

 CC-BY 4.0
CC-BY 4.0
1. Introduction
SARS-CoV-2 is an RNA virus. Its genome is approximately 30,000 bases long, making it the longest known RNA virus genome. In comparison, the influenza genome is 10,000 to 15,000 bases long, and HIV (a retrovirus) is about 10,000 bases long. The first sequences of the SARS-CoV-2 genome were available in late December 2019, all from Wuhan, China, with the first sequence available on 23/12, the second on 26/12, and 16 more on 30/12 [1, 2]. By mid-January 2020, sequencing began outside of China, and by the end of January there were about 250 sequences available from many countries (Thailand, Nepal, Japan, Canada, USA, France, Germany, Italy, Australia…). Sequencing was initially rather slow, since at the end of March 2020 only a few hundred sequences were available. It accelerated considerably in April with a massive confinement on the surface of the globe and tens, then hundreds, of thousands of cases reported in Europe and the United States. On certain days in April–May several thousand new sequences were deposited on the GISAID (Global Initiative on Sharing Avian Influenza Data, www.gisaid.org) web-site, which collects and makes public sequences from around the world. Since then, the number of genomes deposited has varied but remains high, with more than 500 genomes per day from all over the world. Today (30/09/2020) there are more than 100,000 sequences on the GISAID web-site from 120 countries. Some countries have not sequenced much (or did not make their sequences public). For example, until recently there were less than 300 Italian sequences on GISAID despite the impact of the pandemic. There are 700 today. France has not provided many sequences either, especially in comparison with the UK (about 600 versus more than 42,000 on GISAID today). In particular, only very few (11) sequences are available from Eastern France, even though it is a major source of SARS-CoV-2 spread [3]. In this context, tracing the spread of the epidemic in France as well as on a more global scale is an arduous task, even impossible in some regions.
All results on the origin, evolution and spread of SARS-CoV-2 come from computer analysis of these sequences, coupled with associated metadata such as the date and place of sequencing, the sequencing technique, etc. [4]. In some cases, it has been possible to determine the precise origin of a virus found at a given location by contact tracing. For example, this is the case for the first two Thai genomes: It was shown by following the routes of patients that they came from China. It was also possible to trace the Chinese origin of the infection of “patient one” in Codogno, Italy, in February 2020, even though we now know from the analysis of wastewater that the virus was already circulating in Lombardy in December 2019. But this information is very partial. Most of our knowledge comes from sequence analysis [4], which is based on models (for example to describe mutations, their rhythm and regularity over time) as well as on algorithms [5], which are now confronted with extraordinary masses and flows of data. It should be kept in mind that the conclusions drawn from these analyses depend on the models and approximations made by these algorithms, which follow heuristic approaches due to the mass of data and the complexity of the problems [6]. Since the evolution of the virus started less than a year ago, the strains still show few differences, which limits some analyses, for example the search for traces of adaptation [7, 8]. Finally, some analyses are complicated by the poor sequencing of certain regions of the globe (see above) and the quality of certain sequences. Despite these limitations, we now have very clear answers to a number of questions, for example on the natural origin of the virus and the fact that it is not from a laboratory [9]. These advances and their limitations are reviewed here, with a special focus at the end of the article on the global spread of the pandemic using a new phylogeographic approach to correct sampling biases [10].
Phylogeny of SARS-CoV-2 related strains (GISAID, 10/05/2020).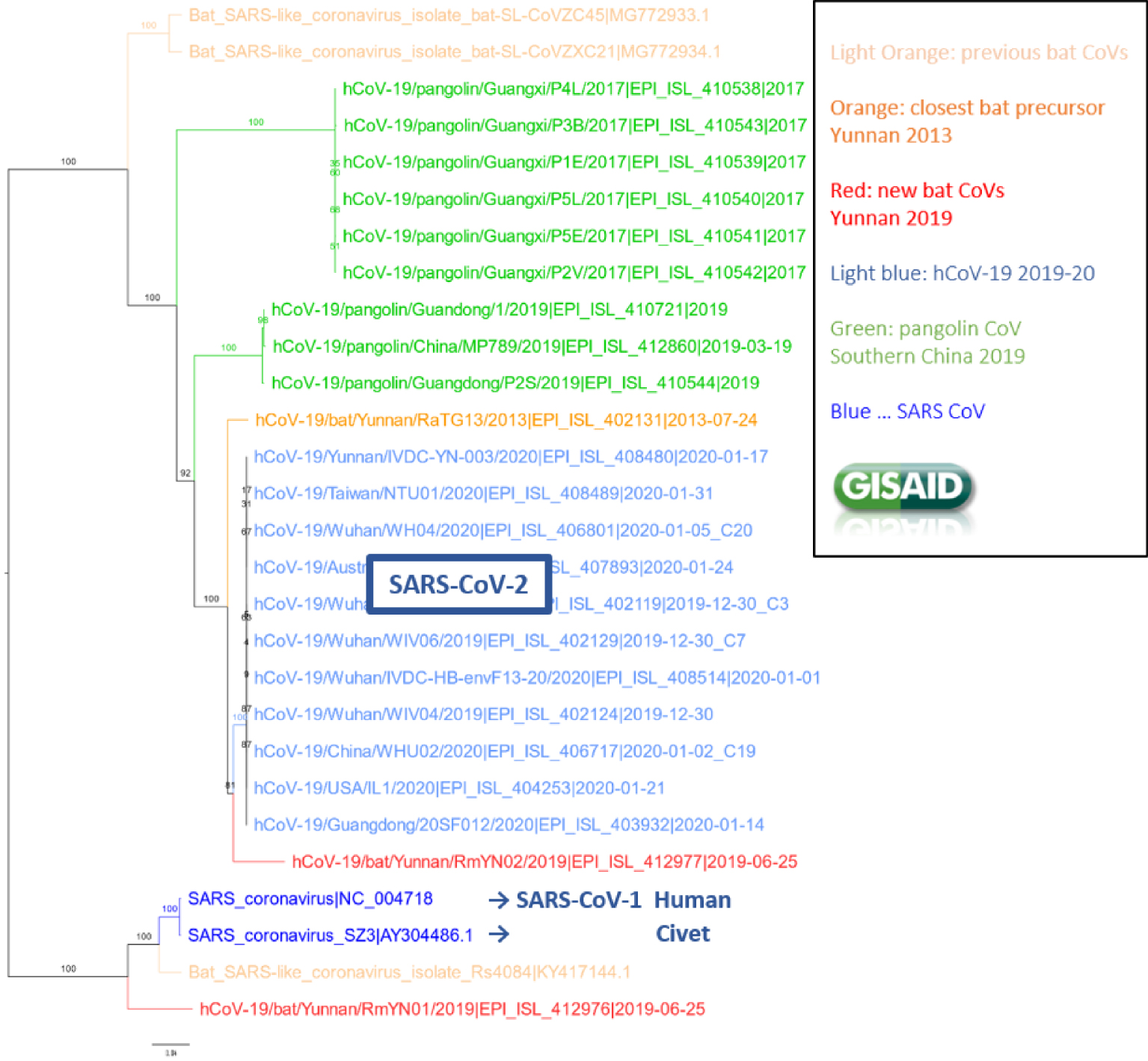
2. Origin and phylogeny
As soon as the first sequences were obtained, phylogenies were constructed to trace the origin of SARS-CoV-2 (which was not yet called that). It is a Betacoronavirus, a member of the Sarbecoviruses, a viral subgenus including the virus responsible for the SARS epidemic in 2003, named SARS-Cov-1 (for severe acute respiratory syndrome-related coronavirus). SARS-CoV-2 is also referred to as hCoV19 by some people, notably GISAID members, who rightly consider that this is probably not the second human epidemic of this type, and that others have preceded it. Sarbecoviruses infect not only humans, but also many mammals, including civets, bats and pangolins. The phylogeny of this virus and its variants (Figure 1) shows that:
- The closest known viruses to SARS-CoV-2 come from two Rhinolophidae or “horseshoe” bats found in Yunnan in 2013 (RaTG13, [1]) and 2019 (RmYN02, [11]). Genome identity is about 96% for one (RaTG13) and 93% for the other (RmYN02), but this rate of identity varies along the sequences. In particular, it is quite low (60–70%) in the region of binding domain (RBD, ∼60 amino acids, Spike protein) to the human protein ACE2, which allows entry into the host cell [9].
- More distant overall (90% identity) is a pangolin virus, whose RBD sequence is very close to SARS-CoV-2, with only one amino acid mutation, compared to about a dozen for bat [9].
- All other strains related to SARS-CoV-2 are much more distant, notably SARS-CoV-1 (80% identity).
Analyses show that recombinations are numerous among Sarbecoviruses [12]. These recombinations are very likely the origin of SARS-CoV-2, but to date this cannot be affirmed because no genomes or significant portions of genomes that are very close to the human form have been found in natural reservoirs. In this respect, SARS-CoV-2 is clearly different from SARS-CoV-1, which is very close (99.6%) to the civet virus [13]. Initially, it was suggested that due to the similarities observed (see above), SARS-CoV-2 would be a recombinant in the RBD region of bat and pangolin viruses. But since then, observation of the evolution of this region in human strains has shown that this region mutates rapidly, with 36 amino acid mutations present among the human strains available today (30/09/2020). The alternative hypothesis of adaptation of the bat virus in this region, rather than recombination with the pangolin virus, is therefore quite credible, especially since it has been shown that SARS-CoV-2 passes easily to mice, with only a few mutations presumably adaptive in the RBD region [14].
