant, il est frappant de constater que divers coronavirus de genres différents ont convergé pour utiliser des peptidases de surface cellulaire pour entrer dans la cellule, et les relations potentielles avec leur pathogénicité restent un champ d’investigation ouvert [21].
3. Le récepteur ACE2
En 2003, Farzan et ses collègues ont identifié l’enzyme 2 de conversion de l’angiotensine (ACE2) comme le principal récepteur du coronavirus du syndrome respiratoire acquis sévère (SARS-CoV) [22]. Il a été montré que le nouveau coronavirus, SARS-CoV-2, utilise le même récepteur [23]. L’ACE2 est une protéine transmembranaire à passage unique de type I présente à la surface des cellules de multiples tissus dans divers organismes. C’est un composant du système rénine–angiotensine (SRA) qui contrôle le volume sanguin et la résistance vasculaire systémique, jouant un rôle crucial dans la protection contre l’hypertension artérielle et les maladies cardiovasculaires [24]. Les autres composants de ce système hormonal sont la rénine, protéase aspartique soluble, et le précurseur de l’angiotensinogène (AGT), ainsi que l’enzyme de conversion de l’angiotensine (ECA) liée à la membrane et différents récepteurs couplés aux protéines G (GPR) spécifiques des divers peptides d’angiotensine dérivés du clivage de l’AGT. Le clivage de l’AGT par la rénine est l’étape limitant la vitesse de réaction [25] et est étroitement régulé pour maintenir une pression sanguine normale, évitant ainsi l’hypertension et d’autres maladies cardiovasculaires. L’AGT est une protéine circulante de 452 acides aminés (aa) qui appartient à la famille des serpins, des inhibiteurs de protéase [26, 27], bien qu’elle n’ait pas de fonction inhibitrice. La rénine clive l’AGT à son extrémité N-terminale pour générer l’angiotensine I (Ang-I) [28, 29], un décapeptide qui est le substrat de l’ECA, qui le convertit en Ang-II (Figure 1). L’hormone octapeptidique Ang II joue un rôle important dans l’homéostasie de la pression artérielle grâce à son interaction avec le GPR AP1, ce qui entraîne une augmentation de la pression artérielle. Le rôle de la carboxypeptidase ACE2 est d’éliminer le résidu phénylalanine C-terminal de l’Ang II pour produire l’Ang (1-7) [30, 31, 32], qui se lie au GPR Mas [33, 34]. L’interaction Ang (1, 7) / GPR Mas entraîne une vasodilatation et une protection contre une pression sanguine excessive, contribuant ainsi à l’homéostasie de la pression vasculaire. L’infection par un virus est connue pour provoquer une régulation négative de l’expression de son récepteur d’entrée, comme c’est le cas par exemple avec le VIH et son récepteur CD4 [35]. La raison en est que de faibles quantités de récepteurs dans les cellules infectées aident le virus à se propager loin du site où il s’est répliqué dans un organisme pour atteindre plus facilement d’autres tissus. La régulation à la baisse (downregulation) du récepteur ACE2 du SRAS-CoV-2 pourrait donc entraîner un déséquilibre du rapport entre les peptides Ang II / Ang(1/7), ce qui pourrait contribuer à sa pathogénicité.
Le système rénine–angiotensine et l’organisation structurale de l’ACE2. (A) Protéolyse par ACE2 des peptides d’angiotensine. La séquence d’acides aminés de l’angiotensinogène (ATG) (tronquée entre les résidus 51 à 481, comme indiqué). L’extrémité amino-terminale de l’ATG est générée co-traductionnellement par une signalase dans la lumière du RE (ciseaux gris). L’ATG sécrété réagit à des signaux redox environnementaux pour exposer son segment N-terminal pour être clivé par la rénine et générer le décapeptide angiotensine I (Ang I) (montré sur fond jaune) [28, 29]. Le site de clivage de la rénine est marqué par le ciseau noir à droite. L’enzyme de conversion de l’angiotensine (ECA) clive ensuite l’Ang I au niveau du site indiqué par le ciseau noir de gauche, pour générer l’Ang II, un peptide très actif qui stimule la vasoconstriction via une interaction avec son GPR AP1 présent au niveau de la membrane plasmique de multiples cellules. Un excès d’Ang II entraîne une hypertension artérielle et des maladies vasculaires, et cet effet est contrebalancé par l’ACE2 (ciseaux violets sous la séquence), qui a une activité carboxypeptidase et clive les résidus C-terminaux de l’Ang I et de l’Ang II, pour générer respectivement l’Ang (1-9) et l’Ang (1-7).L’ACE2 clive le résidu C-terminal de l’Ang II avec une efficacité environ 400 fois supérieure à celle du clivage de l’Ang-I pour produire l’Ang (1-9). L’Ang (1-7) induit une vasodilatation par son interaction avec le GPR Mas. L’activité de l’ACE-2 est donc critique dans le système SRA. Les cercles verts et rouges représentent, respectivement, une asparagine glycosylée et une cystéine qui forme un pont disulfure labile avec une autre cystéine ATG située 120 aa en aval dans la séquence aa (non montré). Il a été montré que ces deux résidus jouent un rôle dans la reconnaissance et le clivage par la rénine [29]. (B) Diagramme linéaire de l’ACE, de l’ACE2 et de la collectrine, montrant que l’ACE2 est une protéine chimérique dont le domaine N-terminal provient de l’ACE et le C-terminal de la collectrine. Les limites du domaine sont indiquées, et les flèches verticales pointent vers différentes boucles du domaine de la collectrine qui sont clivées pour libérer l’ACE2 soluble dans le milieu. ADAM17 clive dans la boucle 652–659, tandis que TMPRSS2 clive dans la boucle 697–716 [36]. (C) Organisation de l’ACE2 sur les membranes. La structure de l’ACE2 a été déterminée par cryo-EM en tant que complexe binaire avec le transporteur transmembranaire d’acides aminés B0AT1 (panneau de gauche, PDB: 6M18) et en tant que complexe ternaire avec le domaine de liaison