


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Chromosomal inversions are rearrangements of DNA segments, often containing many genes, and are an important mechanism by which genome organization evolves [1, 2]. Inversions may be found when comparing genomes of different taxa, but can also be polymorphic, when alternative arrangements coexist and segregate among individuals of a population. Polymorphic inversions are often found to be associated with the variation of so-called complex traits, which involve the coordination of multiple elements of phenotype (e.g. morphology and behaviour). Examples range from dimorphic social organization in several ant species [3] to coloration and behavioral polymorphisms in many birds and butterflies [4, 5, 6, 7, 8], or from dimorphic flower morphology in plants [9] to the ubiquitous sexual dimorphism observed in animals [10]. This association presumably owes to inversions facilitating the coordination of genes controlling the different features of phenotype. Indeed, recombination is strongly reduced, if not suppressed, between differentially oriented segments and thus, inversions spanning multiple loci may protect from recombination alternative combinations of co-adapted alleles at these loci. Such group of tightly-linked loci involved in the variation of a complex trait are referred to as “supergenes” [11]. Despite their unique place in the history of genetics, little is understood about why and how these supergenes and the polymorphisms they control arise.
Polymorphisms at supergene have been proposed to be maintained by drift or by the balance of opposing evolutionary forces [2, 12, 13, 14]. Typically, explaining why certain elements of phenotype are coordinated is relatively well understood. For instance, migration syndromes in trout associate variations in reproductive physiology and migration behaviour in a way that can be envisioned as alternative strategies of maximizing the net product of fecundity and mortality risk [15]. Yet why alternative strategies are maintained as long-term polymorphisms is often less clear. Moreover, how those polymorphisms establish is still unclear. Notably, we still lack a good understanding of why chromosomal variations become associated with complex polymorphisms, and how alternative combinations of co-adapted alleles can form in the face of recombination.
In this paper, we review recent advances on the evolutionary dynamics of supergenes by focusing on the example of a wing-patterning supergene in the butterfly Heliconius numata. This example allows us to highlight key aspects affecting the formation and maintenance of inversion polymorphisms, which could bring insight into factors affecting the maintenance of other polymorphic rearrangements.
2. Genetic architecture of wing-pattern polymorphism in H. numata
The butterfly Heliconius numata is a widespread species across the Eastern Neotropics, found from the northern part of the Orinoco basin in Venezuela to the southern parts of the Atlantic Forests of Brazil (Mata Atlântica), and from the Andean foothills to the Atlantic ocean (Figure 1). H. numata is part of a clade of butterflies, the Heliconiini (Nymphalidae), containing distasteful cyanogenic glycosides protecting them from predation. Heliconius are known for their geographically diverse mimetic wing patterns, which represent key traits for local adaptation. Indeed, in Heliconius, most species display colourful wing patterns acting as toxicity warning signals to predators, and engage in warning signal mimicry with other toxic and non-toxic species [16]. Individual bearing locally-abundant wing patterns are strongly favoured by selection, since it enhances adult survival and allows butterflies to be active at all times. Typically, chemically-defended species display a single mimetic wing pattern in any locality. However, H. numata displays multiple distinct morphs in the same population, sometimes up to six or seven, each one involved in a mimicry relationship (or mimicry “ring”) with a group of similar-looking species belonging to the Heliconiinae, Danainae and Pericopiinae. This mimicry polymorphism is found across the entire range of H. numata, even though the actual set of mimetic wing patterns displayed locally by H. numata vary geographically and finely match the local variations in mimicry communities (Figure 1). Only some peripheral populations seem to be monomorphic, notably the populations from the far south of the Atlantic forests.
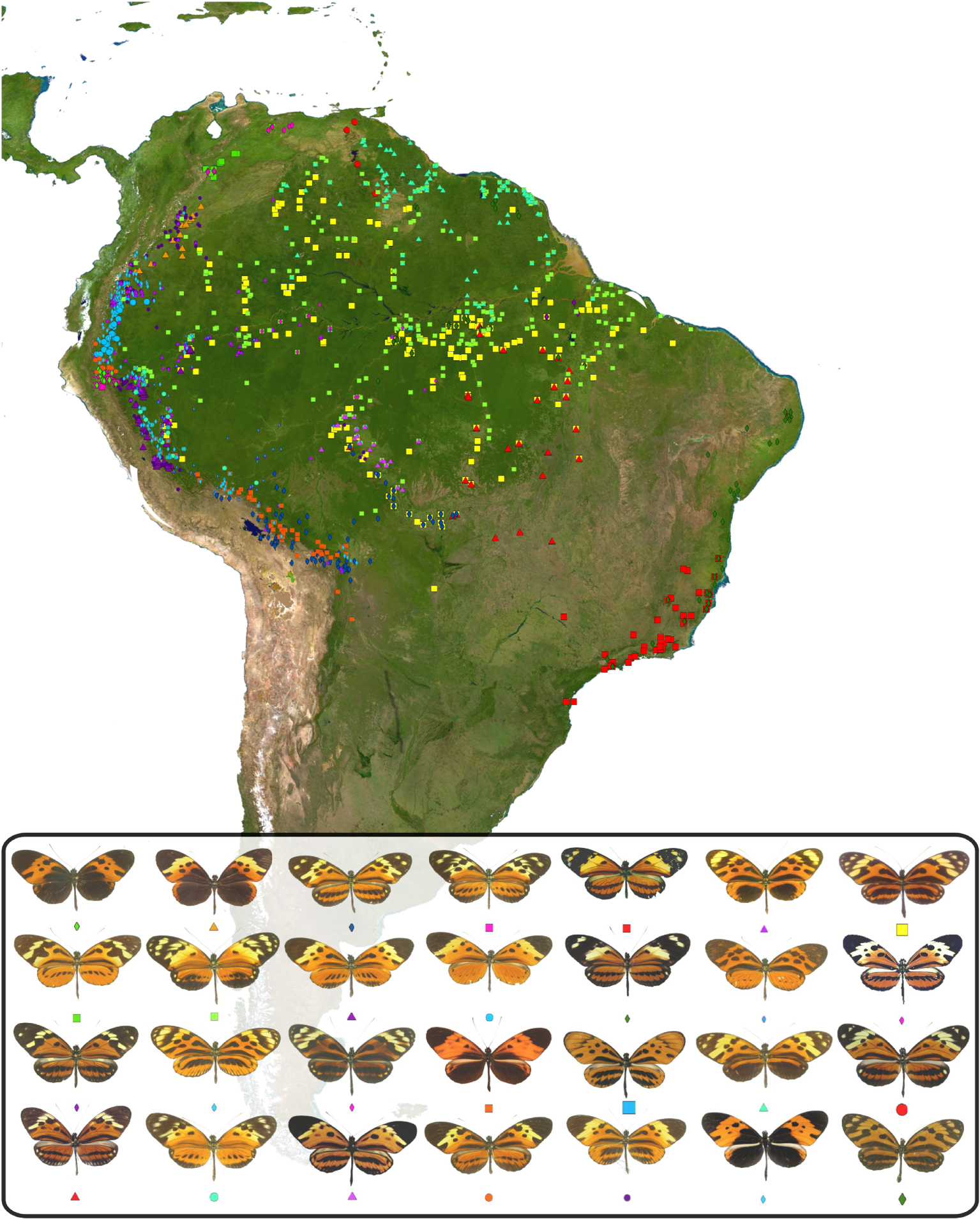
Diversity and geographic range of Heliconius numata. Colored points represent location where a given H. numata morph was observed. Data from [17].