It has been widely read that SARS-CoV-2 is derived from bats and it is believed that the transition to humans is recent. In reality there are ∼4% differences between the two genomes, i.e. about 1200 mutations. Since December 2019 we see the evolution of the virus in humans. Based on the number of mutations observed today compared to the very first sequences, and relating this number to the time elapsed, we find that the genomes evolve at the rate of one or two mutations per month, which is slow compared to influenza or HIV. The 1200 mutations observed thus correspond to 50 to 100 years of evolution and a date between 1970 and 1995 for the common ancestor of SARS-CoV-2 and RaTG13 (100 years of evolution gives the date 1970, because the years in the two branches from the common human/bat ancestor are added together). Further Bayesian analyses indicate similar or even earlier dates, with a part of uncertainty covering the whole 20th century [12]. In any case, these results indicate and confirm that between this common ancestor and SARS-CoV-2 there have been many intermediates, in bats or other mammals such as pangolin, and that these intermediates remain to be discovered. Since coronaviruses have crossed the species barrier three times (both SARS and MERS) in a striking manner over the last 20 years, it is likely that they will do so again, hence the importance of searching for these animal reservoirs.
3. A natural origin
Claims that SARS-CoV-2 is a laboratory product were found early on in the mainstream press. All the results and figures given above demonstrate the contrary. The 1200 mutations separating SARS-CoV-2 from the closest bat strain (RaTG13) are randomly distributed along the genome, whereas bioengineering would have produced an assembly of known fragments (without mutations compared to some known strains), with point mutations in strategic regions, e.g. RBD [9]. Based on local similarities between the SARS-CoV-2 genome and the HIV genome, it has also been claimed that this was an HIV vaccine attempt. But these local similarities do not explain the origin of the rest of the genome, nor are they significant. They relate to a short segment of 38 nucleotides where the two viruses have 87 percent identity. But when comparing the SARS-CoV-2 genome to other genomes, many similarities of this order can be found, for example 89% on a segment of length 44 with a plant genome. We simply see here the effect of evolutionary “bricolage”, which uses and reuses the same solutions to build living things. This is by no means a mark of statistical significance, which is very difficult to estimate.
4. Evolution in the human host
The question of the origin, date and “patient zero” of the human pandemic was raised early on. The sequences alone do not provide complete answers to these questions. In phylogeny the external group method is used to root the group of interest; for example, to root the tree of mammals we use the genome of reptiles or birds that are the closest species. But this method does not work here, the number of mutations observed between the human and bat viruses (1200, cf. above) being out of all proportion to the number of mutations observed among human sequences (a few dozen). In other words, all the human sequences are more or less the same distance from the bat sequence, and it is not possible with this method to designate with certainty the root of the pandemic.
We have better guarantees by relying not only on sequences but also on dates and history. Indeed, on December 30, 2019, the exact same sequence was found in several Chinese patients. It was quickly found in Thailand, Japan and the USA and, for example, it was still present at the end of March in the United Kingdom. It is therefore a good candidate to be the “sequence zero”. It is used as such by many teams and numerous software and websites, including GISAID, NEXSTRAIN (https://nextstrain.org/), etc. As this sequence (WIV04/2019) has been found in different parts of the world, it is impossible to say what its geographical origin is. But the history of the pandemic clearly shows a Chinese origin [1, 2]. Various dating methods indicate a beginning of the spread of the epidemic between mid-October and early December 2019 [7]. It is difficult at the present time to be more certain. As with other viruses, especially HIV, the acquisition of new sequences, possibly from older samples, should allow us to refine these estimates and possibly find an earlier origin of the human pandemic.
The first sequences showed little or no difference. Today, after about 10 months of evolution, the most recent sequences are separated from the “sequence zero” by at most 30 nucleotide mutations and 15 amino acid mutations. These figures are partly uncertain, as it is sometimes difficult to distinguish mutation from sequencing or assembly errors. Some deletions, sometimes long and found in several patients, are observed, notably a deletion of 382b in ORF8 and its regulatory region, sampled about 50 times between Singapore and Taiwan [15]. This deletion has been observed in a similar form in SARS-CoV-1, where it is associated with attenuation of the virus, but to date this attenuation has not been observed in SARS-CoV-2, nor any adaptive effect. In contrast to deletions, no prominent and widely shared insertion between viruses from different patients has been found [16].
Mutations that could result from adaptation to the human host, or that could be attached to greater virulence or severity, were very quickly sought. The rarity of mutations and the short evolutionary time of the virus in humans make these tests difficult. Mutations observed in viruses over short periods of time are generally considered to have a neutral or low impact on the phenotype and are essentially the result of complex random processes related to errors in replication and subsequent spread in the human population. According to this commonly accepted hypothesis, there are no strains that are more virulent or more severe than others. At present, there is very little data to contradict this hypothesis. However, the D614G mutation of the Spike protein (transformation of a D residue into a G residue at position 614) seems to correspond to increased transmissibility, based on the increasing frequency of this mutation in the global data [17]. While the pandemic started with the D614 version and was transmitted in this version to many countries, these countries are now almost all predominantly affected by the G614 version (e.g. in France, G: ∼100%), with the notable exceptions of China (D: 60%, G: 40%), Iceland where D is returning after an almost exclusively G phase, or for example Santa Clara in California, which is essentially D since the beginning of the pandemic (whereas California is essentially G). We can see from this last example that these results must be taken with care, as they are largely the result of founding effects whose impact can last for a long time. In the case of D614G, however, there are additional clues from in vitro experiments that indicate higher titers and infectivity of variant G; but no difference in severity is seen between the two variants, which have the same resistance to neutralization by the serum of convalescent patients [17, 18]. About 100 other Spike mutations have been studied in vitro [18], some of which have an impact on infectivity and antigenic potency, but do not show a substantial increase in prevalence in the global population as seen for the G614 variant.
While coronaviruses recombine abundantly [12], no reliable markers of recombination among human strains have been found so far [4]. These could occur if co-infection by significantly different strains occurs, but the probability of co-infection is low, and the strains currently circulating are too similar for this phenomenon to be detected if it occurs at all. From this point of view, SARS-CoV-2 appears to be clearly distinguishable from influenza, which evolves by reassortment of different subtypes, which can lead to radical changes with major pandemic risks.
Finally, before concluding this section on the evolution of SARS-CoV-2, it is important to point out a tendency in its genome to replace cytosine bases (C) by uracils (U) [8]. This tendency is explained by cytosine metabolism and replication errors [19]. It is relatively weak but significant, with ∼0.1% increase in U on average at the variable sites between December 2019 and April 2020. Detailed analyses [8] show that mutational mechanisms globally produce an excess of U, but that these mutations tend to be counter-selected at the level of synonymous sites and certain di-nucleotides. These evolutionary mechanisms constitute a drift rather than an adaptation to the human host, with a low impact on the proteome. However, they may be key in the design of vaccines based on attenuated forms of the virus [8].
5. Clades and subtypes
The existence of subtypes of SARS-COV-2 was quickly questioned, by analogy with the subtypes of HIV or Dengue fever, for example. The concept of subtype makes sense if it corresponds to clearly distinct sequences with epidemiological characteristics of interest that separate them from other subtypes. For example, HIV subtypes B and C are clearly separated in phylogeny, with strong statistical support [20], and correspond to distinct epidemics affecting mainly Africa for C and Europe and North America for B. The separation between these two subtypes is estimated to be about 100 years [21] and they appear to differ in terms of resistance to treatment or time to AIDS (in the absence of treatment). The same type of clear separation is found for all four dengue subtypes [22].
For SARS-CoV-2 nothing like this is expected today, since the virus appeared in humans at the end of 2019. Several groups have proposed classifications, with the aim of facilitating the monitoring of the epidemic and building a nomenclature that should prove useful in the long run, for example if some strains no longer circulate or others become predominant. The most convincing distinction is associated with the D614G mutation discussed above. Sequences containing the G614 version, together with two mutations at the RNA level, constitute the G clade of GISAID [23], named B1 by the PANGOLIN system [24]. This clade has a clear phylogenetic difference with the other sequences, even if the bootstrap supports are not very high, and is of great potential epidemiological interest, since it seems to correspond to increased transmissibility and the G clade is becoming predominant in most countries and continents [17]. From the G clade are derived the GH and GR sub-clades of GISAID (B1.1 and B1.2 for PANGOLIN), also carrying the G614 mutation of the Spike, whose epidemiological interest, essentially phylogeographic, is less obvious. The sequences outside the G clade constitute the S clade (GISAID, A for PANGOLIN), which contains the “sequence zero” and the first sequences observed in December/January, as well as the L and V clades (B and B2 for PANGOLIN, respectively). The prevalence of these three clades (S, L, V) decreases in favor of G, although one must be wary of sampling bias depending on the country (see above). About 5% of the sequences are unclassified by GISAID.
Forster et al. [25], based on similar classifications (but with only 160 sequences), suggested that some clades may be better adapted to certain populations and that conversely some populations may be resistant to certain variants of the virus. This “news” was picked up by the mainstream press and then tweeted by Donald Trump. The scientific world protested against this study and its hasty interpretation, with a series of responses published in PNAS [10, 26, 27]. Beyond the methodological problems (see below), the study by Forster et al. ignored the impact of the founding effects. As if in the example of Santa Clara above, it was inferred that the inhabitants of this city had different genetic and phenotypic characteristics from the rest of the Californian population. It is therefore important to be cautious about interpreting these classifications and the traits that seem to be associated with them.