au récepteur (RBD) de la S du SRAS-CoV-2 (panneaux de droite, PDB : 6M17) [37]. La membrane plasmique est indiquée dans le panneau de gauche, à titre indicatif. ACE2 est représenté en surface tandis que B0AT1 et le RBD sont représentés par des rubans en bleu et en rose, respectivement. Dans le panneau de gauche, ACE2 est coloré en vert et jaune selon les domaines comme dans le diagramme linéaire, les deux protomères étant représentés dans des nuances différentes de vert et de jaune. De même, les deux protomères du transporteur B0AT1 sont représentés dans des teintes bleues différentes. Notez que les hélices transmembranaires de ACE2 font partie intégrante du transporteur et contribuent à son interface dimèrique. Dans le panneau de droite, ACE2 est coloré selon la conservation de sa séquence d’acides aminés à partir d’un alignement de 24 séquences ACE2 provenant de divers mammifères. Cet alignement montre que la conservation de surface est élevée dans la fente qui porte le site actif. Le RBD se lie loin de cette fente à une surface exposée qui varie selon les espèces.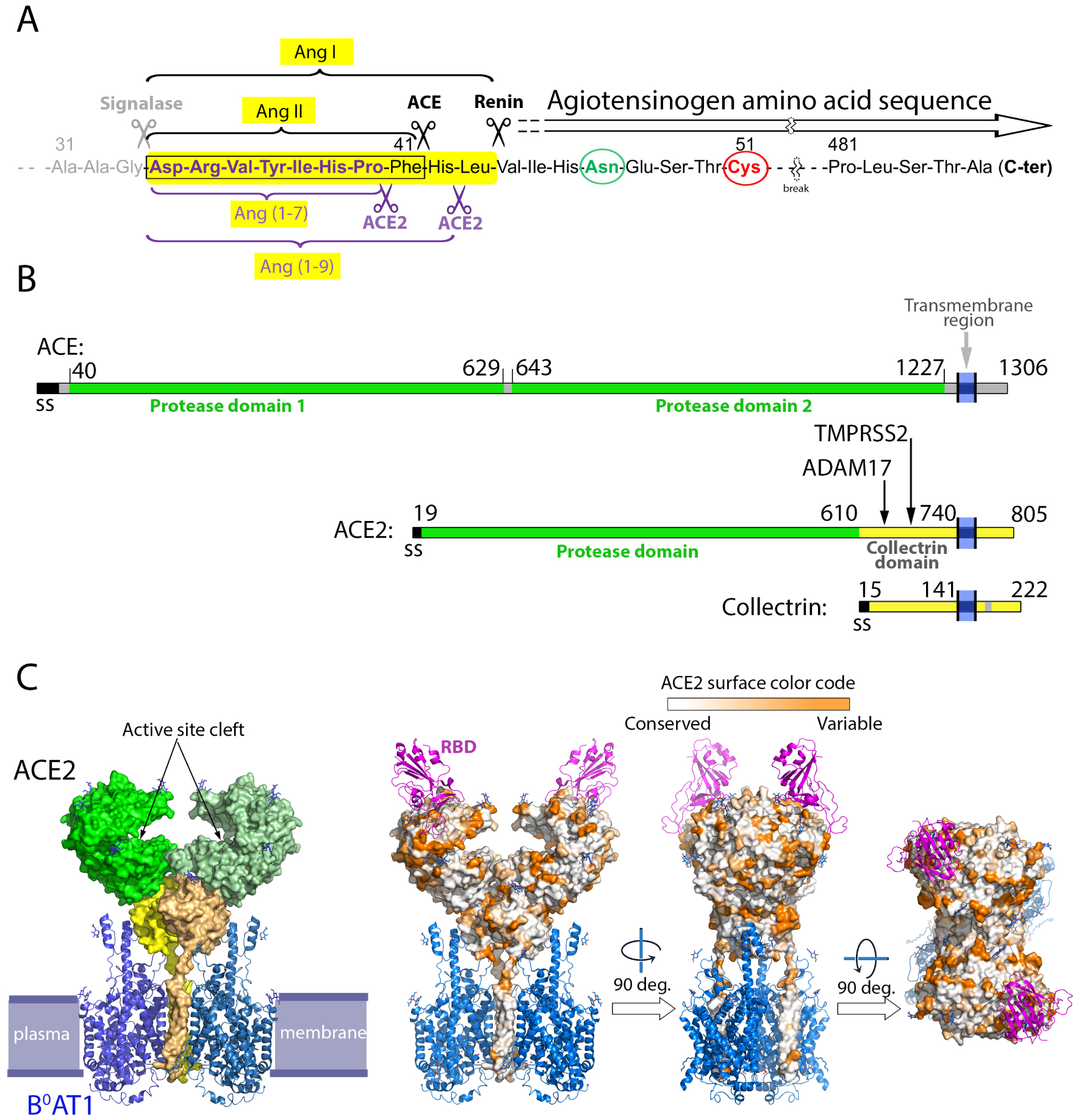
L’ACE2 humaine a été identifiée pour la première fois en 2000 [30, 31] comme un paralogue de l’enzyme de conversion de l’angiotensine (ACE), découverte en 1956 [38]. Le gène correspondant, situé en position p22.2 sur le chromosome X, contient 18 exons et donne naissance à plusieurs isoformes d’ACE2 par épissage alternatif. L’isoforme la plus longue contient 805 acides aminés et est exprimée dans un large éventail de tissus humains, y compris les cellules endothéliales des vaisseaux sanguins, les entérocytes de l’intestin, les cellules alvéolaires du poumon, les cellules épithéliales nasales, tous des organes qui sont la cible du SRAS-CoV-2. L’organisation de cette isoforme de l’ACE2 est représentée sur la Figure 1B. Son ectodomaine est composé de deux domaines structurels [39] : un domaine catalytique N-terminal avec une activité zinc métallopeptidase présentant 42% d’identité de séquence d’acides aminés avec chacun des deux domaines catalytiques de l’ACE [40] (Figure 2). Le domaine protéase de l’ACE2 est suivi d’un domaine de type collectrine, qui s’associe avec le transporteur d’acides aminés neutres B0AT1 (Figure 1C), au moins dans les cellules épithéliales intestinales. Il a été démontré que cette association est nécessaire pour l’expression du transporteur sur la surface luminale des cellules épithéliales intestinales [41, 42]. Le domaine collectrine de l’ACE2 humain partage 50% d’identité de séquence avec la collectrine humaine, une glycoprotéine transmembranaire à passage unique de type I qui se lie aux transporteurs d’acides aminés de type B0, tels que B0A1 et B0A3. Ces transporteurs, qui appartiennent à la famille des transporteurs de solutés 6 (SLC6) [43], ne sont actifs que lorsque la collectrine est liée [44]. Il a également été proposé que l’activité de la collectrine joue un rôle dans l’homéostasie de la pression sanguine [45].
Organisation structurale du spicule des coronavirus. (A) Schéma linéaire de la protéine S indiquant la position des sous-unités individuelles S1 et S2, les segments transmembranaires (TM), les domaines individuels et les sites de clivage. Les numéros des acides aminés correspondent à la séquence SARS-CoV-2, souche originale de Wuhan. Les régions « tête » et « tige » indiquées en B sont également marquées. Le peptide de fusion est indiqué par une région hachée blanche en aval du site de clivage S2′. Une fine boîte violette englobe une région de 140 résidus de S2 (aa 772-912 dans SARS-CoV-2 S), englobant le FP et le site de clivage S2′, qui n’a pas été résolue dans les structures cryo-EM disponibles du coronavirus S2 [46, 47]. (voir également la Figure 3.) (B) Représentation en ruban du spicule tel que modélisée par dynamique moléculaire [48] sur la membrane virale et basée sur des structures de tomographie cryo-électronique de résolution intermédiaire du spicules sur des particules virales inactivées [49, 50, 51]. La chaîne polypeptidique du protomère au premier plan est colorée selon les domaines comme en A. Les lipides de la membrane modélisée par dynamique moléculaire et les chaînes de sucre complètes des glycanes attachés à la protéine sont représentés sous forme de bâtonnets colorés selon le type d’atome (carbone, azote, oxygène et soufre en gris clair, bleu, rouge et jaune, respectivement). Les dimensions approximatives sont indiquées sur le côté, avec la partie flexible « tige » et la partie dynamique plus globulaire « tête » du spicule trimérique étiquetées. L’extrémité C-terminale du segment TM du côté intraviral de la membrane est marquée en rose. (C et D) Structures Cryo-EM de l’ectodomaine du spicule, montrant seulement la portion de tête, avec le protomère du front coloré selon les domaines comme dans A. Le trimère est montré dans la forme fermée avec trois RBDs couchés (C) et avec un RBD debout et les deux autres couchés (D) dans un spicule partiellement ouvert. Sur les virions, la tête du spicule présente un équilibre dynamique entre la conformation fermée et les formes partiellement ouvertes. Les virions présentent également S2 en post-fusion, donc inactive, résultant vraisemblablement de la perte spontanée de S1 et de l’activation prématurée du spicule [49] (voir Figure 3). Les modèles affichés correspondent à PDB : 7BNM (C) et 7BNN (D), déterminés à une résolution d’environ 3,5 Å [52].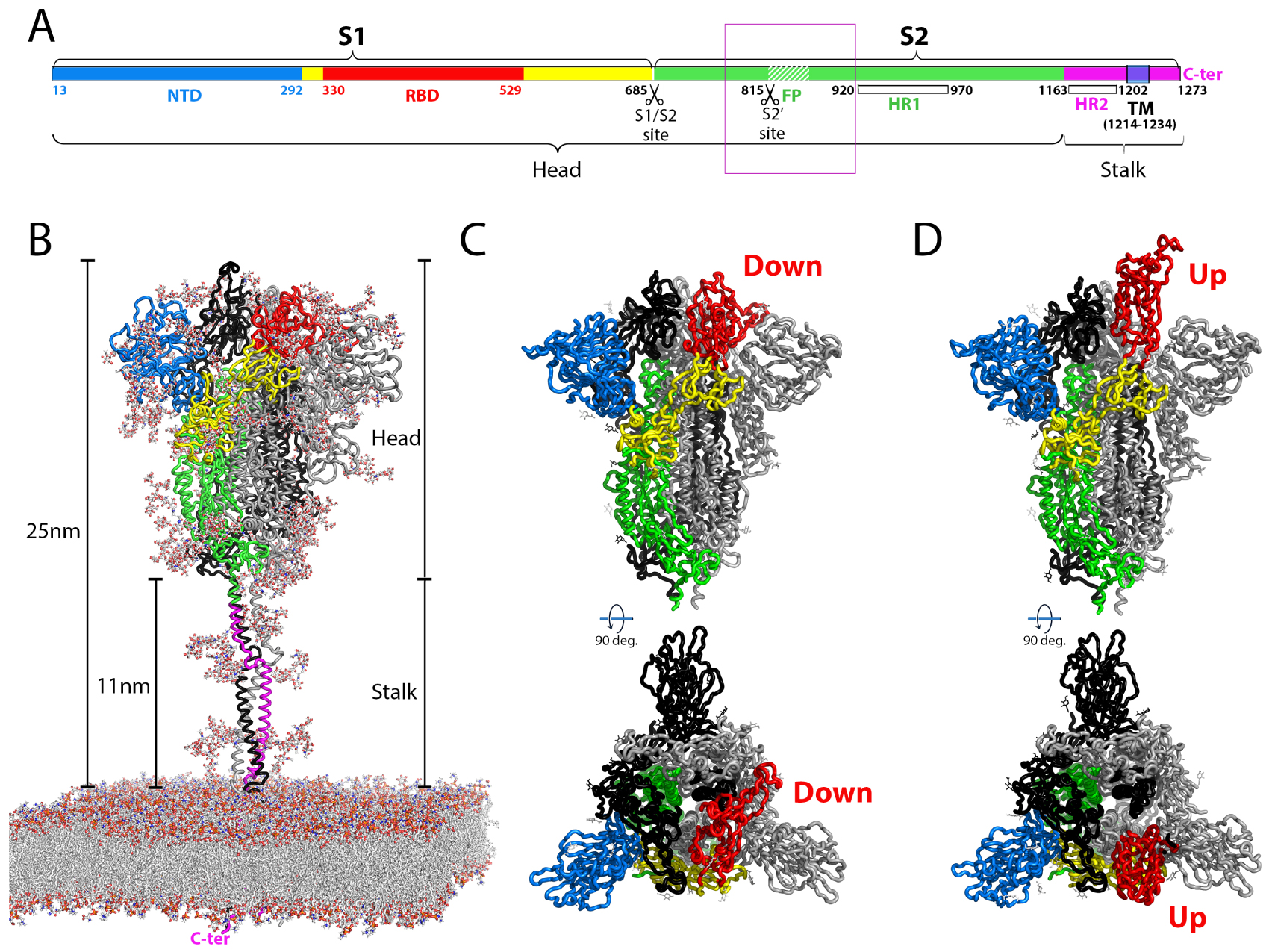