Crosses among morphs showed that mimetic wing patterns in H. numata are inherited as alleles of a single Mendelian locus. However, rare individuals displaying wing patterns that seem to be intermediate between pair of common morphs are sometimes observed in crosses and in the wild, suggesting they are recombinant individuals [18]. Therefore, it was proposed that wing pattern variations are controlled by a supergene, i.e. a set of genes mostly inherited together as a single unit due to strong recombination reduction. This recombination reduction explains how multiple complex wing patterns may coexist and interbreed without producing numerous unfit (non-mimetic) recombinants. This supergene, named P, is sitting on chromosome 15 at the homologous position of the group of loci Yb–Sb–N which control variation in black vs. yellow patterns in the closely related species Heliconius melpomene, and in the more distantly-related H. erato [19]. To better understand the structure of the supergene alleles, we produced de novo genome assemblies for 12 H. numata individuals with various wing patterns [20]. In contrast to what was observed in closely related species, those assemblies showed that three forms of chromosome 15 coexist in populations of H. numata, each one associated with specific wing patterns and displaying different levels of dominance (Figure 2). The ancestral, standard chromosomal form, referred to as Hn0, underlies the recessive alleles associated with morphs silvana or laura. The dominant allele, controlling the morph bicoloratus, is associated with a single 400 kb inversion (P1, 21 genes), and this allelic class is referred to as Hn1. A third allelic class, displaying intermediate dominance, is associated with a diversity of mimetic forms (tarapotensis, arcuella, aurora, numata, superioris, etc.) and shares the same inversion P1, but also bears two additional inversions in adjacent positions, P2 (200 kb, 15 genes) and P3 (1150 kb, 71 genes). This third allelic class, referred to as Hn123, is thus formed by three adjacent inversions.
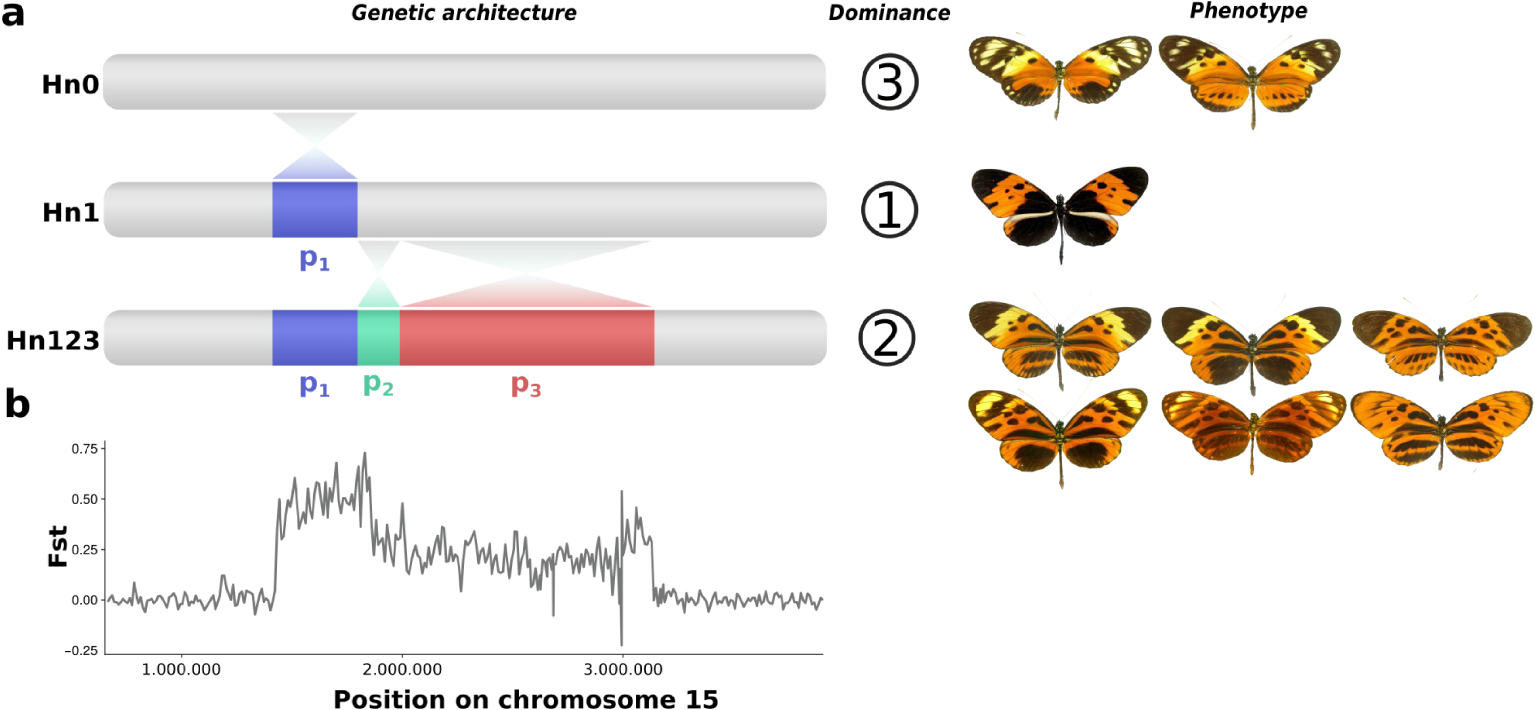
Genetic architecture and divergence at the mimicry supergene. (a) Schematic representation of the three allelic classes of the supergene of H. numata, their dominance relationship and their associated wing patterns (only a subset presented here). (b) Divergence (Fst) between Hn0 and Hn123 along the chromosome 15 (focus on the supergene region).
Since chromosomal regions with opposite orientations rarely form chiasma during meiosis, recombination is strongly reduced among haplotypes carried by different inversions. Alternative supergene rearrangements Hn0, Hn1 and Hn123 therefore do not recombine over most of the their length, as shown by their high level of divergence (Figure 2). Those results all show that the supergene is a derived architecture formed by the coordination of variations at multiple genes which recombine in other species of the clade, but are locked together via inversion polymorphism in H. numata. The supergene is therefore a novel genetic architecture, maintained by a specific selection regime in H. numata. Wing-pattern variations are subject to well-studied selection regimes, especially in Heliconius butterflies [16]. This makes the polymorphism in H. numata a great model to understand the evolution of genetic architectures.
Because recombination is suppressed as soon as inversions form segments in opposite orientations, the divergence between haplotypes carrying the functional wing patterning alleles provides a measure of their relative ages and evolutionary histories. Yet, inverted segments are sufficiently large that they contain sequences showing large differences in their level of genetic variation. Therefore, estimates of inversion ages can be obtained by computing, for different intervals (“windows”) within the inverted segment, phylogenetic relationships among individuals, and determining the most recent coalescence events between inverted and non-inverted haplotypes. Phylogenies for the Heliconius clade containing H. numata and performed with different genomic regions revealed incongruent topologies at the P1 inversion when compared to the rest of the genome [21]. It showed that P1 likely arose about 3.0 Ma in another lineage, ancestral to the extant species H. pardalinus, and was subsequently introgressed into the H. numata lineage via interspecific hybridization [21]. This occurred about 1.30 Ma after H. numata and H. pardalinus lineages had speciated. In contrast to in H. pardalinus where it is fixed, P1 remained polymorphic in H. numata, where it continued to diverge after introgression and formed the Hn1 haplotype. The introgression of P1 was followed in H. numata by the subsequent occurrence of the inversions P2 and P3 in linkage with P1 more than 1.8 million years ago, resulting in the formation of an Hn123 haplotype carrying three inverted segments [20]. Strikingly, the evolution of this novel haplotype did not result in the loss of the other haplotypes (Hn0 and Hn1). Therefore, haplotypes Hn0, Hn1, and Hn123 have been maintained at intermediate frequency for ca. two millions years, implying that a powerful mechanism balances this polymorphism.
3. The maintenance of wing-pattern polymorphisms in H. numata
Brown and Benson [18] noted that polymorphism in H. numata is puzzling because it contradicts mimicry theories [22]. Indeed, mimicry evolves in response to strong selection on the resemblance to warning signals known by local predators. For chemically-defended prey, selection therefore favours the most common forms, imposing strong positive frequency-dependence. This form of selection should oppose polymorphism because rarer variants are expected to suffer higher predation. In H. numata, Chouteau et al. [23] showed that the alternative morphs indeed enjoy different protection levels against predators, with higher protection to the commonest morph in any locality [23]. Frequency-dependence is therefore clearly positive: in Peru, it was estimated that the commonest form—carried by chromosomal rearrangement Hn1—experienced a seven-fold benefit in protection from predation compared to the rarest form—controlled by the ancestral arrangement Hn0 (Figure 3) [23]. Under the sole effect of mimicry selection, Hn0 chromosomes should be rapidly eliminated and replaced by the highly beneficial Hn1 chromosomes. Yet, Hn0 persist at high frequencies, so a powerful force must be acting against the fixation of Hn1 to explain the maintenance of polymorphism. We review below the main factors which may result in such a polymorphism.