6. Virus spread and phylogeography
Phylogeographic methods are based on the phylogeny of sequences and on the geographical characters attached to them, to infer the geographical origin of the ancestral nodes of phylogeny, from the leaves to the root of the tree. We thus obtain scenarios that explain the origin of the pandemic and the successive countries it has contaminated. Early approaches to phylogeography were based on parsimony. Today, probabilistic models of migration are used, within likelihood or Bayesian frameworks [22]. Beyond its questionable interpretations, the study by Forster et al. posed two problems in terms of phylogeography: the method for rooting the tree was unfounded, and the sampling biases, which are considerable depending on the country (see above), were not taken into account [10]. To root the tree the authors used the external group method, which consists in finding the point of the human virus tree closest to the bat virus (RaTG13). We have already explained above why this method cannot work here. As for sampling biases, these have a considerable impact on the reconstructions. For example, with 42,000 sequences from the UK versus 700 from Italy, as currently available on GISAID, one will tend to see a UK origin for most of the ancestral nodes, and conclude that the origin of the Italian epidemic comes from the UK.
Below, we describe a new approach to correct these two limitations and to offset as much as possible the relatively weak phylogenetic signal in the data, due to the very short evolution time since the origin of the pandemic. Our study focuses on the first epidemic wave. All data, programs and options, as well as the overall pipeline are available at https://github.com/evolbioinfo/phylocovid/tree/CRAS.
We have used 11,316 genomes, corresponding to the totality of sequences available on GISAID as of April 25, and covering the first wave of the epidemic in most regions of the world. A total of 70 countries are represented, as well as the two cruise ships Diamond Princess and Grand Princess. To estimate sampling biases in these data, we use the number of cases reported in each country as of April 25, 2020 (www.ecdc.europa.eu/en/publications-data/download-todays-data-geographic-distribution-covid-19-cases-worldwide). The biases are considerable, with for example in Italy 0.4% of all sequences versus 7.4% of reported cases worldwide, while in the UK the same figures are 28.8% and 5.5%. To validate our reconstructions, we use patient travel and contact follow-up data, available at www.gisaid.org for 294 sequences.
Genomes are aligned by COVID-Align [16]. A first tree is inferred from the totality of the data, minus the duplicated sequences, by combining FastME [28] for the initial tree and RAxML-NG [29] to refine this first tree. This first tree is rooted with the “zero” sequence. This very simple rooting method is widely accepted and is used by others, notably GISAID and NEXTSTRAIN. Duplicated sequences are reinserted into the tree at a zero distance from their sister sequences. Outlier sequences (sequencing or dating errors) with an abnormally high rate of evolution compared to the zero sequence are removed (rate > median rate + 3 standard deviations). The tree thusly obtained contains 11,262 sequences. It is poorly resolved, due to the close proximity of the sequences, and highly biased in terms of sampling density depending on the country. We will see below that the phylogeographic reconstruction based on this complete tree is poorly supported.
This tree is used to construct low biased sub-samples, while keeping the “phylogenetic diversity” as high as possible. This measure of biodiversity, commonly used in ecology and species conservation, is simply the sum of the branch lengths of the phylogeny studied [30]. In short, in species conservation the approach underlying this measure is to conserve the essential length of the tree of living organisms, which represents the sum of the genetic inventions carried by the species under consideration, rather than a large number of species, some of which may be genetically similar. Here, the method consist in removing duplicated or very similar sequences, while preserving the essential part of the complete epidemic tree. Steel [31] has shown the optimality of the algorithm consisting in iteratively removing the leaf associated with the shortest branch. This simple and fast algorithm allows us to find the sub-tree of greatest phylogenetic diversity for a given number of sequences, starting from a large initial tree. Here, one first calculates how many leaves from each country should be removed from the complete epidemic tree to approximate as closely as possible the proportions of reported cases. Next, the leaves associated with the shortest branches corresponding to the over-represented countries are randomly removed until a tree of the desired size is obtained. Moreover, for each country we aim to select sequences equally spread in time. However, all (263) sequences dating from December 2019 and January 2020 are kept in order to have as much information on the origin of the pandemic as possible. In this study we chose to keep about 2000 genomes in the tree, to have enough information and unbiased samples. As this algorithm has a random part, we repeated the sub-sampling five times in order to check the stability of our results on different data sets.
Phylogenetic scenario showing the main transmission clusters of SARS-CoV-2 until April 25, 2020. The nodes correspond to transmission clusters sharing the same geographical origin. We display the number of viruses sequenced in these clusters (e.g. 42 in the Italian cluster). For each cluster, we display the clades (S, V, L, G, GR and GH from GISAID) the sequences belong to. S contains the “sequence zero”. The three G clades carry the G614 mutation of the Spike, GR and GH being G-derived clades. The dates are those of the origin of transmission within the cluster (for example between 29/11 and 10/12 2019 for the initial Chinese cluster). The thin arrows show the transmission by a single patient from one country to another (e.g. a Chinese origin for the English cluster of size 31). Thick arrows indicate multiple transmissions and their number (e.g. 13 transmissions from China to small US clusters, of sizes between 1 and 7). The dashed arrows indicate a polytomy whose resolution comes from geographical characters (e.g. Italian outbreak of size 42, with descendants in Spain, USA, France, Brazil and Russia). The smallest clusters ( < 16 sequences) are not represented; 1239 out of 1996 sequences are included in this graph; the complete tree is available on https://github.com/evolbioinfo/phylocovid/tree/CRAS.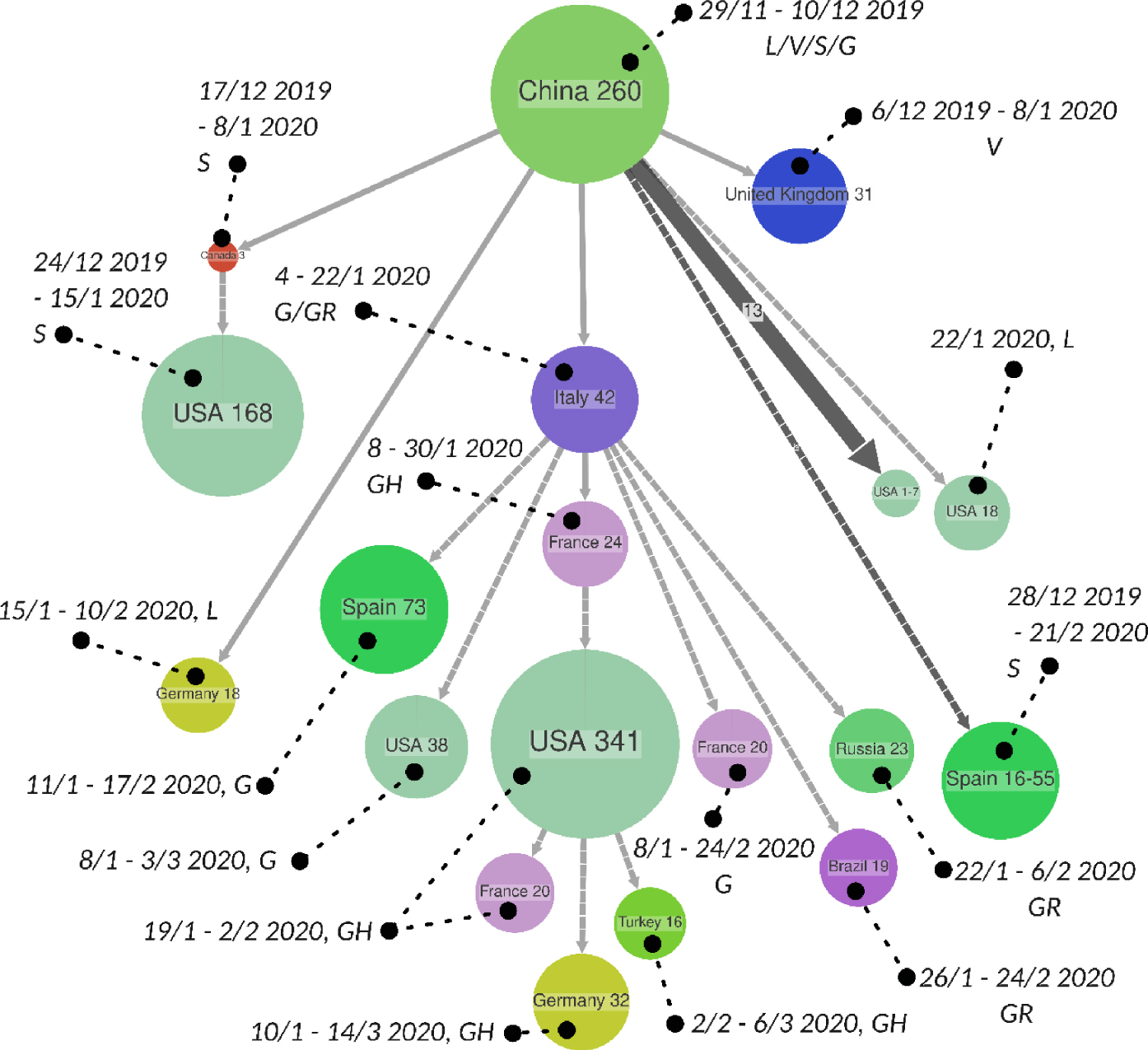
For each sample of size ∼2000, we constructed and rooted a tree with the same method as for the complete tree (see above), and dated this tree using LSD [32]. Although these trees are smaller than the complete tree, they still present a relatively low resolution, with many polytomies (nodes having more than two descendants). To resolve these as much as possible, we used geographic characters, since the sequences did not provide any information. The idea is to group descendants attached to the same geographic character in the same sub-tree. For example, if a polytomy has direct descendants from France and Italy, we will create two sub-trees of this polytomy (now resolved) corresponding to the Italian and French nodes. This procedure for resolving polytomies takes place after the ancestral inference of geographic characters by PastML [33] and does not call into question this phase of inference. It induces a better resolution of the tree, which conforms to the principle of parsimony (and maximum likelihood with standard hypotheses and models). The result for one of the de-biased samples is given in Figure 2 (the scenarios for the other four sub-samples are almost identical, see https://github.com/evolbioinfo/phylocovid/tree/CRAS/data/20200425/figures). To improve the readability of the figure, the PastML options are used, which consist in showing only the main epidemic clusters (i.e. a set of leaves and nodes connected in the tree and associated to the same geographical character [34]), and in grouping in the same arrow similar transmissions between two countries. This graph (Figure 2), which shows only the main epidemic clusters (size ⩾ 16), does not show all the data (1239 sequences out of 1996) and the complexity of the transmission chains.