La protéine S se lie à la surface de l’ACE2 la plus distale de la membrane, loin de la fente du site actif (Figure 1C, panneaux de droite). Les comparaisons de séquences d’acides aminés montrent que le patch de surface reconnu par le RBD est variable à travers les différentes espèces de mammifères, contrairement à la fente hautement conservée qui abrite le site catalytique de l’ACE2. La structure montre également que les deux sites d’interaction avec S, présents dans le dimère (ACE2/B0A1) sont situés de telle sorte que deux RBDs du même spicule trimérique ne peuvent pas se lier simultanément, ce qui implique que deux spicules trimèriques différents se lient à un dimère du récepteur [37]. Cette situation a le potentiel de regrouper les spicules sur le côté du virion faisant face à la membrane cellulaire, où chaque spicule peut lier jusqu’à trois dimères ACE2, chacun étant lié à un second spicule, ce qui entraîne la formation de clusters. Jusqu’à présent, aucune étude ne s’est penchée sur la stœchiométrie de la liaison et sur la question de savoir si le regroupement proposé aurait un effet fonctionnel sur l’entrée du virus, soit en déclenchant la fusion directement au niveau de la membrane plasmique, soit en stimulant l’endocytose avec fusion ultérieure dans les endosomes tardifs.
3.1. La glycoprotéine S du coronavirus
La chaîne polypeptidique de S du sarbecovirus est longue d’environ 1300 acides aminés et comporte de multiples glycanes liés à l’asparagine dans son ectodomaine. Elle forme un trimère ancré à la membrane virale par une hélice transmembranaire C-terminale (Figure 2). S est activé par clivage protéolytique pour générer deux sous-unités : S1 (en gros la moitié N-terminale de S), et S2, la moitié C-terminale ancrée à la membrane. Le SARS-CoV-2 présente une insertion d’une séquence polybasique au niveau du site de clivage S1/S2, ce qui permet la protéolyse de S par la furine, une protéase résidant dans le réseau transgolgien dans les cellules productrices du virus. Le clivage par la furine ne semble pas avoir lieu de manière extensive, et la plupart des trimères de S ne sont que partiellement clivés (c’est-à-dire que le clivage ne se produit que dans un ou deux des trois protomères formant le trimère) [47]. Un tel clivage partiel peut avoir des conséquences fonctionnelles, comme le propose la Figure 3B.
Activation du spicule pour provoquer la fusion membranaire et l’entrée du virus. (A) Diagramme résumant les principales étapes actuellement acceptées du processus de fusion du virus déclenché par la liaison au récepteur. La protéine de fusion est ancrée par son extrémité C-terminale à la membrane virale, et est activée protéolytiquement par clivage en une sous-unité N-terminale (NSU, en jaune), et une sous-unité C-terminale (CSU, en rouge et bleu). Le peptide de fusion, indiqué en vert et étiqueté FP (i), est situé immédiatement en aval du site de clivage. Lors de l’activation protéolytique, il devient l’extrémité N-terminale de la CSU. Ce clivage activateur rend la protéine de fusion métastable, avec le CSU monté sur ressort sous une couronne de NSU (i).Le CSU réagit à des facteurs tels que la liaison à un récepteur ou l’acidification du milieu (c’est-à-dire dans un endosome, selon le virus) pour déclencher la fusion par un réarrangement conformationnel majeur. Dans le cas des coronavirus, le modèle actuel postule que la liaison au récepteur induit le détachement de la NSU (c’est-à-dire la sous-unité S1) du trimère (ii), ce qui permet à la CSU de jaillir et de projeter le FP dans la membrane cellulaire, où il s’insère fermement dans la première étape du réarrangement fusogène (iii). Cette forme « intermédiaire allongée » n’est que transitoire, et se replie en une conformation en « épingle à cheveux » dans laquelle la moitié N-terminale (en rouge) reste liée à la membrane cible sous la forme d’une d’un trousseau trimérique torsadé d’hélices, tandis que la moitié C-terminale du CSU se déplace en sens inverse le long des rainures de la bobine centrale. La formation de l’épingle à cheveux implique donc la juxtaposition des extrémités N- et C-terminales de la CSU, forçant ainsi les deux membranes l’une contre l’autre (iii, iv et v)). La fusion se produit d’abord par la fusion des feuillets externes, un processus appelé « hémi-fusion » (v) et est suivie par la formation d’un pore de fusion (vi), qui s’élargit ensuite pour donner naissance à une seule membrane fusionnée. (Adapté de [53]). (B) Compréhension actuelle du processus d’activation du SRAS-CoV-2 pour conduire la fusion membranaire. Le trimère de spike est représenté avec la sous-unité S1 en surface et S2 sous forme de rubans. Un protomère est en couleurs et les autres en gris et noir. La sous-unité S1 du protomère du front dans le trimère est en jaune pour correspondre à la NSU représentée en A. La sous-unité S2 du front est colorée en rampe bleu-vert-jaune-orange-rouge de son extrémité N- à C-terminale (voir la barre sous le panneau C), soulignant que sa partie N-terminale est avec HR1 montée sur ressort sous la couronne de S1, tandis que sa partie C-terminale est dans la tige, comme indiqué dans les Figures 2A et B. Le récepteur ACE2 en complexe avec B0AT1 dans la membrane cellulaire est représenté en surface avec les deux protomères du dimère ACE2 différenciés en rouge foncé et pâle, tandis que B0AT1 est en gris clair. L’étape centrale (étiquetée « (0) ») montre le trimère du spicule avec la tête fermée, qui est en équilibre avec des formes partiellement ouvertes (comme celle montrée dans la Figure 2C). La liaison de l’ACE2 déplace l’équilibre vers des formes ouvertes (étape étiquetée (1)). Des études structurales ont montré que dans la tête ouverte, les interactions S1/S2 sont plus faibles, en particulier lorsque les trois RBDs sont debout [54]. Il a été proposé que les spicules entièrement clivés par la furine en S1 et S2 sont