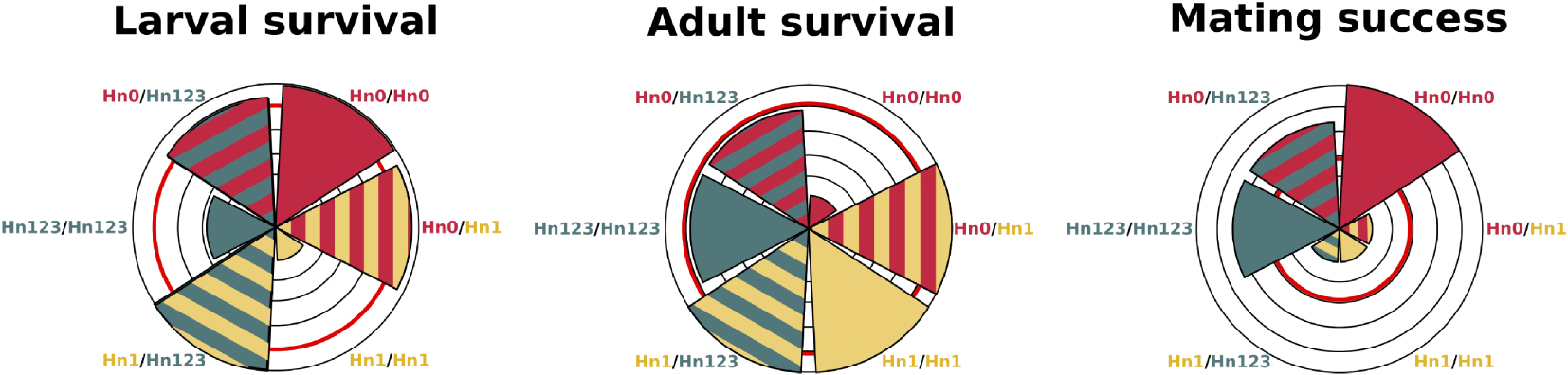
Schematic representation of the variation in fitness components associated with supergene genotypes. Triangle size represents for each genotype the selection coefficient acting on the fitness component, relative to the best genotype. Thus, triangles reaching the limit of the outer circle indicate that the underlying genotype has the most successful phenotype for the focal trait. Smaller triangles represent genotypes that are the most counter selected for the focal trait. The red circle represents the population average: genotypes represented by triangles smaller than the red circle are counter-selected, while genotypes represented by triangles bigger than the red circle are favored by selection. Selection coefficients were estimated using haplotype and genotype frequencies observed in the population of Tarapoto, Peru. Adult survival estimates are based on protection against predator. Selection coefficients were calculated relative to the population mean, and estimated in the H. numata population of Tarapoto, Peru [20]. Predation and mating success data come from [12, 23]. Masquer
Schematic representation of the variation in fitness components associated with supergene genotypes. Triangle size represents for each genotype the selection coefficient acting on the fitness component, relative to the best genotype. Thus, triangles reaching the limit of the outer circle ... Lire la suite
3.1. Variable selection on mimicry
Variable selection on wing patterns could explain the maintenance of polymorphism through fine-scale migration-selection balance. Indeed, in the region of Tarapoto (Peru), a high level of spatial heterogeneity was found in the abundance and distribution of the mimicry rings to which H. numata morphs belong, at a spatial scale under the dispersal distance of those butterflies [13]. However, several other species of Heliconius share with H. numata similar selective landscapes and similar life history traits, but do not display comparable polymorphisms. Furthermore, the heterogeneous landscape of selection is real, but models considering the sole effect of variable selection showed that an unrealistic level of admixture among sites would be required to explain the high frequency polymorphisms observed [24], suggesting other mechanisms must be at play.
3.2. Mate preferences
Chouteau et al. [12] showed that H. numata displays disassortative mate preferences, with females rejecting males of their own morph [12]. This mating pattern is relatively uncommon especially for traits involved in local adaptation. Indeed, local adaptation is expected to favour assortative mate preferences, preventing the formation of under-adapted offspring due to the mating of individuals adapted to different environment [25, 26]. This confirms that although the mosaic of selection on mimicry may favor wing pattern evolution, it cannot be the primary driver of polymorphism. Disassortative mating alone provides a powerful mechanism explaining how multiple forms are maintained in the population, since rare forms enjoy a reproductive advantage via increased mating opportunities. However, why this peculiar mating system evolved remains to be elucidated, and so the proximate origin of H. numata polymorphism.
3.3. Heterozygous advantage
Chouteau et al. [12] showed that populations of H. numata display a deficit of individuals homozygous for the allelic classes involving chromosomal inversions (Hn1 and Hn123) [12]. This lack of homozygotes may result from the above-mentioned disassortative mating, which enhances the formation of heterozygous offspring, but may also result from heterozygous advantage (or homozygous disadvantage) at the supergene. Heterozygous advantage alone may favor the maintenance of polymorphism but, strikingly, may also trigger the evolution of disassortative mating, further favouring polymorphism maintenance [27, 28]. Indeed, choosing mates that maximise the production of heterozygous offspring is beneficial under heterozygote advantage. To investigate this hypothesis of heterozygous advantage acting the supergene, we finely investigated the fitness effect of the alternative supergene alleles [20].
4. Inversions are associated with antagonistic fitness effects
As stated above, the benefits to inversions causing them to rise in frequency are thought to stem from the protection against recombination of combinations of coadaptated mutations conferring accurate mimicry resemblance to other toxic prey. Indeed, only accurate mimicry, i.e. only the right allelic combinations, bring survival and energetic benefits, while recombinants and intermediate phenotypes are unfit [23]. In this context, inversions may be favoured as they lock together good allelic combinations via the suppression of recombination. In H. numata, Chouteau et al. [23] have showed that individuals with a derived supergene allele (Hn1 and Hn123) have wing patterns that are very abundant among the species composing the local prey community in those forests and are therefore less attacked by predators than individuals with the recessive, ancestral wing-pattern (Figure 3) [23]. Derived, inverted alleles are therefore strongly favoured by selection, even as heterozygotes due to their strong allelic dominance (Figure 3) [29]. We can therefore explain why inversions were positively selected when they formed, and increased in frequency.
When investigating fitness consequences of inversions in other life stages, however, we found that inversions had recessive detrimental effects on larval survival [20]. Indeed, homozygous larvae for the rearranged haplotypes Hn1 or Hn123 had a much lower survival than other genotypes: only 6.2% of Hn1/Hn1 larvae and 31.3% of the Hn123/Hn123 larvae survived to the adult stage whereas other genotypes (Hn0/Hn0, Hn0/Hn1, Hn1/Hn123, Hn0/Hn123) had a survival rate of about 80%. This shows the detrimental effect of homozygous inversions on larval survival, generating supergene hetetozygote advantage (Figure 3). Since the relative frequencies of homozygous individuals increase with inversion frequency, the fitness of inversions decline as they rise in frequency. H. numata inversions are therefore under negative frequency-dependant selection and, even though they underlie the most advantageous wing patterns, they cannot replace the standard chromosome arrangement: recessive deleterious factors within inversions become expressed more often as the inversions rise in frequency, to the point of preventing their fixation. As a corollary, the fitness of the standard arrangement increases as inversions increase in frequency, leading to a balanced polymorphism.
Balancing selection acting on these inversions in H. numata thus results from their antagonistic effects: positive selection on adult mimicry but negative frequency-dependent selection on larvae viability and mate choice (Figure 3). The interplay of positive and negative frequency-dependence suggests that there exists an equilibrium frequency where selection regimes neatly oppose each other, resulting in a stable polymorphism. In the region of Tarapoto, Peru, observed allelic frequencies of the 3 main chromosome types are all in the range of 25–40% [12]. When we estimate selection gradients based on observed parameter values for larval survival, adult survival, and mating success, it appears that each of those fitness components displays a different distribution of effects for the three chromosome types (alleles), illustrating how fitness effects might compensate each other (Figure 3). Around observed frequencies, our analyses suggest that selection on larval survival favours Hn0, disassortative mate choice favours Hn0 and Hn123, while mimicry selection favours Hn1. Negative frequency dependence is strongest on the dominant Hn1 haplotype via disassortative mate choice and strong mortality in homozygous larvae.
5. The origin of inversion negative-frequency dependent selection
Mortality in inversion homozygotes may be the result of inversion breakpoints disrupting genes or regulatory sequences. However, while several genes within the supergene region show differential expression when comparing wing-pattern forms, none of the inversions P1, P2 and P3 seem to disrupt a gene [20]. Moreover, we found no transcript missing, disrupted or differentially spliced when comparing Hn0, Hn1 and Hn123 haplotypes.