As expected, at the root of the phylogeographic scenario (Figure 2), a Chinese epidemic cluster is observed, containing the four original clades: S (to which the “sequence zero” belongs), L, V and G (carrying the Spike G614 mutation). Conversely, the most recent outbreaks are all G, GR and GH, the last two being sub-clades derived from G. This scenario shows the role and diversity of the epidemic in the USA, by far the most affected country on April 25, 2020 (34% of reported cases worldwide). The first US clusters date from the end of December–beginning of January, the majority coming from China, but with a major cluster (size 168) coming from Canada, affected very early (17/12–08/01) by basal S strains. Other later USA clusters (January to March) came from Italy and France, the latter being the source of a large number of cases (341) in the USA, all from the GH sub-clade. In turn, there were transmissions from the USA to France (20), Germany (32) and Turkey (16). The main French clusters come from Italy (G and GH). Spain and Germany were both directly affected (S and L, respectively) by viruses coming from China, and by secondary epidemics, coming from Italy for Spain (G), and from the USA via France and Italy for Germany (GH). In this graph (Figure 2) showing only the main outbreaks, there is only one basal English outbreak in the UK (V, at the hinge 2019–2020, size 31), but there are many smaller and more recent ones, notably from Italy (G, 22/01–28/02/2020, size 13; GR, 21/01–22/02/2020, size 12; etc. see full tree). The global scenario (Figure 2) is consistent with more localized studies, for example on Europe [35] where it is shown, as here, that the first Italian epidemic outbreak originated in China and not in Germany as previously thought.
To confirm the accuracy of our phylogenetic reconstructions, we used available contact and travel data for 294 patients. For each, we compared the historical data (for example, a French patient known to have returned from Italy or to have been in contact with people returning from Italy), with the reconstruction produced by PastML (for example, for a French patient, his first ascendant in the tree whose prediction differs from France). The agreement between these very different sources of information is high, about 50% for the five de-biased trees of size ∼2000. When considering the complete tree (11,269 sequences), the agreement is much lower (16%). It should be noted that full agreement is not expected due to the incompleteness of the data. A French patient may have been infected by a German, for example, even if they travelled to Italy. Similarly, a French patient who stayed in France may have been infected with an Italian strain whose carrier was not sampled. A 50% agreement is therefore particularly high and validates the approach as a whole.
7. Conclusions and perspectives
Sequence analysis very clearly indicates a natural origin of SARS-CoV-2 and no significant resemblance to HIV, as has been suggested. However, its origin remains largely unknown due to its remoteness from the closest sequenced animal viruses found in bats and pangolin. New data, from yet unexplored reservoirs or from old samples, should advance our knowledge of the origin of the virus and its date of appearance and circulation in the human population.
At the time of the second wave, work on the evolution of the virus is more important than ever. The sequences of SARS-CoV-2 are mutating and have many variants, both in nucleotides and amino acids. With rare exceptions, the mutations observed since December 2019 have so far not been shown to have an impact on virulence or severity. The most notable exception is the D614G mutation, which is increasing in prevalence worldwide and seems to increase infectivity. All mutations, while not directly affecting virulence or severity, are likely to induce variations in immune responses, which will need to be investigated in potential vaccines or new tests. A greater number of sequences covering longer periods of evolution, with a more exhaustive representation of different human populations, countries and continents, will make it possible to study these mutations (point mutations, deletions, insertions, recombinations, etc.) in terms of selection pressure, convergence, adaptation to the human host, virulence, severity and pandemic risk.
Under the pressure of this pandemic and its massive data, methods, algorithms and models are progressing rapidly, as seen above with phylogeographic analyses. This methodological work, supported by ever more abundant and exhaustive data, should establish molecular epidemiology as a key area in the study and control of future viral pandemics.
Acknowledgements
Many thanks to Amandine Perrin and Etienne Simon-Lorière of the Institut Pasteur for their help and comments, as well as to the GISAID Team and all data contributors who share their viral sequences.
French version
1. Introduction
Le SARS-CoV-2 est un virus à ARN. Son génome comporte environ 30 000 bases, ce qui en fait le plus long des génomes de virus à ARN connus. Par comparaison, le génome de la grippe a une longueur de 10 à 15 000 bases, et le VIH (un rétrovirus) une longueur d’environ 10 000 bases. Les premières séquences du génome du SARS-CoV-2 ont été disponibles fin décembre 2019, toutes issues de Wuhan (Chine), la première le 23/12, la seconde le 26/12, puis 16 autres le 30/12 [1, 2]. A partir de mi-janvier 2020 a commencé le séquençage hors Chine, et fin janvier on disposait d’environ 250 séquences issues de nombreux pays (Thaïlande, Népal, Japon, Canada, Etats-Unis, France, Allemagne, Italie, Australie…). Le séquençage a d’abord été assez lent, puisque fin mars 2020 on ne disposait toujours que de quelques centaines de séquences. Il s’est considérablement accéléré en avril avec le confinement massif à la surface du globe et les dizaines puis centaines de milliers de cas déclarés en Europe et aux Etats-Unis. Certains jours d’avril-mai plusieurs milliers de nouvelles séquences ont été déposées sur le site du GISAID (Global Initiative on Sharing Avian Influenza Data, www.gisaid.org), qui recueille et rend publiques les séquences du monde entier. Depuis, le nombre de génomes déposés varie mais reste élevé, avec plus de 500 génomes par jour issus du monde entier. Aujourd’hui (30/09/2020) il y a plus de 100 000 séquences sur le site du GISAID, provenant de 120 pays. Certains pays ont peu séquencé (ou gardé leurs séquences sans les rendre publiques). Par exemple, jusqu’à tout récemment il y avait moins de 300 séquences italiennes sur le GISAID malgré l’impact de la pandémie. Il y en a 700 aujourd’hui. La France n’a pas non plus fourni beaucoup de séquences, notamment en comparaison du Royaume Uni (environ 600 versus plus de 42 000 sur le GISAID aujourd’hui). En particulier, on ne dispose que de très peu (11) de séquences du Grand Est, alors que c’est un foyer majeur [3]. Dans ce contexte, retracer la diffusion de l’épidémie en France comme à une échelle plus globale est une tâche ardue, voire impossible dans certaines régions.
L’ensemble des résultats sur l’origine, l’évolution et la diffusion du SARS-CoV-2 vient de l’analyse informatique de ces séquences, couplée aux métadonnées associées comme la date et le lieu de séquençage, la technique de séquençage, etc. Dans certains cas on a pu faire du suivi de contacts et déterminer quelle était l’origine précise d’un virus trouvé à un endroit donné. C’est par exemple le cas pour les deux premiers génomes Thaïlandais, dont on a montré en suivant les itinéraires des patients qu’ils venaient de Chine. On a aussi pu retracer l’origine chinoise de l’infection du « patient un » à Codogno en Italie, en février 2020, même si on sait maintenant par l’analyse des eaux usées que le virus circulait déjà en Lombardie en décembre 2019. Mais ces informations sont très partielles. Pour l’essentiel nos connaissances viennent de l’analyse des séquences [4], qui se base sur des modèles (par exemple pour décrire les mutations, leur rythme et leur régularité au cours du temps) ainsi que sur des algorithmes [5], aujourd’hui confrontés à des masses et flux de données hors normes. On doit garder en tête que les conclusions que l’on tire de ces analyses dépendent des modèles et des approximations effectuées par ces algorithmes, qui suivent des approches heuristiques du fait de la masse de données et de la complexité des problèmes [6]. L’évolution du virus ayant commencé sous nos yeux il y moins d’un an, les souches montrent encore peu de différences ce qui limite certaines analyses, par exemple la recherche de traces d’adaptation [7, 8]. Finalement, certaines analyses sont compliquées par le faible séquençage de certaines régions du globe (cf. ci-dessus) et la qualité de certaines séquences. Malgré ces limites, on a aujourd’hui des réponses très claires sur un certain nombre de questions, par exemple sur l’origine naturelle du virus et le fait qu’il n’est pas issu d’un laboratoire [9]. On fait ici un bilan de ces avancées et leurs limites, avec une attention particulière en fin d’article sur la propagation mondiale de la pandémie au moyen d’une nouvelle approche phylogéographique visant à corriger les biais d’échantillonnage [10].