instables et lâchent spontanément S1 lors de l’adoption de la conformation ouverte (étape (−1)), ce qui entraîne un changement de conformation prématuré et l’insertion de la S2 post-fusion dans la membrane virale (étape (−2) [47]). Dans ce panneau, la forme post-fusion contient une connexion dessinée à la main entre les résidus 772 et 912, qui sont désordonnés dans les structures cryo-EM [46, 47] (région marquée aussi dans le diagramme linéaire de la Figure 2A). Notez que la conformation S2 du panneau de gauche correspond à l’étape vi de la Figure 3A, et non à celle de l’étape ii, qui représente le CSU toujours dans la conformation de pré-fusion chargée par ressort. L’étape (1) dans B correspond à une étape entre i et ii dans A, où le spicule a déjà lié le récepteur ACE2. Les forces de traction résultant de l’écoulement ou d’autres fluctuations entre la particule liée et la cellule entraînent alors la libération de S1, une étape qui nécessite l’achèvement du clivage entre S1 et S2 sur le spicule. Ce clivage est effectué par TMPRRSS2 à la surface de certaines cellules, ou par la cathepsine lors de l’endocytose des virions après liaison au récepteur [55]. Dans ce contexte, le détachement de S1 conduit à l’insertion de la boucle de fusion dans la membrane de la cellule lors de la projection de l’hélice HR1, comme indiqué dans l’étape iii de (A). La partie HR2 complète alors la conformation en épingle à cheveux en se liant aux rainures du trousseau hélicoïdal trimérique formée par HR1, structure représentée dans l’étape (3). (C) Projection de l’hélice HR1. Les quatre panneaux représentés illustrent la transition entre les étapes 1 et 3 de la Figure 2B, avec seulement S2 représenté en rubans colorés en rampe de l’extrémité N- à C-terminale selon le code couleur de la barre en dessous. Pour simplifier, un seul des trois protomères du trimère S2 est représenté. Le panneau de gauche montre HR1 ainsi que les segments en amont du site de clivage de S2′ placés sous la couronne S1 dans la forme de pré-fusion. La sous-unité S1 et le récepteur ACE2 sont schématisés et colorés selon la Figure 3A dans les étapes i et ii. La dissociation de la couronne S1 liée au récepteur induit la HR1 à jaillir et à atteindre la membrane cible (deuxième panneau). Simultanément, le site de clivage de S2′, qui est enfoui et inaccessible dans la forme montée à ressort de S2 en pré-fusion, devient exposé pour être clivé soit par TMPRSS2 à la surface cellulaire, soit par les cathepsines dans les endosomes. Les flèches noires courbes indiquent le repliement ultérieur de S2 dans l’épingle à cheveux stable post-fusion, rapprochant les deux membranes comme indiqué dans la Figure 3A, étapes iv à vi. On ne sait pas si le clivage au site S2′ a lieu avant ou après la formation de l’épingle à cheveux, et les deux options sont représentées (troisième et quatrième panneau). Il a été montré que le FP est nécessaire à l’extrémité N-terminale de S2′ pour que la fusion membranaire ait lieu, mais le site de clivage n’est exposé qu’après le changement de conformation. Son emplacement dans la forme non clivée est trop éloigné pour atteindre la membrane cible (comme marqué). Nous postulons que la région immédiatement en amont de HR1 est également impliquée dans l’insertion dans la membrane cible. Des analyses de sa séquence d’acides aminés indiquent que cette région, appelée « peptide de fusion interne » dans la figure, a une forte propension à former une hélice amphipathique. Sa liaison au feuillet externe de la membrane cible permettrait d’exercer le bon effet de levier pour amener les deux membranes en apposition, même si le clivage en S2′ se produit après le changement de conformation. Sinon, le trimère S2 se retournerait et finirait par s’insérer dans la membrane virale, comme on l’a observé sur les particules virales et comme indiqué sur la Figure 3B, panneau de gauche. L’effet du segment IFP n’a cependant pas encore été testé expérimentalement.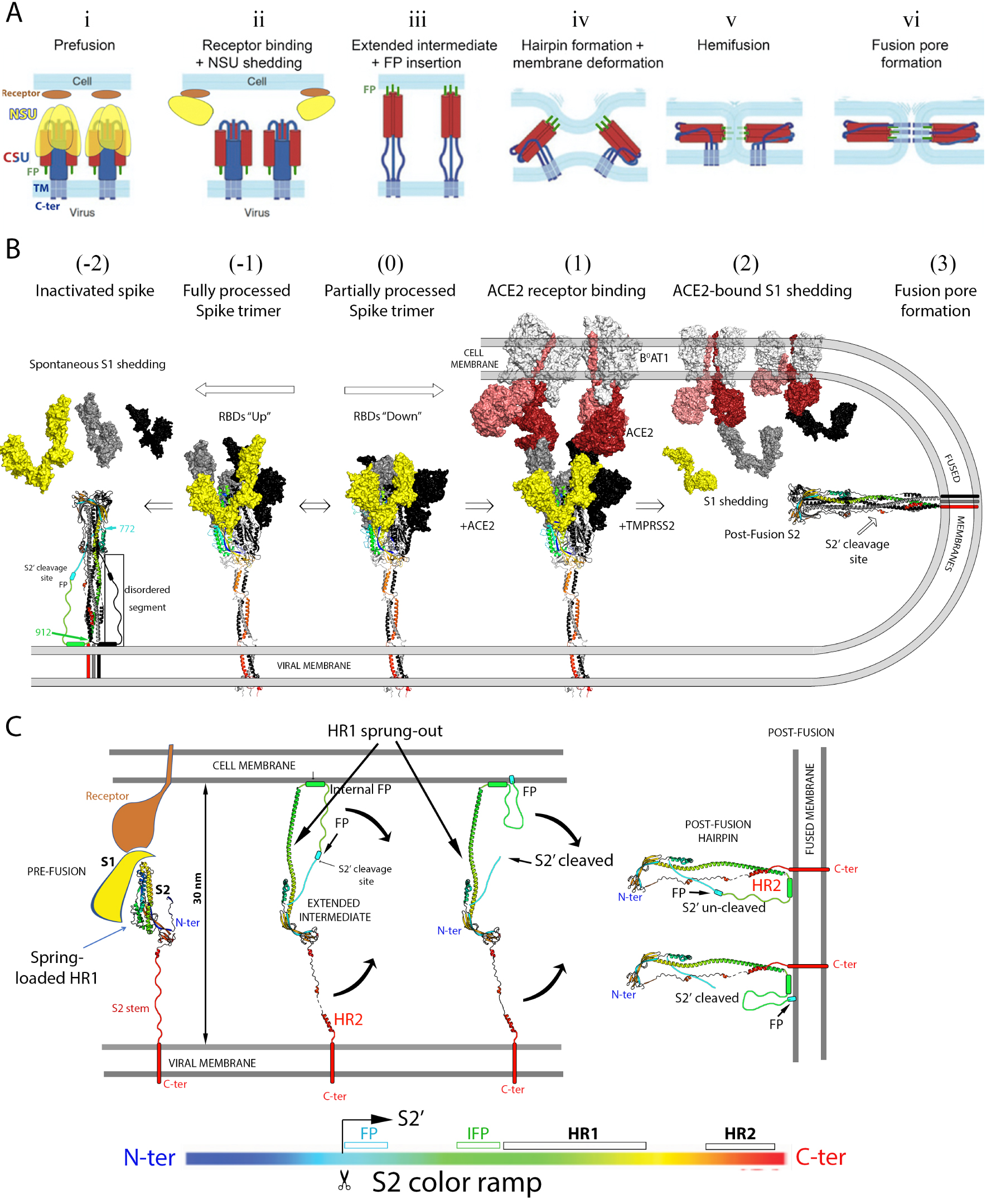
4. L’insertion du site de clivage de la furine
Il a été montré que le site polybasique de S du SARS-CoV-2 est impliqué dans les interactions avec la neuropiline 1 [56, 57]. Les neuropilines lient les facteurs de croissance endothéliaux vasculaires (VEGFs) au niveau de la séquence carboxyl-terminale générée par le clivage par la furine du précurseur du VEGF [58]. Les neuropilines sont présentes dans l’épithélium olfactif et pourraient jouer un rôle dans l’anosmie observée chez les patients atteints de Covid-19 [59] et potentiellement dans l’invasion du système nerveux central par le virus [60]. Le même site polybasique de la furine a également été proposé pour interagir avec le récepteur nicotinique de l’acétylcholine (nAChR) [61], par analogie avec un motif similaire présent dans la glycoprotéine du virus de la rage et dans plusieurs neurotoxines de serpent [62]. Aucune preuve n’a cependant été apportée jusqu’à présent d’une interaction directe entre le nAChR et le site polybasique. Une preuve indirecte réside dans le fait que l’ivermectine (IVM), un composé antiparasitaire qui est également un effecteur allostérique positif du nAChR neuronal α7 [63], atténue les symptômes neurologiques dans le modèle hamster de Covid 19 [64]. L’IVM ne réduit la charge virale chez les hamsters infectés, mais uniquement les symptômes pathologiques du Covid 19, contredisant un rapport précédent [57] qui suggérait que l’IVM avait un effet antiviral direct. En tout état de cause, quel que soit le rôle de la séquence polybasique, des études indépendantes ont démontré que les mutants du SRAS-CoV-2 dans lesquels elle est supprimée sont moins pathogènes que le virus parent [65, 66].