A recessive load associated with inversions may also result from their capture of haplotypes harbouring deleterious variants. To investigate this, we estimated the prevalence of putatively deleterious elements in inverted and non-inverted sequences. We first evaluated the abundance of transposable elements (TEs), whose insertion may affect gene function and regulation. Consistent with our prediction, we found that the three inversions at the P supergene are enriched in TEs compared to the rest of the genome, and that this accumulation has led to a notable increase (+9.5% in average) in size of inverted haplotypes compared to standard haplotypes [20]. Second, we examined the frequency of non-synonymous mutations, which modify amino acid identity and can affect protein functioning. We found that P1, P2, and P3 are all enriched in non-synonymous mutation compared to non-inverted ancestral segments and to the whole genome. The formation and subsequent evolution of inversions have therefore led to the capture of a large amount of deleterious variants across their entire length.
The capture of this genetic load is predicted because of two major effects of inversions on their own evolution. First, an inversion is a single mutational event affecting simultaneously an entire segment of chromosome, with a length comprised between 200 kb and 1150 kb in the case of this supergene. Under normal recombination, many deleterious recessive mutations segregate in populations, but an inversion rising in frequency (e.g. if favored by selection for any reason) will simultaneously bring to high frequency a single haplotype including all the linked deleterious mutations contained in this haplotype [30]. This phenomenon equates to a drastic demographic bottleneck on the inversion region induced by its formation. This situation never happens among deleterious mutations in a recombining genome (with otherwise equal demographic parameters) and is specific to positively-selected inversions. The joint effect of deleterious mutations expressed in homozygotes may be strong enough to prevent the fixation of an inversion. Second, recombination is suppressed in inversion heterozygotes and inversions at intermediate frequency are rarely homozygous—especially with heterozygote advantage. Inversions have therefore reduced opportunity to recombine compared to the rest of the genome, and this lowers the efficiency of selection. This may contribute to an accumulation of deleterious variants which would otherwise be purged [31]. Both processes may explain our observation of a genetic load carried by inverted segments.
6. Discussion
These results shed new light into how the ecological and genetic components of balancing selection allow inversion polymorphisms to establish. New inversions may be under strong positive selection via the capture of coadapted mutations and their protection against recombination, which should lead to their rapid fixation (Figure 3). But inversions also capture upon formation recessive deleterious variants in initial linkage with the positively-selected mutations [30]. Recombination suppression locks all those mutations together, forming a fixed load protected from purging, and, by reducing selection efficiency, may also promote the accumulation of further deleterious variants. The expression of a recessive genetic load associated with inversions inevitably translates into negative frequency-dependent selection favouring their maintenance at intermediate frequency.
These results bring key insights into the formation of supergenes underlying the co-variation of multiple traits, such as mimicry polymorphisms or sexual dimorphisms. The build-up of antagonistic fitness effects found here is likely to be a general feature of the formation of inversion polymorphisms and their evolution through time. The recombination suppression effect of chromosomal rearrangements might be advantageous when it protects alternative combinations of co-adapted alleles from being reshuffled, but may also lead to the capture and accumulation of deleterious mutations, preventing these rearrangements to spread to fixations. The two sides of this same medal are somehow intimately tied together, and may explain why many inversions segregate at intermediate frequency in many species, including humans, and are sometime involved in complex polymorphisms [2]. Taken together, these results on the origin of wing pattern polymorphism in a tropical butterfly open new perspective for understanding how the occurrences of chromosomal rearrangements jointly shape the evolution and the ecology of species.
Data and materials availability
No data were generated by this study.
Author contributions
PJ and MJ wrote and edited the paper.
Conflicts of interest
Authors declare no competing interests.
Funding
This research was supported by Agence Nationale de la Recherche (ANR) grants ANR-12-JSV7-0005 and ANR-18-CE02-0019-01 and European Research Council grant ERC-StG-243179 to MJ.
Acknowledgments
The authors thank Mathieu Chouteau, Annabel Whibley and Violaine Llaurens for their participation in this research, MGX (Montpellier) for sequencing, and INIBICO (Peru) for insectary work. Research grants: ANR-12-JSV7-0005 and ANR-18-CE02-0019-01. Research permits (Peru): 236-2012-AG-DGFFS-DGEFFS, 201-2013-MINAGRI-DGFFS/DGEFFS and 002-2015-SERFOR-DGGSPFFS.
French version
1. Introduction
Les inversions chromosomiques sont des réarrangements de segments d’ADN contenant souvent de nombreux gènes, et qui constituent un mécanisme important par lequel l’organisation du génome évolue [1, 2]. Les inversions peuvent être découvertes en comparant les génomes de différents taxons, mais elles peuvent aussi être polymorphes, lorsque des différents arrangements coexistent et ségrègent parmi les individus d’une population. Les inversions polymorphes sont souvent associées à la variation de traits dits complexes, qui impliquent la coordination de multiples éléments du phénotype (par exemple, la morphologie et le comportement). Comme exemple, citons le dimorphisme d’organisation sociale chez plusieurs espèces de fourmis [3] ou les polymorphismes de coloration et de comportement chez de nombreux oiseaux et papillons [4, 5, 6, 7, 8], ou encore le dimorphisme de morphologie florale chez de nombreuses plantes [9], et même le dimorphisme sexuel observé chez de nombreux animaux [10]. Cette association entre réarrangements chromosomiques et variation de traits complexes est vraisemblablement due au fait que les inversions facilitent la coordination des gènes contrôlant les différentes caractéristiques du phénotype. En effet, la recombinaison est fortement réduite, voire supprimée, entre les segments d’ADN présentant une orientation différente et, par conséquent, les inversions couvrant plusieurs locus peuvent protéger de la recombinaison des combinaisons alternatives d’allèles co-adaptés à ces locus. De tels groupes de locus étroitement liés et impliqués dans la variation d’un caractère complexe sont appelés « supergènes » [11]. Malgré la place unique qu’ils occupent dans l’histoire de la génétique, pourquoi et comment apparaissent ces supergènes et les polymorphismes qu’ils contrôlent restent des questions non résolues.
Il a été proposé que les polymorphismes au niveau des supergènes soient maintenus par la dérive ou par l’équilibre entre des forces évolutives opposées [2, 12, 13, 14]. Sur le plan général, on comprend relativement bien comment plusieurs éléments du phénotype se coordonnent. Par exemple, les syndromes de migration chez la truite associent des variations de la physiologie de la reproduction et du comportement migratoire d’une manière qui peut être envisagée comme des stratégies alternatives de maximisation du produit de la fécondité et du risque de mortalité [15]. Cependant, la raison pour laquelle les stratégies alternatives sont maintenues sous forme de polymorphismes à long terme est souvent moins claire. De plus, la façon dont ces polymorphismes s’établissent n’est toujours pas claire. Notamment, nous ne comprenons toujours pas bien pourquoi les variations chromosomiques sont associées à des polymorphismes complexes et comment des combinaisons alternatives d’allèles co-adaptés peuvent se former malgré la recombinaison.
Dans cet article, nous passons en revue les avancées récentes de notre compréhension de la dynamique évolutive des supergènes, en nous concentrant sur l’exemple d’un supergène contrôlant la coloration des ailes chez le papillon Heliconius numata. Cet exemple nous permet de mettre en évidence les aspects clés affectant la formation et le maintien des polymorphismes d’inversion, ce qui pourrait apporter un éclairage sur les facteurs affectant le maintien d’autres réarrangements polymorphes.