Phylogénie des souches apparentées au SARS-CoV-2 (GISAID, 10/05/2020).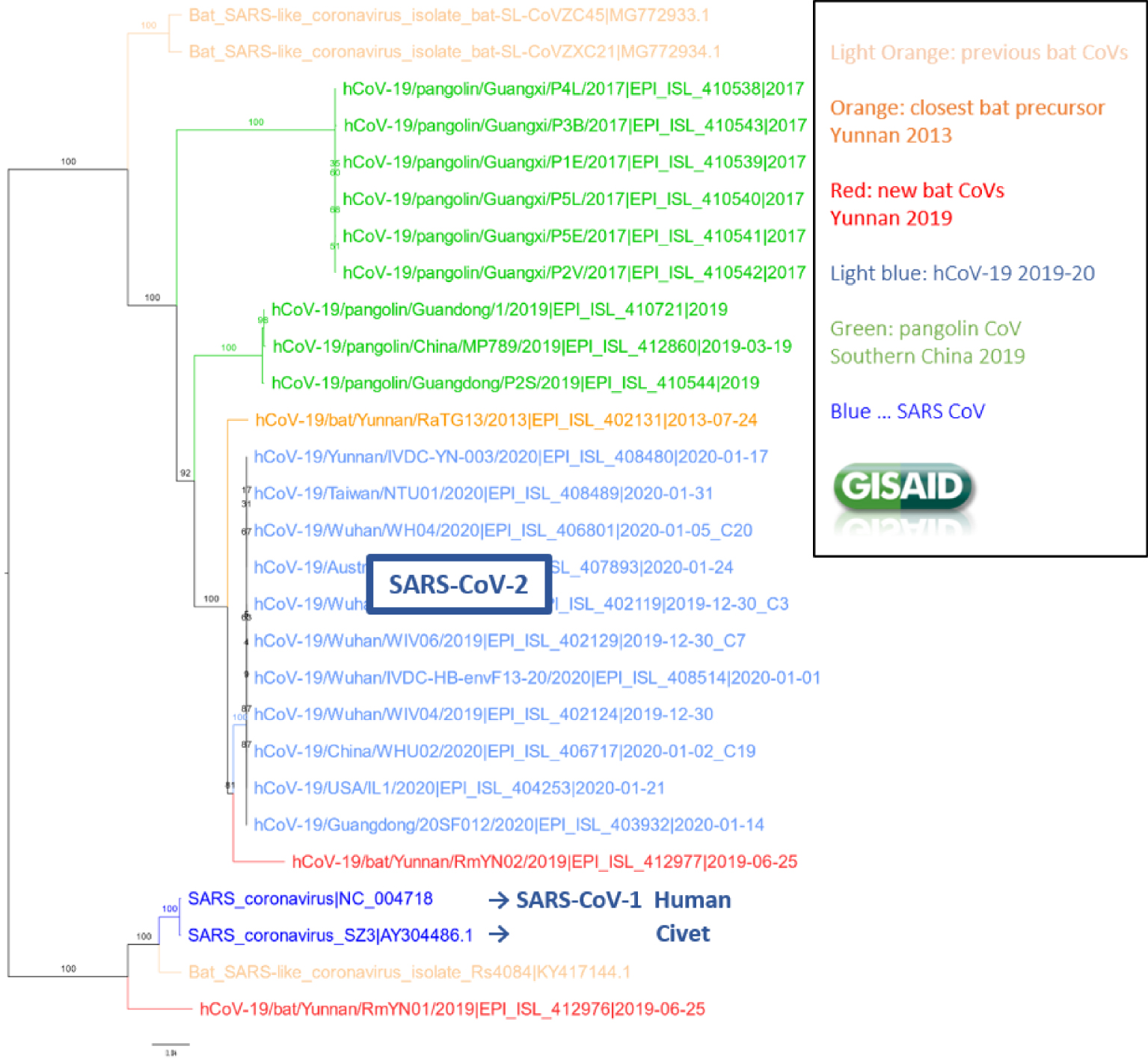
2. Origine et phylogénie
Dès l’obtention des premières séquences on a construit des phylogénies pour retrouver l’origine du SARS-CoV-2 (qui ne s’appelait pas encore ainsi). C’est un Betacoronavirus, membre des Sarbecovirus, sous-genre viral incluant le virus responsable de l’épidémie du SRAS en 2003, et nommé SARS-Cov-1 (pour severe acute respiratory syndrome-related coronavirus). Le SARS-CoV-2 est aussi appelé hCoV19 par certains, le GISAID notamment, qui considèrent à juste titre que ce n’est sans doute pas la 2ème épidémie humaine de ce type, et que d’autres l’ont précédée. Les Sarbecovirus infectent non seulement les humains, mais également de nombreux mammifères, notamment les civettes, les chauves-souris et les pangolins. La phylogénie de ce virus et ses variants (Figure 1) montre que :
- Les virus connus les plus proches du SARS-CoV-2 viennent de deux chauves-souris Rhinolophe ou « fer à cheval », trouvées dans le Yunnan en 2013 (RaTG13, [1]) et 2019 (RmYN02, [11]). L’identité entre les génomes est d’environ 96% pour l’une (RaTG13) et 93% pour l’autre (RmYN02), mais ce taux d’identité varie le long des séquences. En particulier, il est assez faible (60–70%) dans la région RBD (Region Binding Domain, ∼60 acides aminés, au sein de la protéine Spike) de liaison à la protéine humaine ACE2, qui permet l’entrée dans la cellule hôte [9].
- Plus éloigné globalement (90% d’identité), se trouve un virus de pangolin, dont la région RBD est à l’inverse très proche du SARS-CoV-2, avec une seule mutation en acide aminé, contre une douzaine pour la chauve-souris [9].
- Toutes les autres souches apparentées au SARS-CoV-2 sont beaucoup plus éloignées, notamment le SARS-CoV-1 (80% d’identité).
Les analyses montrent qu’au sein des Sarbecovirus, les recombinaisons sont nombreuses [12]. Celles-ci sont très possiblement à l’origine du SARS-CoV-2, mais à ce jour on ne peut l’affirmer car on n’a pas trouvé dans les réservoirs naturels de génomes ou portions significatives de génomes qui soient très proches de la forme humaine. En ceci le SARS-CoV-2 se distingue nettement du SARS-CoV-1 qui est lui très proche (99,6%) du virus de la civette [13]. Dans un premier temps, on a suggéré qu’en raison des similarités observées (cf. ci-dessus), le SARS-CoV-2 serait un recombinant dans la région RBD de virus de chauve-souris et de pangolin. Mais depuis, l’observation de l’évolution de cette région dans les souches humaines a montré que celle-ci mute rapidement, avec 36 mutations en acide aminé présentes parmi les souches humaines disponibles aujourd’hui (30/09/2020). L’hypothèse alternative d’une adaptation du virus de chauve-souris dans cette région, plutôt qu’une recombinaison avec le virus de pangolin, est donc tout à fait crédible, d’autant qu’on a montré que le SARS-CoV-2 passait facilement à la souris, avec seulement quelques mutations vraisemblablement adaptatives dans la région RBD [14].
On a beaucoup lu que le SARS-CoV-2 est issu de la chauve-souris et on tend à penser que le passage à l’humain est récent. En réalité il y a 4% de différences entre les deux génomes, soit environ 1200 mutations. Depuis décembre 2019 on voit évoluer le virus chez l’humain. En se basant sur le nombre de mutations observées aujourd’hui par rapport aux toutes premières séquences, et en rapportant ce nombre au temps écoulé, on trouve que les génomes évoluent au rythme d’une ou deux mutations par mois, ce qui est lent si on le compare à la grippe ou au VIH. Les 1200 mutations observées correspondent donc à 50 à 100 ans d’évolution et une date comprise entre 1970 et 1995 pour l’ancêtre commun du SARS-CoV-2 et de RaTG13 (100 ans d’évolution donnent la date de 1970, car on additionne les années dans les deux branches issues de l’ancêtre commun homme/chauve-souris). Des analyses bayésiennes plus poussées indiquent des dates analogues voire plus anciennes, avec une part d’incertitude recouvrant tout le 20ème siècle [12]. Quoiqu’il en soit, ces résultats indiquent et confirment qu’entre cet ancêtre commun et le SARS-CoV-2 il y a eu de nombreux intermédiaires, chez la chauve-souris ou d’autres mammifères comme le pangolin, et que ces intermédiaires restent à découvrir. Les coronavirus ayant franchi trois fois (les deux SARS et le MERS) de manière marquante la barrière d’espèces au cours des 20 dernières années, il est probable qu’ils le fassent à nouveau, d’où l’importance de rechercher ces réservoirs animaux.
3. Une origine naturelle
On a très tôt trouvé dans la presse grand-public des allégations selon lesquelles le SARS-CoV-2 serait un produit de laboratoire. Tous les résultats et chiffres donnés ci-dessus démontrent le contraire. Les 1200 mutations qui séparent le SARS-CoV-2 de la souche la plus proche chez la chauve-souris (RaTG13) sont réparties aléatoirement le long du génome, alors que le génie biologique aurait produit un assemblage de fragments connus (sans mutations par rapport à certaines souches connues), avec des mutations ponctuelles dans des régions stratégiques, par exemple RBD [9]. En se basant sur des similarités locales entre le génome du SARS-CoV-2 et celui du VIH, on a aussi prétendu qu’il s’agissait d’une tentative de vaccin contre le VIH. Mais ces similarités locales n’expliquent pas l’origine du reste du génome, et elles ne sont pas significatives. Elles portent sur un court segment de 38 nucléotides où les deux virus ont 87% d’identité. Mais en comparant le génome du SARS-CoV-2 à d’autres génomes on trouve de nombreuses similarités de cet ordre, par exemple 89% sur un segment de longueur 44 avec un génome de plante. On voit simplement ici l’effet du bricolage de l’évolution, qui utilise et réutilise les mêmes solutions pour bâtir le vivant. Mais en aucun cas la marque d’une significativité statistique, bien difficile à estimer.