5. La protéine S sur la membrane virale
Le trimère S dépasse d’environ 25 nm de la membrane virale, présentant une « tête » proéminente, globulaire mais dynamique, et une région « tige » flexible (Figure 2A). La structure tridimensionnelle (3D) de la tête du spicule a été déterminée par microscopie électronique cryogénique (cryo-EM) et moyennage de particules uniques pour plusieurs coronavirus, dont les premiers ont été rapportés en 2016 [67, 68] (revus dans [69]) à des résolutions d’environ 3 Å ou mieux, y compris celle du spicule du SRAS-CoV-2 [47, 70, 71]. Le spicule complet a été visualisé sur les virions de SARS-CoV-2 par des analyses de tomographie cryo-électronique de particules virales inactivées utilisant la moyennation des sous-tomogrammes pour atteindre une résolution supérieure à 1 nm [49, 50, 51]. S1, qui reste attaché de manière non covalente à S2 dans le trimère clivé, contient deux domaines structurels, un domaine N-terminal (NTD) et le RBD. Le repliement de ces deux domaines est indépendant du reste de la chaîne polypeptidique (Figure 2). Les études structurales ont montré que le RBD présente un équilibre dynamique entre deux conformations, « couché » et « debout ». La tête trimérique du spicule a donc été visualisée sous quatre formes différentes, une forme complètement fermée avec les trois RBD couchés, une conformation ouverte avec les trois RBD debout, et des formes intermédiairement ouvertes avec un ou deux RBD debout et les autres couchés [54, 52]. Ce n’est que dans la conformation debout que le RBD peut engager ACE2, ce qui déplace l’équilibre vers la forme ouverte du spicule.
5.1. La sous-unité S2 : une protéine de fusion de « classe I »
S2 est responsable de la catalyse de la réaction de fusion membranaire nécessaire à l’entrée. Cette réaction suit la dissociation de S1 du spicule lors de la liaison avec ACE2, ce qui libère S2 des contraintes dérivées de son repliement dans le contexte du précurseur S. Des études structurales ont permis de caractériser trois classes de protéines de fusion membranaire virale (examinées dans [72]). S2 appartient à la classe I, comme les protéines de fusion de nombreux virus enveloppés, tels que les paramyxovirus, les virus de la grippe, les rétrovirus et d’autres familles virales. Toutes ces protéines subissent un changement de conformation pour passer d’une forme métastable de pré-fusion à une forme hautement stable de post-fusion, afin d’entraîner la fusion membranaire. L’énergie stockée dans la forme métastable est couplée à la liaison de la membrane cible pour catalyser la fusion lipidique. En particulier, il a été démontré que les protéines des trois classes adoptent une conformation en « épingle à cheveux » très stable dans la forme post-fusion. Dans les protéines prototypiques de classe I, telles que la HA2 du virus de la grippe ou la gp41 du VIH-1, la moitié N-terminale forme une longue « hélice N » qui constitue Un trousseau trimérique torsadé d’hélices alpha parallèles, tandis que la moitié C-terminale est antiparallèle à l’hélice N (dans certains cas sous forme d’une « hélice C », comme dans la gp41) le long des rainures du trousseau. L’ « épingle à cheveux » qui en résulte est telle que ses extrémités N et C-terminales se trouvent du même côté d’une longue tige trimérique. Un segment hydrophobe, appelé « peptide de fusion » (FP), est situé immédiatement en amont de l’hélice N. Ce segment est responsable de l’insertion dans la membrane cellulaire pendant les premières étapes du réarrangement conformationnel fusogène, une étape absolument nécessaire pour conduire le processus de fusion, comme indiqué dans la Figure 3A. Les protéines de fusion prototypiques de classe I, comme celles du virus de la grippe ou des rétrovirus, ont le peptide de fusion à l’extrémité N-terminale, et l’épingle à cheveux est formée exclusivement par la région située entre le FP et l’ancre TM C-terminale. Dans la protéine du spicule des coronavirus, comme dans son homologue des paramyxovirus, ce n’est pas le cas car le peptide de fusion se trouve à l’extrémité N-terminale de S2′, qui est interne à S2 (voir Figure 2A), et les régions en amont du site de clivage de S2′ sont également présentes dans l’épingle à cheveux. Les similitudes structurelles entre la protéine S2 du coronavirus et la protéine de fusion (F) des paramyxovirus suggèrent qu’elles ont eu une origine commune [46], formant une sous-classe distincte par rapport aux protéines prototypiques de classe 1. Il existe deux régions à heptades répétées dans la partie S2′, appelées HR1 et HR2 (marquées sur la Figure 2A), qui ont un fort potentiel pour former des hélices alpha. Dans le trimère S de pré-fusion, la région HR1 est montée sur ressort sous la forme d’un faisceau de segments hélicoïdaux courts sous une « couronne » formée par la sous-unité S1 (Figure 3), tandis que HR2 fait partie de la tige flexible connectant à la membrane virale (Figure 2B). Pour stabiliser le spicule dans la forme pré-fusion utilisée dans les vaccins actuels, des mutations en proline ont été introduites aux connexions entre les hélices courtes de la HR1 chargée par ressort [73, 74, 75]. Les prolines étant des résidus de rupture d’hélice, le HR1 muté ne peut pas se déployer pour former une longue hélice, comme le montre la Figure 3C, ce qui donne un trimère globalement stabilisé qui n’est pas fusogène.