2. Architecture génétique du polymorphisme des motifs alaires chez H. numata
Le papillon Heliconius numata est une espèce répandue dans la zone néotropicale, que l’on trouve depuis la partie nord du bassin de l’Orénoque au Venezuela jusqu’aux parties sud des forêts atlantiques du Brésil (Mata Atlântica), et depuis les contreforts des Andes jusqu’à l’océan Atlantique (Figure 1). H. numata fait partie d’un clade de papillons (la tribu des Heliconiini au sein des Nymphalidae) contenant des glycosides cyanogéniques au mauvais goût les protégeant de la prédation. Les Heliconius sont connus pour leurs motifs de coloration alaire mimétiques géographiquement diversifiés, et importants dans l’adaptation locale. En effet, chez les Heliconius, les motifs alaires colorés jouent un rôle de signal d’avertissement aux prédateurs, car ces derniers apprennent à éviter les proies qu’ils savent toxiques. Les motifs alaires sont donc protecteurs et impliqués dans des relations de mimétisme entre espèces (toxiques et non toxiques) qui arborent les mêmes signaux vis-à-vis des mêmes prédateurs [16]. En effet, les motifs de coloration les plus abondants sont bien connus des prédateurs locaux et donc favorisés par la sélection naturelle. Les papillons qui les portent sont bien identifiés comme toxiques, et bénéficient ainsi d’un risque de prédation réduit et de la possibilité d’être actifs à tout moment. En général, les espèces protégées par leurs composés toxiques affichent un seul motif de coloration mimétique dans une localité donnée. Cependant, H. numata présente, dans chaque localité, plusieurs morphes distincts, parfois jusqu’à six ou sept, chacun étant impliqué dans une relation de mimétisme avec certaines espèces d’apparence similaire appartenant aux Heliconiinae, Danainae et Pericopiinae (appelé « cercle mimétique »). Ce polymorphisme de coloration des ailes se retrouve sur l’ensemble de l’aire de répartition de H. numata, et varie donc géographiquement : chaque population de H. numata arbore différents motifs correspondant aux communautés mimétiques rencontrées localement (Figure 1). Seules quelques populations périphériques semblent être monomorphes, notamment les populations de l’extrême sud des forêts Atlantiques du Brésil.
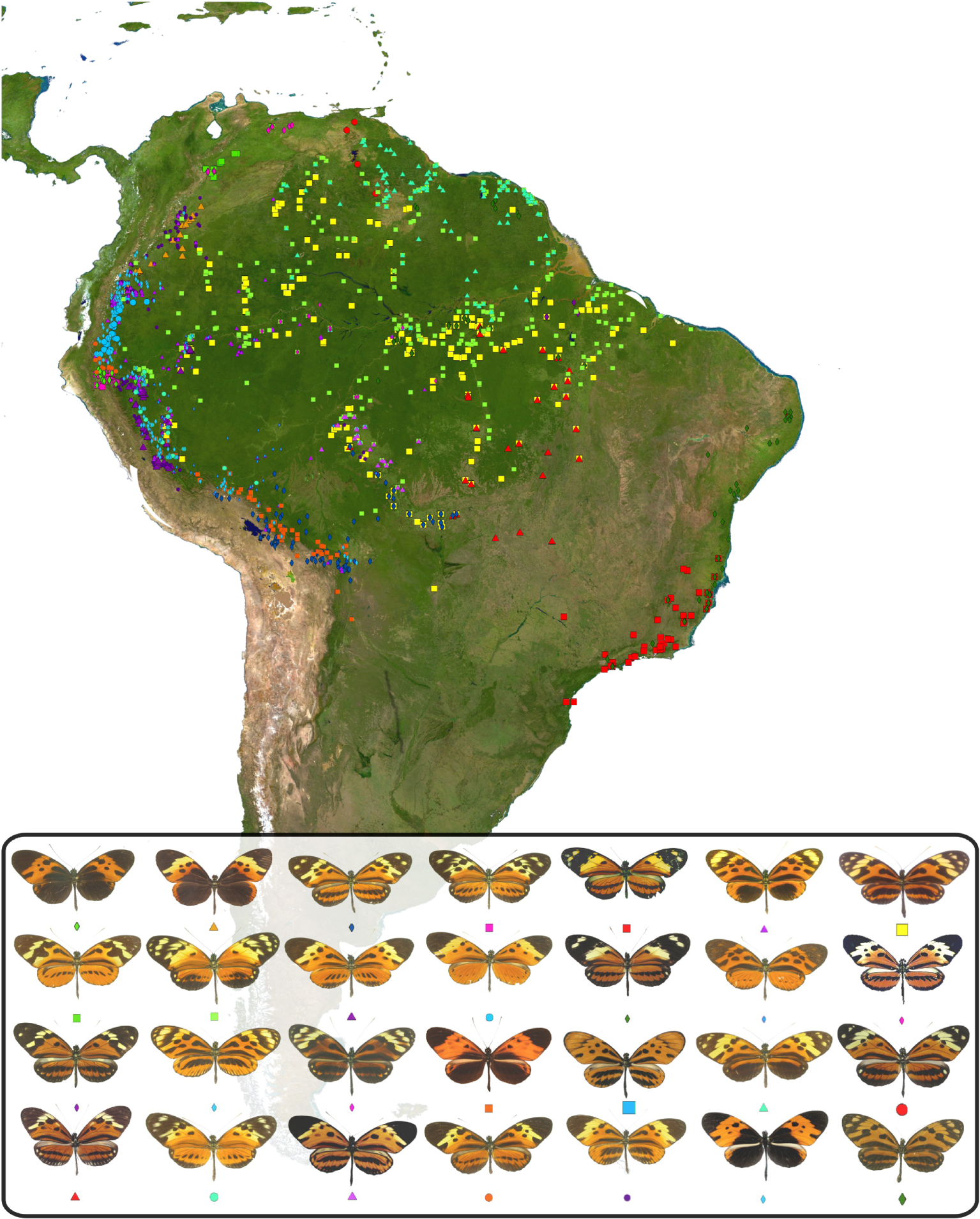
Diversité et distribution de Heliconius numata. Les points colorés représentent les localités où un morphe donné de H. numata a été observé. Données provenant de [17].
Les croisements entre morphes ont montré que les motifs alaires mimétiques chez H. numata sont hérités comme les allèles d’un locus mendélien unique. Cependant, de rares individus présentant des motifs alaires qui semblent être intermédiaires entre morphes communs sont parfois observés dans les croisements et dans la nature, ce qui suggère qu’il s’agit d’individus recombinants [18]. Par conséquent, il a été proposé que les variations des motifs alaires de H. numata soient contrôlées par un supergène, c’est-à-dire un groupe de gènes hérités ensemble comme une seule unité en raison d’une forte réduction de la recombinaison. Cela expliquerait comment de multiples motifs alaires complexes en coexistence puissent se croiser sans produire toute une palette de recombinants mal adaptés (non-mimétiques). Ce supergène, nommé P, se situe sur le chromosome 15 à la position homologue du groupe de locus Yb–Sb–N du génome de l’espèce étroitement apparentée Heliconius melpomene, et du locus Cr de l’espèce plus éloignée H. erato, chez lesquelles cette région contrôle les changements noir/jaune de certains éléments du motif [19]. Pour mieux comprendre la structure des allèles du supergène, nous avons produit des assemblages génomiques de novo pour 12 individus H. numata portant des motifs alaires différents [20]. Contrairement à ce qui a été observé chez des espèces étroitement apparentées, ces assemblages ont montré que trois formes du chromosome 15 coexistent dans les populations de H. numata, chacune étant associée à des motifs alaires spécifiques et présentant un niveau de dominance différent (Figure 2). Les allèles récessifs du supergène, associés aux morphes silvana ou laura, sont portés par la forme chromosomique standard, appelée Hn0, considérée comme la structure ancestrale, semblable aux espèces apparentées. L’allèle dominant, contrôlant la coloration bicoloratus, est associé à la structure montrant une inversion de 400 kilobases (P1, 21 gènes), et cette classe allélique est appelée Hn1. Une troisième classe allélique, présentant une dominance intermédiaire, est associée à une diversité de colorations mimétiques (tarapotensis, arcuella, aurora, numata, superioris, etc.) et montre la même inversion P1, mais également deux inversions adjacentes supplémentaires : P2 (200 kb, 15 gènes) et P3 (1150 kb, 71 gènes). Cette troisième classe allélique, appelée Hn123, est donc formée par trois inversions adjacentes.