4. Evolution chez l’hôte humain
On s’est très tôt posé la question de l’origine, de la date et du « patient zéro » de la pandémie humaine. Les seules séquences ne permettent pas de répondre complétement à ces questions. En phylogénie on utilise la méthode du groupe externe pour enraciner le groupe d’intérêt ; par exemple, pour enraciner l’arbre des mammifères on utilise un génome de reptile ou d’oiseau qui en sont les espèces les plus proches. Mais cette méthode ne fonctionne pas ici, le nombre de mutations observées entre le virus humain et celui de la chauve-souris (1200, cf. ci-dessus) étant sans commune mesure avec le nombre de mutations observées parmi les séquences humaines (quelques dizaines). Autrement dit toutes les séquences humaines sont peu ou prou à la même distance de celle de la chauve-souris et on ne peut pas avec cette méthode désigner avec certitude la racine de la pandémie.
On a de meilleures garanties en s’appuyant non seulement sur les séquences mais aussi sur les dates et l’histoire. En effet, le 30 décembre 2019 on a trouvé chez plusieurs patients chinois exactement la même séquence. Celle-ci a rapidement été trouvée en Thaïlande, au Japon et aux USA et, par exemple, elle était toujours présente fin mars au Royaume Uni. C’est donc une bonne candidate pour être la « séquence zéro ». Elle est utilisée comme telle par de nombreuses équipes et de nombreux logiciels ou sites web, notamment GISAID, NEXSTRAIN (https://nextstrain.org/), etc. Comme cette séquence (WIV04/2019) a été trouvée en différents points du globe, il est impossible de dire quelle est son origine géographique. Mais l’histoire de la pandémie démontre clairement une origine chinoise [1, 2]. Diverses méthodes de datation indiquent un début de la diffusion de l’épidémie entre mi-Octobre et début décembre 2019 [7]. Il est difficile à l’heure actuelle d’avoir plus de certitudes. Comme avec d’autres virus, VIH notamment, l’acquisition de nouvelles séquences, possiblement prélevées dans des échantillons anciens, devrait permettre d’affiner ces estimations et vraisemblablement trouver une origine plus précoce de la pandémie humaine.
Les premières séquences ne montraient que très peu voire aucune différence. Aujourd’hui, après environ 10 mois d’évolution, les séquences les plus récentes sont séparées de la « séquence zéro » par au plus une trentaine de mutations en nucléotides et une quinzaine en acides aminés. Ces chiffres sont pour une part incertains, car il est parfois difficile de distinguer mutation et erreur de séquençage ou d’assemblage. On observe quelques délétions, parfois longues et trouvées chez plusieurs patients, notamment une délétion de 382b dans l’ORF8 et sa région régulatrice, échantillonnée une cinquantaine de fois entre Singapour et Taiwan [15]. Cette délétion a été observée sous une forme proche dans le SARS-CoV-1, où elle est associée à une atténuation du virus, mais à ce jour cette atténuation n’a pas été constatée chez le SARS-CoV-2, non plus qu’un quelconque effet adaptatif. A l’inverse des délétions, on n’a pas trouvé d’insertion marquante et largement partagée entre virus de patients différents [16].
On a très rapidement cherché des mutations qui pourraient résulter d’une adaptation à l’hôte humain, ou être attachées à une plus grande virulence ou sévérité. La rareté des mutations et la faible durée d’évolution du virus chez l’humain rendent ces analyses difficiles. On considère généralement que les mutations observées chez les virus sur des périodes courtes ont un impact neutre ou faible sur le phénotype, et qu’elles sont essentiellement issues de processus aléatoires complexes liés aux erreurs de réplication puis à la diffusion au sein de la population humaine. Suivant cette hypothèse communément admise, il n’existerait pas de souches plus virulentes ou plus sévères que les autres. Pour l’instant très peu de données contredisent cette hypothèse. Cependant, la mutation D614G de la protéine Spike (transformation d’un résidu D en G à la position 614) semble correspondre à une transmissibilité accrue, si l’on se base sur la fréquence croissante de cette mutation dans les données mondiales [17]. Alors que la pandémie a commencé avec la version D614 et s’est transmise dans cette version à de nombreux pays, ceux-ci sont aujourd’hui presque tous majoritairement affectés par la version G614 (par exemple en France, G : ∼100%), à l’exception notable de la Chine (D : 60%, G : 40%), de l’Islande où on assiste à un retour du D après une phase presque exclusivement G, ou par exemple de Santa Clara en Californie, qui est essentiellement D depuis le début de la pandémie (alors que la Californie est essentiellement G). On voit sur ce dernier exemple qu’il faut prendre ces résultats avec précautions, car ils résultent largement d’effets fondateurs dont l’impact peut perdurer longtemps. Dans le cas de D614G, on a cependant des indices complémentaires, venant d’expériences in vitro, qui indiquent des titres et une infectiosité plus élevés du variant G ; mais on ne voit aucune différence de sévérité entre les deux variants, qui ont la même résistance à la neutralisation par le sérum de malades convalescents [17, 18]. Une centaine d’autres mutations du Spike ont été étudiées in vitro [18], dont certaines ont un impact sur l’infectiosité et le pouvoir antigénique, sans pour autant présenter d’augmentation substantielle de prévalence dans la population mondiale comme on le voit pour le variant G614.
Alors que les coronavirus recombinent abondamment [12], on n’a pas trouvé pour l’instant de marqueurs fiables de recombinaisons parmi les souches humaines [4]. Celles-ci pourraient se produire en cas de co-infection par des souches significativement différentes, mais la probabilité de co-infection est faible, et les souches qui circulent actuellement sont trop similaires pour que ce phénomène puisse être détecté si tant est qu’il se produise. De ce point de vue, le SARS-CoV-2 semble se distinguer nettement de la grippe, qui évolue par réassortiment de sous-types différents, pouvant aboutir à des changements radicaux présentant des risques pandémiques majeurs.
Finalement, avant de conclure cette partie sur l’évolution du SARS-CoV-2, il faut souligner une tendance de son génome à remplacer les bases cytosines (C) par des uraciles (U) [8]. Cette tendance s’explique par le métabolisme de la cytosine et les erreurs de réplications [19]. Elle est relativement faible mais significative, on observe ∼0.1% d’augmentation de U en moyenne sur les sites variables entre décembre 2019 et avril 2020. Des analyses détaillées [8] montrent que les mécanismes mutationnels produisent globalement un excès de U, mais que ces mutations tendent à être contre-sélectionnées au niveau des sites synonymes et de certains di-nucléotides. Ces mécanismes évolutifs constituent une dérive plutôt qu’une adaptation à l’hôte humain, avec un impact faible sur le protéome. Cependant, ils peuvent être clefs dans le design de vaccins basés sur des formes atténuées du virus [8].
5. Clades et sous-types
On s’est rapidement posé la question de l’existence de sous-types du SARS-COV-2, par analogie avec les sous-types du VIH ou de la Dengue, par exemple. Le concept de sous-type prend tout son sens s’il correspond à des séquences bien distinctes possédant des caractères épidémiologiques d’intérêt qui les séparent des autres sous-types. Par exemple, les sous-types B et C du VIH sont nettement séparés dans la phylogénie, avec des forts supports statistiques [20], et correspondent à des épidémies distinctes touchant essentiellement l’Afrique pour le C et l’Europe et l’Amérique du Nord pour le B. On estime la séparation entre ces deux sous-types à une centaine d’année [21] et ils semblent présenter des différences en termes de résistance aux traitements ou de temps précédant la phase SIDA (en l’absence de traitement). On a le même type de séparation nette pour les quatre sous-types de la Dengue [22].
Pour le SARS-CoV-2 on ne s’attend aujourd’hui à rien de tel, puisque le virus est apparu chez l’humain fin 2019. Plusieurs groupes ont proposé des classifications, avec l’objectif de faciliter le suivi de l’épidémie et de bâtir une nomenclature qui devrait s’avérer utile à terme, par exemple si certaines souches ne circulent plus ou d’autres deviennent prédominantes. La distinction la plus convaincante est associée à la mutation D614G discutée plus haut. Les séquences comportant la version G614, accompagnée de deux mutations au niveau de l’ARN, constituent le clade G du GISAID [23], nommé B1 par le système PANGOLIN [24]. Ce clade a une différence phylogénétique claire avec les autres séquences, même si les supports bootstrap ne sont pas très élevés, et il présente un grand intérêt épidémiologique potentiel, puisqu’il semble correspondre à une transmissibilité accrue et que le clade G devient prédominant dans la plupart des pays et continents [17]. Du clade G sont dérivés les sous-clades GH et GR du GISAID (B1.1 et B1.2 pour PANGOLIN), portant aussi la mutation G614 du Spike, dont l’intérêt épidémiologique, essentiellement phylogéographique, est moins évident. Les séquences situées en dehors du clade G constituent le clade S (GISAID, A pour PANGOLIN), qui contient la « séquence zéro » et les premières séquences observées en décembre/janvier, ainsi que les clades L et V (respectivement B et B2 pour PANGOLIN). La prévalence de ces trois clades (S, L, V) diminue au profit du G, même s’il faut se méfier des biais d’échantillonnage suivant les pays (cf. ci-dessus). Environ 5% des séquences sont non classées par le GISAID.