Mutations de la protéine S apparaissant dans des variants préoccupants du SRAS-CoV-2 récemment apparus. (A) Diagramme linéaire de la protéine S mature, coloré pour correspondre au diagramme de la Figure 3A. La barre grise en dessous représente le polypeptide complet, y compris sa séquence signal. Sur cette barre sont représentées les mutations apparaissant sur les variants préoccupants du virus annotés immédiatement en dessous, comme indiqué.Notez le regroupement des mutations dans le NTD, le RBD et près du site de clivage par la furine. (B) La tête du spicule représentée en surface avec les protomères colorés différemment selon la clé étiquetée « chaîne ». Les mutations apparaissant dans les variants sont indiqués sur la surface colorée selon le variant. Le trimère de la rangée supérieure est dans la conformation fermée, dans laquelle la surface de liaison de l’ACE2 est principalement enfouie. Le panneau inférieur montre une tête de spicule ouverte avec le domaine protéase de ACE2 lié (rubans jaunes). Notez que certaines des mutations se situent directement sur la surface d’interaction avec ACE2. Les panneaux de droite montrent une représentation « à livre ouvert », avec la sous-unité du front (étiquetée A) tournée de 180 degrés et montrant les sous-unités B et C de l’arrière, pour mettre en évidence les surfaces enfouies dans le trimère (colorées différemment comme indiqué dans la clé). Notez que certaines des mutations sont sur ces surfaces internes, qui sont susceptibles d’affecter la dynamique du trimère S et de jouer un rôle dans des changements allostériques.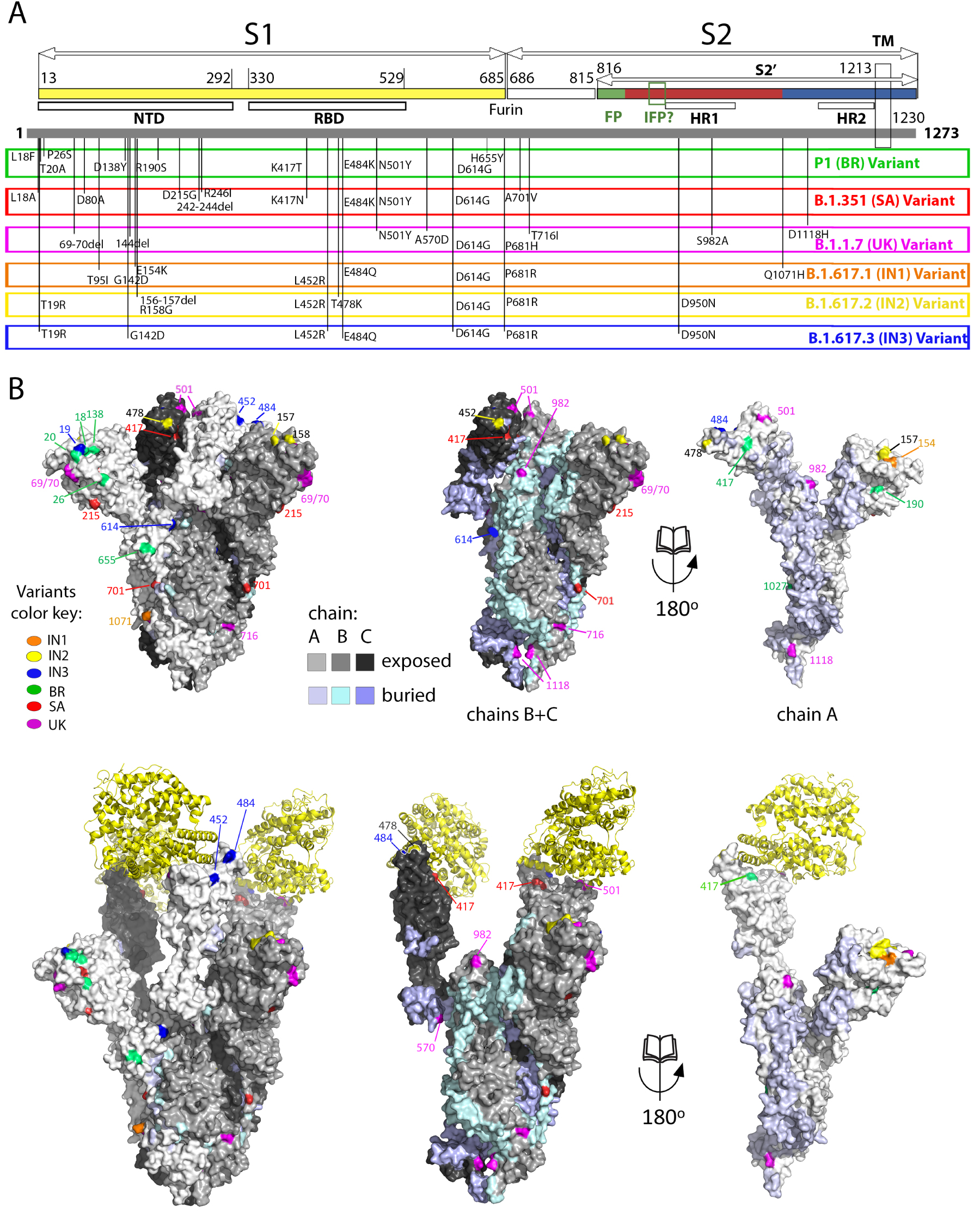
5.2. Mécanisme de fusion membranaire
Le trimère de S devient métastable après maturation protéolytique en S1 et S2. La liaison ultérieure à l’ACE2 sur les cellules déplace l’équilibre vers la forme ouverte, dans laquelle les interactions S1/S2 se sont avérées plus faibles [54]. Comme chaque spicule est lié à plus d’un récepteur, les contraintes mécaniques ont le potentiel d’exercer des forces tirant les sous-unités S1 dans des directions opposées, arrachant la tête du spicule et livrant la sous-unité S1 au récepteur (Figure 2B). La perte de la couronne S1 sur le spicule activé provoque à son tour l’érection de l’hélice HR1, qui projette le FP sur une distance d’environ 15 nm pour atteindre et s’insérer de manière stable dans la membrane cellulaire (Figure 3C). Cette forme insérée de S2, un long trimère qui relie les deux membranes à une distance d’environ 30 nm, est transitoire. La partie HR2, qui est initialement située à distance, près de la membrane virale, finit par se lier aux rainures du trousseau torsadé de HR1 pour former le trimère d’épingles à cheveux observé dans la forme post-fusion (illustrée sur la Figure 3C, panneau de droite, où seul un protomère du trimère est représenté pour plus de clarté). Dans sa conformation finale, S2 force donc la membrane virale et la membrane cible l’une contre l’autre, en surmontant la répulsion entre les deux feuillets externes, comme dans les étapes iv et v de la Figure 3A. Cette répulsion survient lorsque deux membranes sont amenées à moins de 1 nm de distance en raison de la déshydratation des têtes lipidiques requises pour permettre un contact direct [76, 77]. La conformation en épingle à cheveux de la protéine de fusion catalyse donc la réaction de fusion membranaire en forçant l’apposition étroite des bicouches lipidiques externes, abaissant ainsi la barrière cinétique de la fusion, qui est une réaction globalement exothermique. La fusion membranaire s’effectue par une hémi-fusion initiale (étape v de la Figure 3A) dans laquelle seuls les feuillets externes de la membrane fusionnent, suivie de la formation d’un pore par fusion des deux feuillets (étape vi), pore qui s’élargit ensuite pour former une seule membrane fusionnée.
Analyse de la conservation des acides aminés de la protéine spike à partir d’un alignement de 28 différents sarbecovirus infectant différentes espèces de mammifères prédits pour lier l’ACE2. La sélection a été basée sur la conservation des cystéines 480 et 488, qui forment un pont disulfure qui conforme la surface de liaison ACE2 du RBD. Les codes d’accession Uniprot des séquences utilisées sont : P0DTC2, A0A023PTS3, A0A0P0INJ4, A0A0U2IWM2, A0A2D1PX29, A0A2D1PXA9, A0A3G5BJ32, A0A3Q8AKM0, A0A4Y6GL47, A0A6B9WHD3, A0A6G6A2R8, A0A6G8I328, A0A6G9KF73, A0A6G9ZWX7, A0A6G9ZWY9, A0A6G9ZX63, A0A6G9ZXI6, A0A6G9ZXM6, A0A6G9ZY67, A0A6G9ZYB3, A0A6G9ZYE7, A0A6H0C589, A0A6H0C6F4, A0A6H0C6Q6, A0A6H0C790, A0A6H0C7A7, A0A6H0C7Y4, A0A6H0JGD1. (A) La tête du spicule est montrée dans les conformations fermées (panneaux de gauche) et ouvertes (à droite) dans la même orientation, une vue latérale dans la première ligne et une vue de dessus dans la deuxième. Les panneaux tout à gauche et à droite montrent, à titre indicatif, le trimère coloré en domaines comme dans la Figure 2. Les deux panneaux centraux ont la surface de reconnaissance de ACE2 en bleu (étiquetée « patch ACE2 »), montrant que cette surface n’est pas accessible dans le spicule fermé. La conservation est codée par un gradient de blanc (conservé) jusqu’à marron (variable) comme indiqué dans la barre horizontale en haut. La comparaison entre les deuxièmes et troisièmes colonnes, et entre les quatrièmes et cinquièmes colonnes, montre que le patch ACE2 fait partie d’une surface variable, en accord avec l’observation que la surface du récepteur ACE2 qu’il reconnaît varie également selon les espèces (voir Figure 1C, panneaux de droite). Les vues de dessus montrent également que dans le spicule ouvert, une surface conservée de S2 (en vert dans la sixième colonne) devient exposée. Cette surface, qui correspond à l’hélice HR1 montée sur ressort, n’est pas immunogène car elle n’est essentiellement pas exposée sur les particules infectieuses. Cette Figure a été préparée en utilisant les modèles atomiques PDB : 7BNM (pointe fermée) ET 6A98 (pointe ouverte). (B) Équilibre entre les conformations