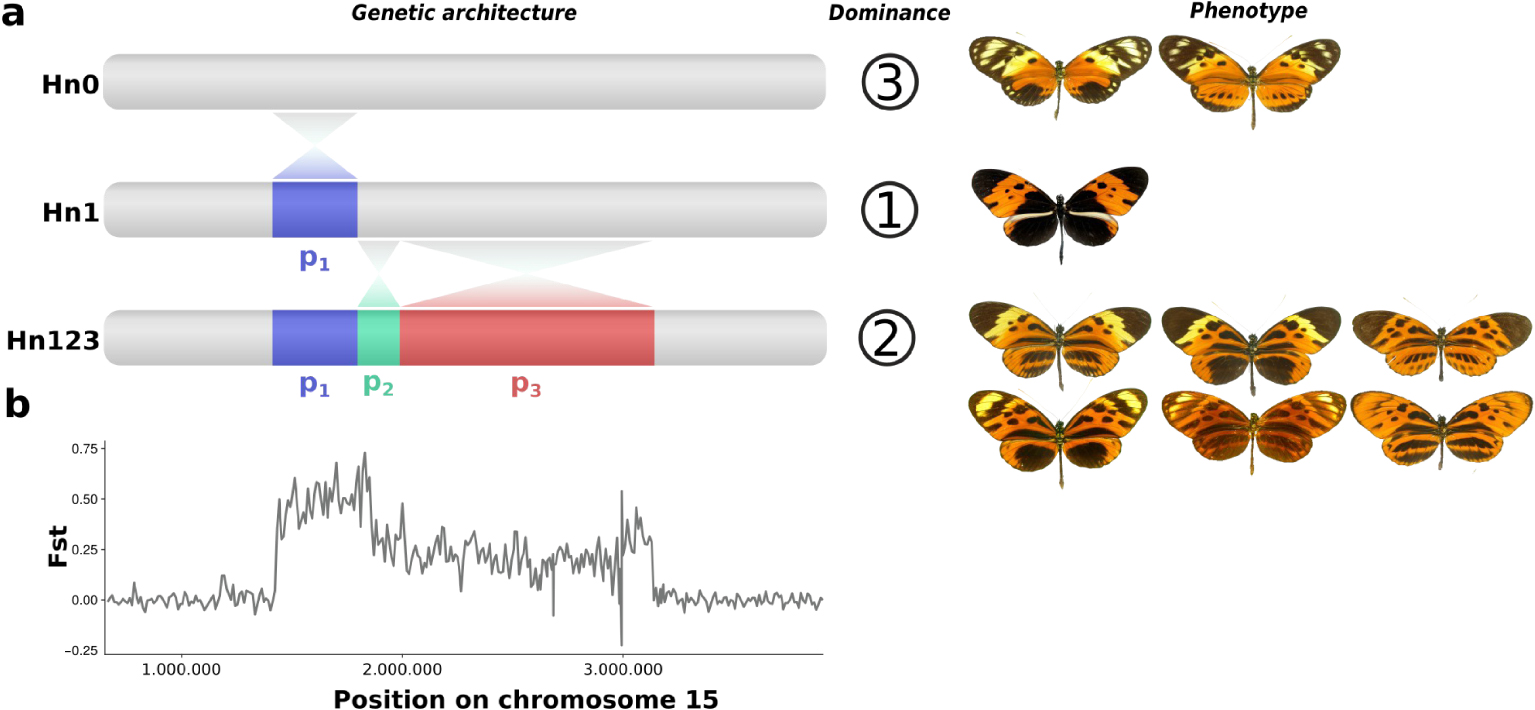
Architecture génétique et divergence nucléotidique au supergène contrôlant le mimétisme. (a) Représentation schématique des trois classes alléliques du supergène de H. numata, de leur relation de dominance et de certains motifs alaires qui leur sont associés. (b) Divergence (Fst) entre Hn0 et Hn123 le long du chromosome 15 (gros plan sur la région du supergène).
Les régions chromosomiques ayant des orientations opposées forment rarement des chiasmas au cours de la méiose, la recombinaison est fortement réduite entre les haplotypes d’orientations différentes …. Les structures Hn0, Hn1 et Hn123 du supergène, arrangées différemment, ne recombinent donc pas sur la majeure partie de leur longueur, comme le prouve leur haut niveau de divergence (Figure 2). Ces résultats montrent que le supergène est une architecture nouvelle, dérivée, impliquant la coordination des variations de plusieurs gènes. Ces gènes recombinent entre eux chez les autres espèces du clade, mais sont verrouillés ensemble par le polymorphisme d’inversion chez H. numata. Cette architecture génétique étant distincte de celle des autres espèces, elle est probablement maintenue chez H. numata par un régime de sélection particulier. Comme les variations des colorations des ailes sont soumises à des régimes de sélection bien étudiés, notamment chez les papillons Heliconius [16], H. numata apparaît comme un excellent modèle pour comprendre l’évolution des architectures génétiques.
Comme la recombinaison est supprimée dès que les inversions forment des segments d’orientations opposées, la divergence entre les haplotypes contrôlant différents motifs alaires fournit une mesure de l’âge de ces haplotypes et de leur histoire évolutive. Or, les segments inversés sont suffisamment grands pour contenir des séquences présentant de grandes différences dans leur niveau de variation génétique. Par conséquent, des estimations des âges d’inversion peuvent être obtenues en calculant les relations phylogénétiques entre individus, pour différents intervalles (« fenêtres ») à l’intérieur des segments inversés, et en déterminant les événements de coalescence les plus récents entre les haplotypes inversés et non inversés. Les phylogénies réalisées sur différents segments génomiques pour le clade Heliconius contenant H. numata ont révélé des topologies discordantes entre l’inversion P1 par rapport et le reste du génome [21]. Ces résultats montrent que P1 est probablement apparue il y a environ 3,0 millions d’années dans une autre lignée, ancestrale à l’espèce actuelle H. pardalinus. Cette inversion a ensuite été introduite dans la lignée H. numata par hybridation interspécifique [21]. Cette introgression s’est produite environ 1,30 millions d’années après la divergence des lignées de H. numata et de H. pardalinus. Contrairement à H. pardalinus où elle est fixée, P1 est restée polymorphe chez H. numata, où elle a continué à diverger après l’introgression et a formé l’haplotype Hn1. L’introgression de P1 a été suivie chez H. numata par l’apparition ultérieure des inversions P2 et P3 en liaison avec P1 il y a plus de 1,8 million d’années, entraînant la formation d’un haplotype Hn123 présentant trois segments inversés [20]. L’évolution de ce nouvel haplotype n’a pas entraîné la perte des autres haplotypes (Hn0 et Hn1). Par conséquent, les haplotypes Hn0, Hn1 et Hn123 ont été maintenus à une fréquence intermédiaire depuis environ deux millions d’années, ce qui implique qu’un mécanisme puissant maintient ce polymorphisme.
3. Le maintien des polymorphismes des motifs alaires chez H. numata
Brown et Benson [18] avaient noté que le polymorphisme de H. numata était déroutant car il semble contredire les théories du mimétisme [22]. En effet, le mimétisme évolue en réponse à une forte sélection sur la ressemblance avec les signaux d’avertissement connus des prédateurs locaux. Pour les proies possédant une défense chimique, la sélection favorise donc les formes les plus communes, imposant une forte sélection fréquence-dépendante positive. Cette forme de sélection s’oppose en principe au polymorphisme car les morphes plus rares subissent une prédation plus importante. Chez H. numata, Chouteau et al. [23] ont montré que chez H. numata, les différents morphes jouissent en effet de niveaux de protection différents contre les prédateurs, la protection étant meilleure pour les morphes les plus communs, dans toutes les localités [23]. La fréquence-dépendance est donc clairement positive : au Pérou, il a été estimé que la forme la plus commune, portée par l’arrangement Hn1, bénéficiait d’une protection contre la prédation sept fois supérieure à la forme la plus rare contrôlée par l’arrangement ancestral Hn0 (Figure 3) [23]. Sous le seul effet de la sélection sur le mimétisme, l’arrangement Hn0 devrait être rapidement éliminé et remplacé par les l’arrangement Hn1. Pourtant, Hn0 persiste à une fréquence élevée, donc une force puissante doit agir contre la fixation de Hn1, expliquant le maintien du polymorphisme. Nous passons en revue ci-dessous les principaux facteurs qui peuvent aboutir à un tel polymorphisme.