Forster et al. [25] en se basant sur des classifications analogues (mais avec seulement 160 séquences) ont avancé que certains clades pouvaient avoir une meilleure adaptation à certaines populations, et que réciproquement certaines populations seraient résistantes à certains variants du virus. Cette « nouvelle » a été reprise par la presse grand publique, puis tweetée par Donald Trump. Le monde scientifique s’est élevé contre cette étude et son interprétation hâtive, avec une série de réponses publiées dans PNAS [10, 26, 27]. Au-delà des problèmes méthodologiques (cf. ci-dessous), l’étude de Forster et al. ignorait l’impact des effets fondateurs. Comme si dans l’exemple de Santa Clara ci-dessus, on en déduisait que les habitants de cette cité avaient des caractéristiques génétiques et phénotypiques différentes du reste de la population Californienne. Il importe donc d’être prudent sur l’interprétation de ces classifications et des caractères qui semblent leur être associés.
6. Propagation du virus et phylogéographie
Les méthodes de phylogéographie se basent sur la phylogénie des séquences et sur les caractères géographiques attachés à celles-ci, pour inférer l’origine géographique des nœuds ancestraux de la phylogénie, depuis les feuilles jusqu’à la racine de l’arbre. On obtient ainsi des scénarios qui expliquent l’origine de la pandémie et les pays successifs qu’elle a contaminés. Les premières approches de phylogéographie étaient basées sur la parcimonie. On utilise aujourd’hui des modèles probabilistes de migration, en se plaçant dans un cadre de vraisemblance ou Bayésien [22]. Au-delà de ses interprétations contestables, l’étude de Forster et al. posait deux problèmes en termes de phylogéographie : la méthode pour enraciner l’arbre n’était pas fondée, et on ne prenait pas en compte les biais d’échantillonnage qui sont considérables suivant les pays (cf. ci-dessus) [10]. Pour enraciner l’arbre les auteurs utilisaient la méthode du groupe externe qui consiste à trouver le point de l’arbre des virus humains le plus proche du virus de la chauve-souris (RaTG13). Nous avons déjà expliqué plus haut pourquoi cette méthode ne peut pas fonctionner ici. Pour ce qui est des biais d’échantillonnage, ceux-ci ont un impact considérable sur les reconstructions. Ainsi, avec 42 000 séquences venant du Royaume Uni contre 700 d’Italie, comme disponible actuellement sur le GISAID, on aura tendance à voir une origine britannique pour la plupart des nœuds ancestraux, et en conclure que l’origine de l’épidémie italienne vient du Royaume Uni.
Nous décrivons ci-dessous nos résultats pour corriger ces deux limitations et contrebalancer autant que possible le signal phylogénétique relativement faible des données, du fait du temps d’évolution très court depuis l’origine de la pandémie. Notre étude porte sur la première vague épidémique. L’ensemble des données, des programmes et options, ainsi que le pipeline global sont disponibles sur https://github.com/evolbioinfo/phylocovid/tree/CRAS.
Nous avons utilisé 11 316 génomes, correspondant à la totalité des séquences disponibles sur le GISAID au 25 avril, et couvrant la première vague de l’épidémie dans la plupart des régions du monde. Au total 70 pays sont représentés, ainsi que les deux bateaux Diamond Princess et Grand Princess. Pour estimer les biais d’échantillonnages dans ces données, nous avons utilisé le nombre de cas déclarés dans chaque pays, à la date du 25 avril 2020 (www.ecdc.europa.eu/en/publications-data/download-todays-data-geographic-distribution-covid-19-cases-worldwide). Les biais sont considérables, avec par exemple en Italie 0.4% de l’ensemble des séquences contre 7.4% des cas déclarés à l’échelle mondiale, alors qu’au Royaume Uni ces mêmes chiffres sont de 28.8% et 5.5%. Pour valider nos reconstructions, nous utilisons les données de voyages et de suivis de contacts des patients, disponibles sur www.gisaid.org pour 294 séquences.
Les génomes sont alignés par COVID-Align [16]. Un premier arbre est inféré à partir de la totalité des données, moins les séquences dupliquées, en combinant FastME [28] pour l’arbre initial puis RAxML-NG [29] pour raffiner ce premier arbre. Celui-ci est enraciné avec la séquence « zéro ». Cette méthode d’enracinement très simple fait consensus et est utilisée par d’autres, notamment le GISAID et NEXTSTRAIN. Les séquences dupliquées sont réinsérées dans l’arbre à distance zéro de leur séquences sœurs. Les séquences aberrantes (erreurs de séquençage ou de datation) présentant un taux d’évolution anormalement élevé par rapport à la séquence zéro sont retirées (taux > taux médian + 3 écarts types). L’arbre ainsi obtenu contient 11 262 séquences. Il est peu résolu, du fait de la grande proximité des séquences, et fortement biaisé en terme de densité d’échantillonnage suivant les pays. On verra ci-dessous que la reconstruction phylogéographique sur la base de cet arbre complet est peu supportée.
Cet arbre est utilisé pour construire des sous-échantillons peu biaisés, tout en conservant une diversité phylogénétique aussi élevée que possible. Cette mesure de biodiversité, communément utilisée en écologie et conservation des espèces, est tout simplement la somme des longueurs de branche de la phylogénie étudiée [30]. Pour aller vite, en conservation des espèces l’approche sous tendue par cette mesure est de conserver l’essentiel de la longueur de l’arbre du vivant, qui représente la somme des inventions génétiques portées par les espèces considérées, plutôt qu’un nombre élevé d’espèces dont certaines peuvent être très proches. Ici, la méthode va consister à retirer les séquences dupliquées ou très similaires, tout en conservant l’essentiel de l’arbre épidémique complet. Steel [31] a montré l’optimalité de l’algorithme consistant à retirer itérativement la feuille associée à la branche la plus courte. Cet algorithme simple et rapide permet de trouver le sous-arbre de plus grande diversité phylogénétique pour un nombre de séquences données, à partir d’un arbre initial de grande taille. Ici, on calcule dans un premier temps combien on doit retirer dans l’arbre épidémique complet de feuilles de chaque pays pour approcher autant que possible les proportions de cas déclarés. Ensuite, on retire aléatoirement les feuilles associées aux branches les plus courtes et correspondant aux pays surreprésentés, jusqu’à ce qu’on obtienne un arbre de la taille désirée. Ce calcul est mensualisé pour suivre les courbes épidémiques. On conserve cependant toutes les (263) séquences datant de décembre 2019 et janvier 2020, de manière à avoir le plus d’informations possibles sur l’origine de la pandémie. Dans cette étude nous avons choisi de conserver environ 2000 génomes dans l’arbre, pour avoir suffisamment d’informations et des échantillons peu biaisés. Comme cet algorithme a une part aléatoire, nous avons répété le sous-échantillonnage cinq fois de manière à vérifier la stabilité de nos résultats sur des jeux de données différents.
Scénario phylogéographique montrant les principaux foyers de transmission du SARS-CoV-2 jusqu’au 25 avril 2020. Les nœuds correspondent à des foyers (ou clusters) de transmission partageant une même origine géographique. Les chiffres donnent le nombre de virus séquencés dans ces foyers (par exemple 42 dans le foyer italien). Pour chaque foyer on donne l’appartenance des séquences aux clades S, V, L, G, GR et GH du GISAID. S contient la séquence « zéro ». Les trois clades G portent la mutation G614 du Spike, GR et GH étant des clades dérivés du G. Les dates sont celles de l’origine de la transmission au sein du foyer (par exemple entre le 29/11 et le 10/12 2019 pour le foyer chinois initial). Les flèches minces montrent la transmission par un seul patient d’un pays à un autre (par exemple une origine chinoise pour le foyer anglais de taille 31). Les flèches épaisses indiquent des transmissions multiples et leur nombre (par exemple 13 transmissions depuis la Chine vers des foyers USA de petite taille, entre 1 et 7). Les flèches hachurées indiquent une polytomie dont la résolution vient des caractères géographiques (par exemple le foyer italien de taille 42, avec des descendants en Espagne, USA, France, Brésil et Russie). Les plus petits foyers ( < 16 séquences) ne sont pas représentés ; 1239 sur 1996 séquences sont incluses dans ce graphique ; l’arbre complet est disponible sur https://github.com/evolbioinfo/phylocovid/tree/CRAS.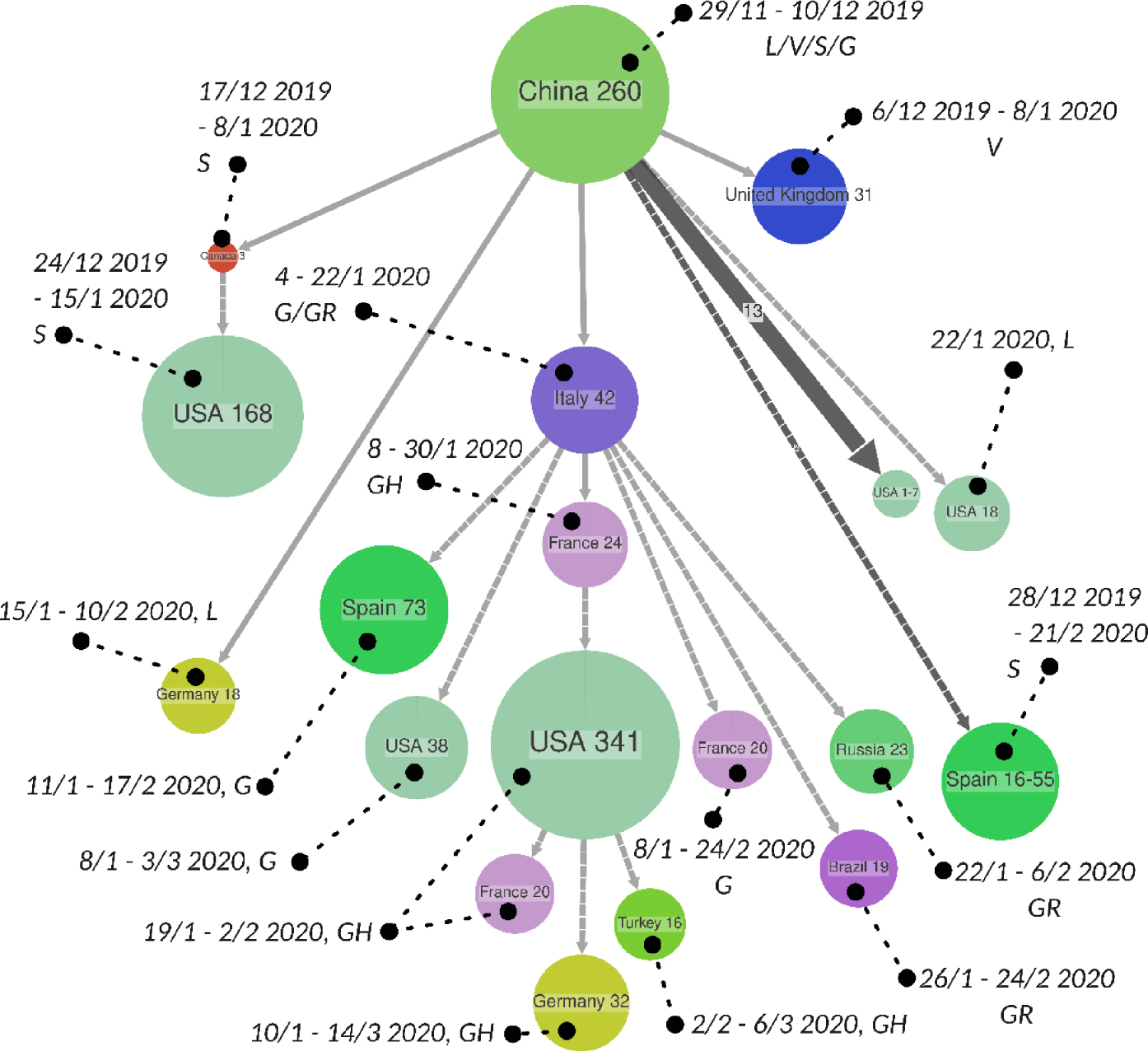