fermée, partiellement ouverte et ouverte du spicule. Les trimères latéraux sont représentés comme guide pour définir le code couleur utilisé, avec les RBDs en trois nuances de bleu : bleu clair, moyen et foncé correspondant respectivement au gris clair, moyen et foncé du reste du même protomère. Pour faciliter la description, les protomères ont été étiquetés A, B et C. Les quatre trimères du milieu affichent, en plus, le patch ACE2 en jaune, et les mutations dans les variants énumérées dans la Figure 4 sont indiquées dans le même code couleur que dans la Figure 4.Notez que ces mutations correspondent à des régions qui varient d’un sarbecovirus à l’autre (comparez avec les rangées 2 et 5 au-dessus). La deuxième rangée en B a les trois RBDs couchés, le troisième panneau a le RBD du protomère A debout et les couchés, le quatrième panneau a les RBDs A et C debout, et le quatrième panneau les trois RBDs debout, résultant en un spicule ouvert. Les doubles flèches entre les panneaux indiquent la réversibilité entre les différentes conformations. Notez que la conformation debout des trois RBDs est instable et semble conduire à un détachement irréversible de S1, comme indiqué dans la Figure 3. Elle est représentée par la flèche unique pointant vers le bas dans le cinquième panneau. Les modèles atomiques utilisés dans ce panneau sont : PDB 7BNM (spicule fermé), 7BNN (un RBD debout), 7BNO (2 RBD debout), 6A98 (spicule ouvert, trois RBD debout).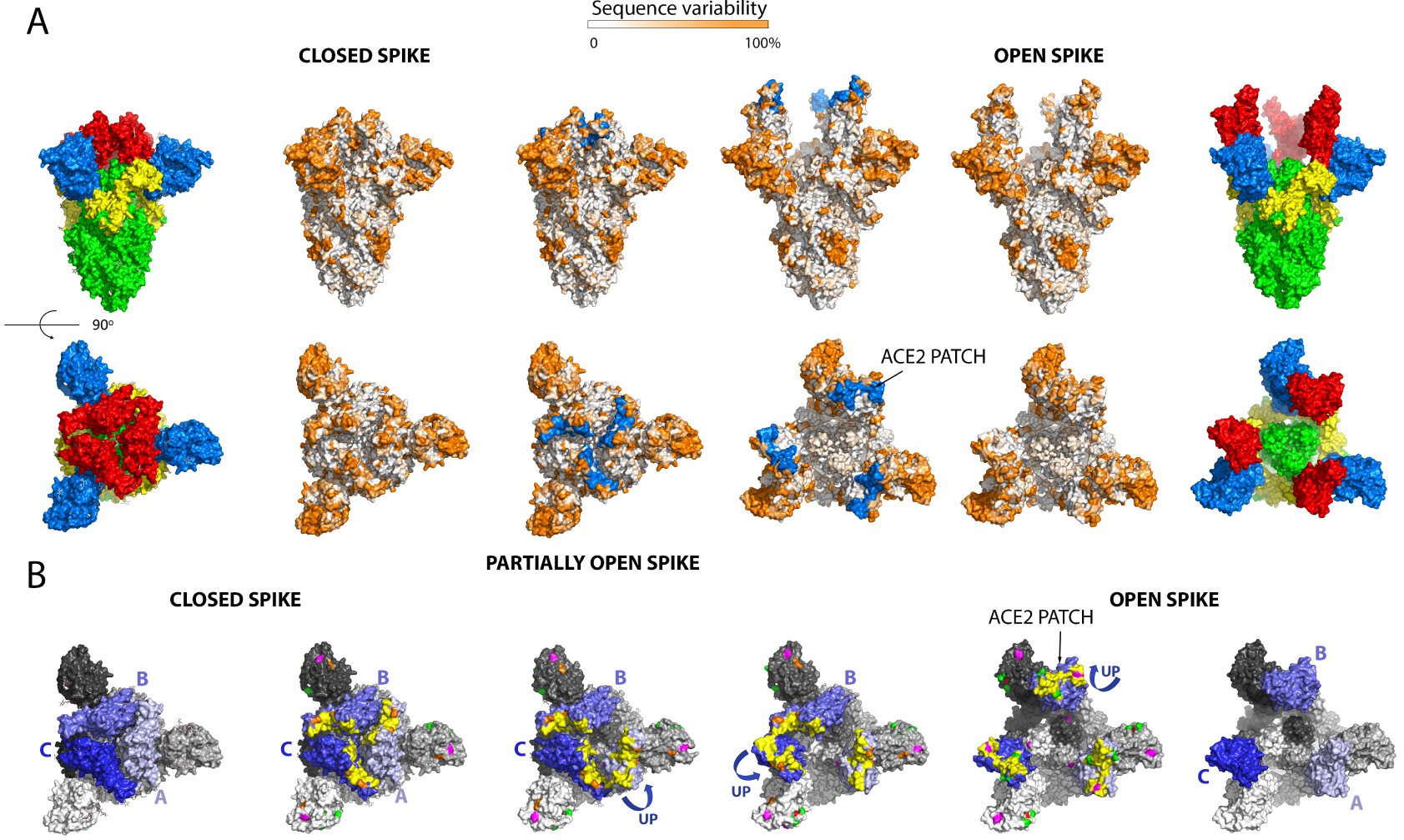
6. La protéine S dans les variants préoccupants du SRAS-CoV-2
De nouveaux variants du SRAS-CoV-2 présentant des mutations augmentant la transmission interindividuelle ou permettant une évasion immunitaire sont apparus récemment et déplacent les souches virales circulantes initiales [78, 79, 80, 81]. Les modifications génomiques de ces variants ont été associées à une transmission interindividuelle accrue et/ou à un échappement à l’immunité post-infection ou post-vaccination. Bien que les mutations des variants se produisent dans tout le génome viral, des mutations clés dans la protéine S semblent contrôler la transmission du virus et/ou modifier des sites ciblés par les anticorps neutralisants. Le premier variant identifié présentait la mutation D614G dans S [82], une mutation qui s’est maintenant fixée dans le virus en circulation et dont on a montré qu’elle augmentait la transmissibilité du virus [83, 78, 84, 85, 86, 87]. Comme cela a été démontré récemment pour le variant B.1.1.7 [88], les nouveaux variants peuvent également provoquer des symptômes plus graves que ceux du virus de la souche Wuhan originale. Les nouveaux variants sont souvent désignés par le pays ou la région où ils ont été détectés pour la première fois : B.1.1.7 est appelé variant du Royaume-Uni, B.1.351 variant de l’Afrique du Sud (SA), P.1 variant du Brésil (BR), B.1.429 variant de la Californie. De nouveaux variants sont également apparus en Inde, qui connaît une forte poussée de l’épidémie de Covid 19. Bien que la souche prédominante qui y circule soit B.1.1.7, une nouvelle lignée, B.1.617, prend le relais et se diversifie en plusieurs sous-lignées [89, 90, 91, 92, 93, 94, 95]. Ce variant a maintenant atteint d’autres pays, notamment le Royaume-Uni où environ la moitié des nouveaux cas appartiennent au variant 1.617.2.
La Figure 4 énumère les positions sur S des mutations qui sont apparues dans les principaux variants préoccupants, y compris les trois nouvelles sous-lignées indiennes. Ces mutations se regroupent dans plusieurs régions de la tête du spicule, en particulier dans le domaine N-terminal. On observe également un regroupement dans le RBD et près du site de clivage S1/S2. Certaines mutations sont dans HR1 et dans la région d’inversion de chaîne dans l’épingle à cheveux post-fusion (entre les moitiés rouge et bleue de S2′, colorées comme dans le diagramme de la Figure 3A). Bien que la majorité des mutations se situent à la surface externe du trimère, un certain nombre d’entre elles affectent les résidus de contact enfouis à l’interface entre promères du trimère (voir Figure 4B, panneaux de droite). C’est le cas des mutations dans S2 et de celles qui dans S1 près du site de clivage S1/S2 (à partir du résidu 570). Une analyse de la conservation de la séquence en acides aminés de S parmi les sarbecovirus infectant d’autres mammifères et prédits pour utiliser l’ACE2 comme récepteur (Figure 5) a montré que l’emplacement des mutations des variants à la surface du spicule est dans des régions variables de la protéine S. Et même si la surface la plus variable correspond au NTD, la surface de liaison de l’ACE2 du RBD l’est aussi (comparez les 4e et 5e colonnes de la Figure 5A), ce qui est conforme à l’observation selon laquelle le patch d’ACE2 reconnu par le RBD est également variable chez les mammifères (Figure 1C). Le spicule ouvert expose une région conservée de S2 au sommet qui correspond à S2 chargé de ressort (Figure 5A, panneaux de droite). Cette région ne semble pas être immunogène, très probablement parce que le spicule complètement ouvert n’est exposé que de façon transitoire, immédiatement avant son changement de conformation irréversible ver la forme post-fusion, comme indiqué par les flèches dans la Figure 5B. L’emplacement et l’orientation relative de la surface de liaison ACE2 de la RBD (surface indiquée en jaune dans la Figure 5B, en particulier dans le 5e panneau) sont également compatibles avec un effet d’ « arrachage » lorsque plus d’un récepteur ACE2 est lié, car ils peuvent tirer le RBD (et S1) dans des directions opposées, perturbant ainsi le spicule et induisant le changement de conformation pour provoquer la fusion des membranes.