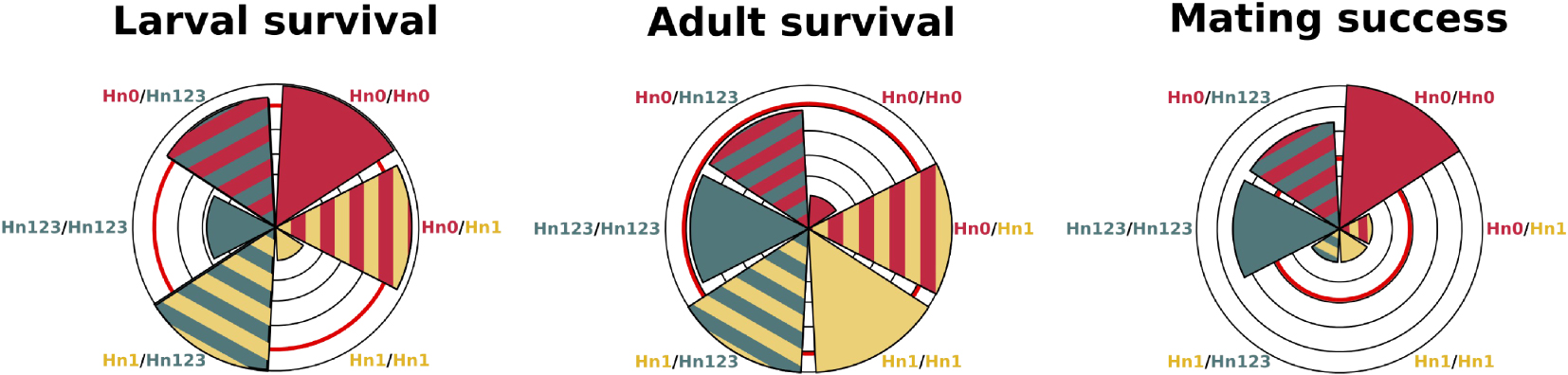
Représentation schématique de la variation des composantes de fitness associées aux génotypes du supergène. La taille des triangles représente pour chaque génotype le coefficient de sélection agissant sur la composante de fitness, par rapport au meilleur génotype. Ainsi, les triangles atteignant la limite du cercle extérieur indiquent que le génotype sous-jacent possède le meilleur phénotype pour le trait étudié. Les triangles plus petits représentent les génotypes qui sont les plus contre-sélectionnés pour le trait focal. Le cercle rouge représente la moyenne de la population : les génotypes représentés par les triangles plus petits que le cercle rouge sont contre-sélectionnés, tandis que les génotypes représentés par les triangles plus grands que le cercle rouge sont favorisés par la sélection. Les coefficients de sélection ont été estimés en utilisant les fréquences d’haplotypes et de génotypes observées dans la population de Tarapoto, au Pérou. Les estimations de la survie des adultes sont basées sur la protection contre les prédateurs. Les coefficients de sélection ont été calculés par rapport à la moyenne de la population, et estimés dans la population H. numata de Tarapoto, Pérou [20]. Les données sur la prédation et le succès de l’accouplement proviennent de [12, 23]. Masquer
Représentation schématique de la variation des composantes de fitness associées aux génotypes du supergène. La taille des triangles représente pour chaque génotype le coefficient de sélection agissant sur la composante de fitness, par rapport au meilleur génotype. Ainsi, les triangles ... Lire la suite
3.1. Variabilité de la sélection sur le mimétisme
Le maintien du polymorphisme pourrait s’expliquer si la sélection sur les motifs alaires varie géographiquement, c’est-à-dire par un équilibre migration-sélection. En effet, dans la région de Tarapoto (Pérou), on observe une forte hétérogénéité spatiale dans la distribution des cercles mimétiques auxquels appartiennent les morphes de H. numata, à une échelle spatiale inférieure à la distance de dispersion de ces papillons [13]. Cependant, plusieurs autres espèces d’Heliconius partagent avec H. numata des paysages sélectifs et des traits d’histoire de vie similaires, mais ne présentent pas de polymorphisme comparable. En outre, le paysage hétérogène de la sélection est réel, mais les modèles théoriques considérant l’effet de la sélection variable ont montré que seul un niveau irréaliste de migration entre localités pourrait expliquer les polymorphismes observés [24], ce qui suggère que d’autres mécanismes doivent entrer en jeu.
3.2. Préférences d’accouplement
Chouteau et al. [12] ont montré que H. numata présente des préférences d’accouplement dites hétérogames, les femelles rejetant les mâles de leur propre phénotype [12]. Ce modèle d’accouplement est peu commun, en particulier pour des traits impliqués dans l’adaptation locale. En effet, la sélection sur les traits d’adaptation locale favorise en général les préférences d’accouplement homogames, car les accouplements hétérogames génèrent une descendance mal adaptée [25, 26]. Comme abordé plus haut, cela suggère que la variation géographique de la sélection sur le mimétisme, bien qu’elle puisse favoriser l’évolution du motif alaire, ne peut pas être le principal moteur du polymorphisme. L’accouplement hétérogame constitue à lui seul un mécanisme expliquant le maintien de formes multiples dans la population, car les formes rares bénéficient d’un avantage reproductif grâce à de plus nombreuses possibilités d’accouplement. Restent cependant à élucider la raison pour laquelle ce système d’accouplement particulier a évolué, et donc l’origine profonde du polymorphisme de H. numata.
3.3. Avantage hétérozygote
Chouteau et al. [12] ont montré que les populations de H. numata présentent un déficit d’individus homozygotes pour les classes alléliques impliquant des inversions chromosomiques (Hn1 et Hn123) [12]. Ce déficit peut résulter des préférences hétérogames mentionnées ci-dessus, qui augmentent la proportion d’hétérozygotes dans la population, en comparaison d’une situation dite panmictique (accouplement au hasard). Mais il peut aussi résulter d’un avantage aux hétérozygotes (ou d’un désavantage aux homozygotes) au niveau du supergène. L’avantage aux hétérozygotes seul peut maintenir le polymorphisme au locus concerné, mais il peut de plus provoquer l’évolution de l’hétérogamie, laquelle favorise aussi le polymorphisme [27, 28]. En effet, les préférences sexuelles qui maximisent la production d’une descendance hétérozygote sont bénéfiques en cas d’avantage aux hétérozygotes. Pour étudier l’hypothèse d’un avantage aux hétérozygotes agissant sur le supergène, nous avons finement étudié les effets des allèles alternatifs du supergène sur la fitness [20].
4. Les inversions sont associées à des effets de fitness antagonistes
Comme indiqué ci-dessus, nous pensons que l’avantage des inversions entraînant une augmentation de leur fréquence provient de la suppression de la recombinaison, ce qui préserve les combinaisons de mutations coadaptées conférant une ressemblance précise à d’autres proies toxiques. En effet, seules les combinaisons alléliques produisant des formes mimétiques apportent des avantages en termes de survie et d’énergie, tandis que les recombinants et les phénotypes intermédiaires sont contre-sélectionnés [23]. Dans ce contexte, une inversion qui survient sur un segment renfermant de bonnes combinaisons alléliques est favorisée car il les protège de la recombinaison. Chez H. numata, Chouteau et al. [23] ont montré que chez H. numata, les motifs alaires associés aux allèles dérivés au supergène (Hn1 et Hn123) sont ceux qui permettent de mimer les groupes de mimétisme les plus abondants dans la communauté locale de papillons, dans ces forêts. Chez les papillons adultes, les allèles réarrangés sont donc bien mieux protégés que l’allèle ancestral et récessif (Hn0 ; Figure 3) [23] et donc fortement favorisés par la sélection, même lorsqu’ils sont hétérozygotes, en raison de leur forte dominance allélique (Figure 3) [29]. Nous pouvons donc expliquer pourquoi les inversions ont été sélectionnées positivement dès qu’elles se sont formées, et ont par conséquent augmenté en fréquence.
Cependant, lorsque nous avons étudié les conséquences des inversions sur la survie à d’autres stades de vie, nous avons constaté que les inversions avaient des effets délétères récessifs sur la survie des larves [20]. Les larves homozygotes pour les haplotypes réarrangés Hn1 ou Hn123 ont montré un taux de survie beaucoup plus faible que les autres génotypes : seulement 6,2% des larves Hn1/Hn1 et 31,3% des larves Hn123/Hn123 ont survécu jusqu’au stade adulte alors que les autres génotypes (Hn0/Hn0, Hn0/Hn1, Hn1/Hn123, Hn0/Hn123) avaient un taux de survie d’environ 80%. Ceci montre l’effet nocif des inversions à l’état homozygotes sur la survie des larves, engendrant un avantage aux hétérozygotes au supergène (Figure 3). Puisque les fréquences relatives des individus homozygotes augmentent avec la fréquence des inversions, la fitness des inversions diminue au fur et à mesure que ces inversions augmentent en fréquence. Les inversions de H. numata font donc l’objet d’une sélection fréquence-dependante négative et, même si elles sont associées aux motifs alaires les plus avantageux, elles ne peuvent pas remplacer l’arrangement chromosomique ancestral : l’effet délétère récessif des inversions s’exprime plus souvent à mesure que la fréquence des inversions augmente, au point d’empêcher leur fixation. En corollaire, la fitness de l’arrangement ancestral augmente à mesure que la fréquence des inversions augmente, ce qui conduit à un polymorphisme équilibré.