Pour chaque échantillon de taille ∼2000, nous avons construit et enraciné un arbre avec la même méthode que pour l’arbre complet (cf. ci-dessus), et daté cet arbre à l’aide de LSD [32]. Bien que ces arbres soient plus petits que l’arbre complet, ils présentent encore une résolution relativement faible, avec de nombreuses polytomies (nœuds ayant plus de deux descendants). Pour résoudre celles-ci autant que possible, nous avons utilisé les caractères géographiques, puisque les séquences n’apportaient aucune information. L’idée est de regrouper dans un même sous-arbre les descendants attachés à un même caractère géographique. Par exemple, si une polytomie a des descendants directs issus de France et d’Italie, on créera deux sous-arbres de cette polytomie (maintenant résolue) correspondant aux nœuds italiens et français. Cette procédure de résolution des polytomies prend place après l’inférence ancestrale des caractères phylogéographiques par PastML [33], et ne remet pas en cause cette phase d’inférence. Elle induit une meilleure résolution de l’arbre, qui est conforme au principe de parcimonie (et de maximum de vraisemblance avec des hypothèses et modèles standards). Le résultat pour l’un des échantillons dé-biaisés est donné dans la Figure 2 (les scénarios pour les quatre autres sous-échantillons sont quasi-identiques, cf. https://github.com/evolbioinfo/phylocovid/tree/CRAS/data/20200425/figures). Pour améliorer la lisibilité de la figure, on utilise les options de PastML qui consistent à ne montrer que les principaux foyers épidémiques (ou clusters, c.à.d. un ensemble de feuilles et de nœuds connectés dans l’arbre et associés au même caractère géographique [34]), et à regrouper dans une même flèche les transmissions analogues entre deux pays. Ce graphique (Figure 2), qui ne conserve que les principaux foyers épidémiques (taille ⩾ 16), ne montre pas toutes les données (1239 séquences sur 1996) et la complexité des chaînes de transmission. Comme attendu, on observe à la racine du scénario phylogéographique (Figure 2) un foyer épidémique chinois, qui contient les quatre clades originels S (auquel appartient la séquence « zéro »), L, V et G (porteur de la mutation Spike G614). A l’inverse les foyers les plus récents sont tous G, GR et GH, les deux derniers étant des sous clades dérivés du G. On voit dans ce scénario le rôle et la diversité de l’épidémie aux USA, de loin le pays le plus touché au 25 avril 2020 (34% des cas déclarés mondiaux). Les premiers foyers USA datent de fin décembre–début janvier, la majorité venant de Chine, mais avec un foyer majeur (taille 168) issu du Canada, touché très tôt (17/12–08/01) par des séquences basales S. D’autres foyers USA plus tardifs (janvier à mars) sont issus d’Italie et de France, cette dernière étant à l’origine d’un grand nombre de cas (341) aux USA, tous du sous clade GH. En retour, on observe des transmissions depuis les USA vers la France (20), l’Allemagne (32) et la Turquie (16). Les principaux clusters Français sont issus d’Italie (G et GH). L’Espagne et l’Allemagne ont à la fois été touchées directement (S et L, respectivement) par des virus venus de Chine, et par des épidémies secondaires, venant d’Italie pour l’Espagne (G), et des USA via la France et l’Italie pour l’Allemagne (GH). Dans ce graphique (Figure 2) ne montrant que les principaux foyers, on ne voit qu’un foyer anglais basal au Royaume Uni (V, à la charnière 2019–2020, taille 31), mais il en existe de nombreux autres plus petits et plus récents, notamment venant d’Italie (G, 22/01–28/02/2020, taille 13 ; GR, 21/01–22/02/2020, taille 12 ; etc. voir arbre complet). Le scénario globale (Figure 2) est conforme à des études plus localisées, par exemple sur l’Europe [35] où on montre comme ici que le premier foyer épidémique italien est originaire de Chine et non d’Allemagne comme on avait pu le penser.
Pour confirmer la précision de nos reconstructions phylogéographiques, nous avons utilisé les données de contacts et de voyages disponibles pour 294 patients. Pour chacun, nous avons comparé la donnée historique (par exemple, un patient français dont on sait qu’il revient d’Italie ou qu’il a été en contact avec des personnes revenant d’Italie), avec la reconstruction produite par PastML (par exemple, pour un patient français, son premier ascendant dans l’arbre dont la prédiction diffère de la France). L’accord entre ces sources d’informations bien différentes est élevé, d’environ 50% pour les cinq arbres dé-biaisés de taille ∼2000. Lorsqu’on considère l’arbre complet (11 269 séquences), l’accord est bien moindre (16%). Il faut noter qu’on ne s’attend pas à un accord total du fait de l’incomplétude des données. Un patient français peut par exemple avoir été infecté par un Anglais, même s’il a voyagé en Italie. De même, un patient français qui est resté en France peut avoir été infecté par une souche italienne dont le porteur n’a pas été échantillonné. Un accord de 50% est donc particulièrement élevé et valide l’approche dans son ensemble.
7. Conclusions et perspectives
L’analyse des séquences indique très clairement une origine naturelle du SARS-CoV-2 et aucune ressemblance significative avec le VIH, comme cela a pu être suggéré. Pour autant son origine reste largement inconnue, du fait de son éloignement avec les virus animaux séquencés les plus proches, trouvés chez la chauve-souris et le pangolin. De nouvelles données, issues de réservoirs encore inexplorés ou d’échantillons anciens, devraient permettre de faire progresser nos connaissances sur l’origine du virus ainsi que sa date d’apparition et de circulation dans la population humaine.
A l’heure de la deuxième vague les travaux sur l’évolution du virus sont plus importants que jamais. Les séquences du SARS-CoV-2 mutent et présentent de nombreux variants, en nucléotides comme en acides aminés. A de rares exceptions près, on n’a pas prouvé à ce jour que les mutations observées depuis décembre 2019 aient un impact sur la virulence ou la sévérité. L’exception la plus notable est la mutation D614G dont la prévalence augmente très nettement à l’échelle du globe et qui augmente l’infectiosité. L’ensemble des mutations, même si elles n’ont pas d’impact direct sur la virulence ou la sévérité, induisent vraisemblablement des variations dans les réponses immunitaires, qui devront être étudiées dans le cadre de vaccins potentiels ou de nouveaux tests. Un plus grand nombre de séquences portant sur des périodes plus longues d’évolution, avec une représentation plus exhaustive des différentes populations humaines, des pays et des continents, permettront d’asseoir l’étude de ces mutations (ponctuelles, délétions, insertions, recombinaisons…) en termes de pression de sélection, convergence, adaptation à l’hôte humain, virulence, sévérité et risque pandémique.
Sous la pression de cette pandémie et de ses données massives, les méthodes, les algorithmes et les modèles progressent rapidement, comme on l’a vu ci-dessus avec les analyses phylogéographiques. Ces travaux de nature méthodologiques, nourris par des données toujours plus abondantes et exhaustives, devraient asseoir l’épidémiologie moléculaire comme un domaine clé dans l’étude et la lutte contre les pandémies virales à venir.
Remerciements
Un grand merci à Amandine Perrin et Etienne Simon-Lorière de l’Institut Pasteur pour leur aide et leurs commentaires, ainsi qu’à l’équipe du GISAID et tous les contributeurs de données qui partagent leurs séquences virales.