Analyse de la surface du RBD, localisation des principaux épitopes et mutations dans les nouveaux variants. (A, B) La surface du RBD colorée selon son accessibilité au solvant dans la conformation fermée du spicule (A) et selon la conservation des acides aminés (B) comme indiqué dans la barre colorée. (C) La surface de liaison de ACE2 a été superposée en jaune sur la même vue qu’en A. Les chaînes de glycanes interagissant avec le RBD sur le spicule fermé sont représentées sous forme de bâtonnets colorés selon le type d’atome : bleu pour l’azote, rouge pour l’oxygène et gris clair pour les atomes de carbone du glycan N343 attaché au protomère représenté, et jaune pour ceux émanant des protomères adjacents (N149 dans un NTD voisin et N343 dans le RBD adjacent), montrant qu’ils masquent une partie de la surface non enfouie dans le trimère fermé par des atomes de la chaine polypeptidique comme indiqué dans le panneau A. Il est à noter que les simulations par dynamique moléculaire suggèrent que le glycane N343 joue un rôle de barrière dans le basculement du RBD vers la conformation debout [96]. Le panneau inférieur montre une vue de dessus sans mettre en évidence le patch ACE2. (D) Les mutations apparaissant dans les COV sont marquées sur la même vue qu’en C (sans les chaînes de glycanes, pour plus de clarté). L’emplacement de l’épitope des principales classes d’anticorps neutralisants est indiqué par des ellipses codées en couleur selon la classe d’anticorps. Les anticorps neutralisants les plus puissants appartiennent aux classes 1 et 2, qui interfèrent avec la liaison de l’ACE2. Ceux de la classe 1 nécessitent que le RBD soit dans la conformation debout pour se lier, car leurs épitopes comprennent un patch enterré dans le spicule fermé (comparer avec A, panneau de gauche). En revanche, ceux de la classe 2 peuvent se lier à la pointe dans les deux conformations, ouverte ou fermée. Notez que les mutations sur les variants sont à la périphérie de la zone de liaison de ACE2, ce qui apparaît plus clairement dans la vue du haut (panneau inférieur).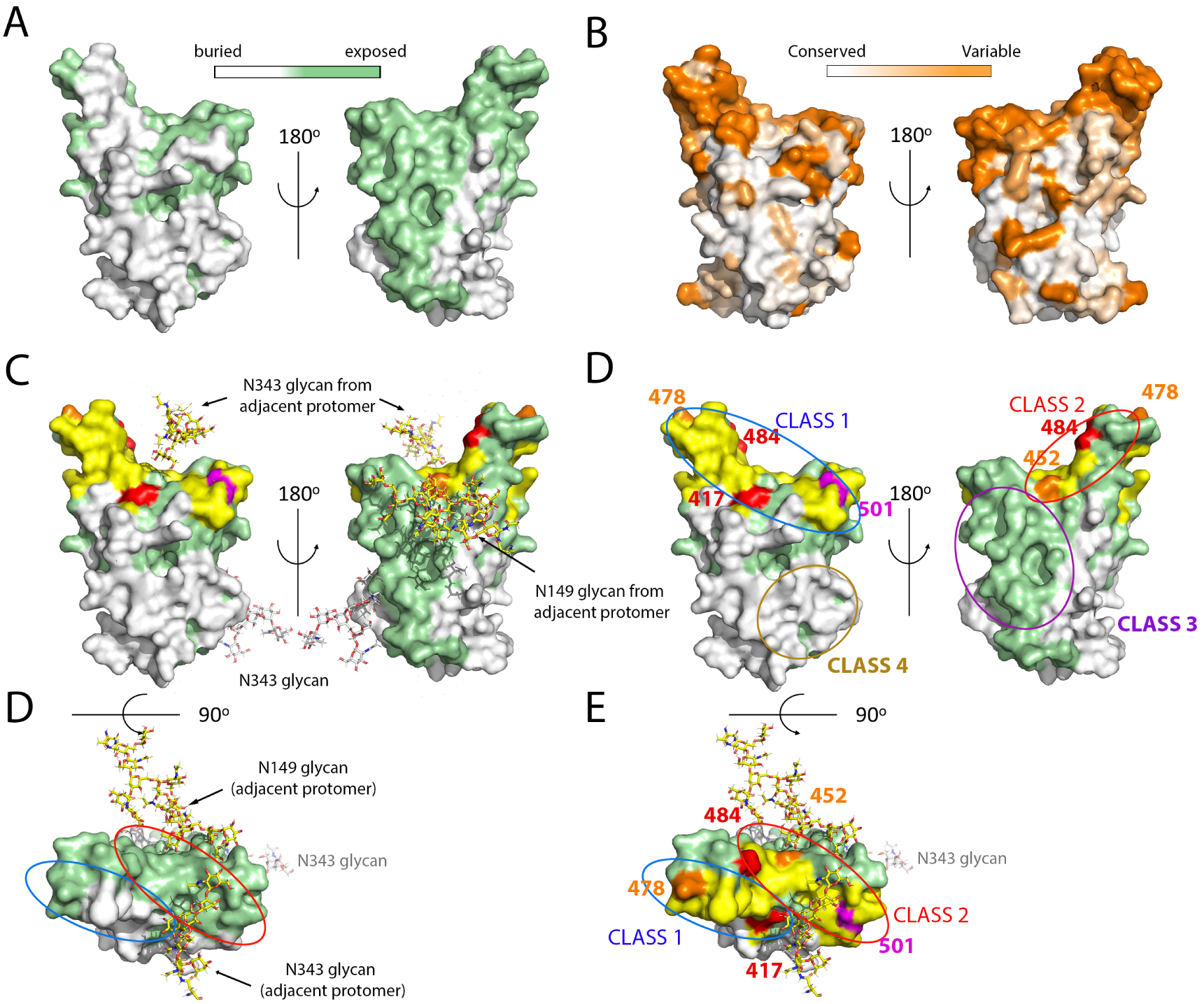
7. Mutations des variants dans le RBD et échappement aux anticorps neutralisants
Bien que la réponse immunitaire contre le spicule soit dirigée vers de multiples sites [97], il a été démontré que le RBD était la cible de la grande majorité des anticorps neutralisants humains les plus puissants contre le SRAS-CoV-2. De multiples rapports décrivant le clonage et l’isolement d’anticorps monoclonaux (AcM) provenant de personnes convalescentes et de personnes vaccinées, ainsi que des études structurales par radiocristallographie et/ou cryo-EM ont été rapportés [98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 8, 108]. Ces études attestent de la forte puissance de neutralisation des anticorps monoclonaux spécifiques du RBD. Globalement, ces anticorps ont été classés en quatre grandes catégories, en fonction de la localisation de leur épitope à la surface du RBD [98]. Les anticorps les plus fortement neutralisants, agissant avec une efficacité proche du picomolaire, ciblent le patch de liaison de l’ACE2 (surface jaune dans la Figure 6) [103]. Ces derniers sont divisés en deux classes : ceux de la classe 1 nécessitent que la RBD soit debout pour se lier, tandis que ceux de la classe 2 ne discriminent pas la conformation du spicule, que RBD soit couché ou debout. L’emplacement des épitopes correspondants est marqué dans la Figure 6D, qui montre que l’épitope des anticorps de classe 1 est partiellement enfoui dans la conformation fermée du spicule, alors que celui de classe 2 ne l’est pas (comparer le panneau supérieur de D avec A, ainsi que les panneaux inférieurs de C et D). Deux autres catégories d’anticorps ont été identifiées dans lesquelles l’épitope est en dehors du patch ACE2 : les anticorps de classe 3 ciblent une surface exposée, tandis que l’épitope de la classe 4 est enfoui dans le spicule fermé et nécessite au moins deux RBD debout pour la liaison [109, 110, 111, 112]. Les anticorps de classe 4 sont parmi ceux qui présentent la réactivité croisée la plus large parmi les sarbecovirus [101], comme on peut le voir en comparant les panneaux D et B de la Figure 6. Les mutations des variants récents entraînent toutes une réduction de la liaison des anticorps des classes 1 et 2, ce qui suggère que ces derniers constituent la forme dominante contrôlant la circulation du virus dans la population en général.
Les mutations sur le RBD apparues dans les variants récents ne sont pas distribuées de manière aléatoire : elles forment une couronne tout autour de la périphérie de la surface de liaison de l’ACE2 (Figure 6D, panneau inférieur). Ceci est en contraste avec la variabilité observée lors de la comparaison entre sarbecovirus, dans laquelle toute la surface est variable. La notion tirée de cette analyse est que le virus échantillonne toutes les mutations qui sont encore compatibles avec la liaison à l’ACE2 humaine tout en interférant avec la liaison des anticorps. Il reste à voir jusqu’à quel point le virus peut poursuivre cette tendance et échapper à la réponse immunitaire humaine tout en conservant sa virulence.
8. Remarques finales
Cette revue couvre la plupart des données structurales disponibles sur la protéine du spicule du SRAS-CoV-2, son interaction avec ACE2, et les inférences mécanistiques qui peuvent être tirées de ces études. La localisation des mutations dans les nouveaux variants du SRAS-CoV-2, distribuées sur toute la surface du spicule, suggère en outre un effet épistatique des mutations et qu’un effet allostérique pourrait contrôler une exposition différentielle des trois sites de clivage à la furine dans le trimère, ce qui semble avoir une importance fonctionnelle. Les mutations observées peuvent également affecter de multiples façons le changement de conformation déclenché par la liaison au récepteur. La biologie du spicule du coronavirus est ainsi loin d’avoir livré tous ses secrets. Néanmoins, la quantité d’informations recueillies sur cette protéine en 18 mois de pandémie a été sans précédent, tout comme la pandémie elle-même. Ce n’est qu’en comprenant la fonction du spicule dans ses moindres détails mécanistiques — ainsi que ceux des autres protéines virales impliquées dans d’autres étapes du cycle viral — que nous pourrons espérer être mieux préparés à faire face aux futures épidémies de virus zoonotiques émergents tels que les coronavirus.
Remerciements
Je remercie mes collègues de l’unité de virologie structurale pour les nombreuses discussions, et les membres de la Task Force Coronavirus de l’Institut Pasteur pour leur soutien. Toutes les illustrations structurales utilisées ont été préparées avec The PyMOL Molecular Graphics System, Version 2.1 Schrödinger, LLC. La représentation de la conservation de la séquence d’acides aminés sur à la surface des protéines ACE2 et S a été réalisé avec EsPript [113]. Je remercie P. Guardado-Calvo pour ses conseils sur l’utilisation de ce logiciel.