La sélection balancée agissant sur ces inversions chez H. numata résulte donc de leurs effets antagonistes n’agissant pas de la même manière aux différentes fréquences : sélection positive sur le mimétisme des adultes mais sélection fréquence-dépendante négative sur la viabilité des larves et le choix du partenaire (Figure 3). L’interaction de la fréquence-dépendance positive et négative suggère qu’il existe une fréquence d’équilibre où les régimes de sélection s’opposent parfaitement les uns aux autres, résultant en un polymorphisme stable. Dans la région de Tarapoto, au Pérou, les fréquences alléliques observées des 3 principaux types de chromosomes sont toutes de l’ordre de 25–40% [12]. En estimant les gradients de sélection sur la base des valeurs observées des paramètres de survie des larves, survie des adultes et succès d’accouplement, il apparaît que chacune de ces composantes de la fitness présente une distribution différente d’effets pour les trois types de chromosomes (allèles). Cela illustre la manière dont les composantes de la fitness peuvent se compenser mutuellement (Figure 3). Autour des fréquences observées dans les populations naturelles, nos analyses suggèrent que la sélection sur la survie des larves favorise Hn0, le choix de partenaire favorise Hn0 et Hn123, tandis que la sélection sur le mimétisme favorise Hn1. La fréquence-dépendance négative est la plus forte sur l’haplotype dominant Hn1 via le choix d’accouplement dissassortatif et de la forte mortalité chez les larves homozygotes.
5. L’origine de la sélection fréquence- dépendante négative sur les inversions
La mortalité chez les individus homozygotes pour les inversions pourrait résulter de la perturbation de gènes ou de séquences régulatrices par les points de cassure des inversions. Cependant, bien que plusieurs gènes de la région du supergène présentent une expression différente entre individus de morphes différents, aucun des points de cassure de P1, P2 et P3 ne semble perturber un gène [20]. De plus, nous n’avons trouvé aucun transcrit manquant, perturbé ou épissé de manière différentielle en comparant les haplotypes Hn0, Hn1 et Hn123.
Un fardeau génétique récessif associé aux inversions pourrait également être dû à la « capture », par ces inversions, de mutations délétères. Pour étudier cela, nous avons estimé la prévalence d’éléments considérés comme délétères dans les séquences inversées et non inversées. Nous avons d’abord évalué l’abondance des éléments transposables (TE), dont l’insertion peut affecter la fonction et la régulation des gènes. Conformément à notre prédiction, nous avons constaté que les trois inversions du supergène P sont enrichies en TE par rapport au reste du génome, et que cette accumulation a conduit à une augmentation notable (+9,5% en moyenne) de la taille des haplotypes inversés par rapport aux haplotypes ancestraux [20]. Deuxièmement, nous avons examiné la fréquence des mutations non synonymes, qui modifient l’identité des acides aminés et peuvent affecter le fonctionnement des protéines. Nous avons constaté que P1, P2 et P3 sont tous enrichis en mutations non synonymes par rapport aux segments ancestraux non inversés et par rapport au génome entier. La formation puis l’évolution des inversions ont donc conduit à la capture d’une grande quantité de variants délétères sur toute leur longueur.
La capture de ce fardeau génétique peut s’expliquer par deux effets propres à l’évolution des inversions. Premièrement, une inversion se forme par un événement unique qui affecte simultanément tout un segment de chromosome, d’une longueur comprise entre 200 kb et 1150 kb dans le cas du supergène de H. numata. Or, de nombreuses mutations récessives délétères ségrègent dans les populations naturelles. Ainsi, une inversion a de grandes chances de contenir dès sa formation plusieurs mutations délétères qui se trouvent être liées chez l’individu concerné. En augmentant en fréquence (par exemple, si elle est favorisée par la sélection pour une raison quelconque) cette inversion portera aussi à une fréquence élevée un haplotype unique comprenant probablement plusieurs mutations délétères [30]. Cette situation ne se produit jamais pour les mutations délétères dans les régions génomiques qui recombinent, et est spécifique aux inversions favorisées par la sélection. L’effet cumulé des mutations délétères capturées par une inversion peut être suffisamment fort chez les homozygotes pour empêcher la fixation d’une inversion. Deuxièmement, la recombinaison est supprimée chez les individus hétérozygotes et les inversions à faible fréquence sont rarement homozygotes. Les régions inversées ont donc moins de possibilités de recombinaison que le reste du génome, ce qui réduit l’efficacité de la sélection contre les mutations délétères. Cela peut contribuer à une accumulation de variants délétères qui seraient normalement « purgés » par la sélection [31]. Ces deux processus pourraient expliquer l’observation d’un fardeau génétique associé aux segments inversés.
6. Discussion
Ces résultats apportent un nouvel éclairage sur la manière dont les composantes écologiques et génétiques de la sélection balancée permettent aux polymorphismes d’inversions de s’établir. Les nouvelles inversions peuvent être fortement favorisées par la sélection si elles capturent des combinaisons de mutations coadaptées qu’elles protègent contre la recombinaison, ce qui devrait conduire à leur fixation rapide (Figure 3). Néanmoins, les inversions capturent également lors de leur formation des variants délétères récessifs en liaison avec les mutations sélectionnées positivement [30]. La suppression de la recombinaison verrouille toutes ces mutations ensemble, formant un fardeau de mutation qui ne peut être purgé par la sélection, et, en réduisant l’efficacité de la sélection, peut également favoriser l’accumulation d’autres variants délétères. L’expression d’un fardeau génétique récessif associée aux inversions se traduit inévitablement par une sélection fréquence-dépendante négative qui, s’il est suffisamment fort, favorise le maintien des inversions à une fréquence intermédiaire.
Ces résultats apportent des éléments clés sur la formation de supergènes contrôlant la variation de traits complexes, tels que les polymorphismes de mimétisme ou les dimorphismes sexuels. L’existence d’effets de fitness antagonistes constatée ici est susceptible d’être une caractéristique générale de la formation des polymorphismes d’inversion et de leur évolution dans le temps. L’effet de suppression de la recombinaison des réarrangements chromosomiques peut être avantageux lorsqu’il protège d’un remaniement les combinaisons alternatives d’allèles co-adaptés, mais il peut aussi conduire à la capture et à l’accumulation de mutations délétères, empêchant ces réarrangements de se propager jusqu’à fixation. Ce double jeu des inversions chromosomiques pourrait expliquer pourquoi de nombreuses inversions ségrègent à une fréquence intermédiaire chez de nombreuses espèces, y compris l’homme, et sont parfois impliquées dans des polymorphismes complexes [2]. Pris ensemble, ces résultats sur l’origine du polymorphisme de coloration des ailes chez un papillon tropical ouvrent de nouvelles perspectives pour comprendre comment les réarrangements chromosomiques façonnent l’évolution et l’écologie des espèces.
Disponibilité des données et des matériaux
Aucune donnée n’a été générée par cette étude.
Contributions des auteurs
PJ et MJ ont rédigé et édité l’article.
Conflit d’intérêt
Les auteurs ne déclarent aucun intérêt concurrent.
Financement
Cette recherche a été soutenue par les subventions de l’Agence Nationale de la Recherche (ANR) ANR-12-JSV7-0005 et ANR-18-CE02-0019-01 et la subvention du Conseil Européen de la Recherche ERC-StG-243179 à MJ.
Remerciements
Les auteurs remercient Mathieu Chouteau, Annabel Whibley et Violaine Llaurens pour leur participation à cette recherche, MGX (Montpellier) pour le séquençage, et INIBICO (Pérou) pour l’élevage. Subventions de recherche : ANR-12-JSV7-0005 et ANR-18-CE02-0019-01. Permis de recherche (Pérou) : 236-2012-AG-DGFFS-DGEFFS, 201-2013-MINAGRI-DGFFS/DGEFFS et 002-2015-SERFOR-DGGSPFFS.