

1 Introduction
Systems biology proposes to study biological systems as systems, rather than study their elements one or a few at a time, as has been the approach in molecular and cellular biology for the past decades. The emergence of systems biology needs to be placed in both a historical and a contemporary context.
1.1 Historical context
Over the past 150 years, there have been four major groups of theories that contributed to the deciphering of biological information: the cell theory has established the cell as the basic unit of biological information for all living forms; the theories of evolution told us how biological information has changed over time; the theories of heredity told us how biological information is inherited from generation to generation; the theories of biochemistry told us how biological information is organized and structured in macromolecules such as nucleic acids (DNA and RNA) and proteins.
During the past 15 years, biology has entered a transition phase with the Human Genome Project, which is telling us how biological information is organized on a global scale. The question is what is the next logical step in deciphering biological information? We suggest that it is a systems approach to biology, which will tell us how information functions to create biological systems with their systems or emergent properties. For example, the human immune system is composed of some 1012 cells that interact with one another and the universe of foreign molecular patterns (e.g., viruses and bacteria) to generate specific molecular (antibodies) and cellular (T cells) responses. Its emergent properties are the immune responses (reactions to foreign entities) and tolerance (the failure to react against self-components).
Systems biology studies all of the elements in a system in response to internal and external signals in order to understand the emergent properties. Systems biology requires the development and application of powerful new technologies and computational tools to carry out systems approach and, accordingly, requires a cross-disciplinary environment, including biologists, chemists, computer scientists, engineers, mathematicians, and physicists. We believe systems biology will be a powerful engine driving biology in the 21st century to collect new knowledge and develop useful applications for monitoring and improving the environment, agriculture, nutrition and human health. In addition, this will require the development of a new conceptual and epistemological framework founded on the lessons of the history of science, and integration of the ethical, legal issues in new practices for the organization and conduct of science.
1.2 Contemporary context
We conjectured that biological systems are self-organized around a conjunction of two complementary spaces [1]. The first one is made of biological entities such as DNA, RNA, proteins and small molecules, assembled into supramolecular structures; this architecture is mostly conserved, with a certain degree of variation of its components, and can be directly related to the underlying genetic information, which is primarily digital in nature. The second one is that of fluctuating biochemical reactions generating homeostatic steady states linked to the environment, and it changes primarily in a continuous analog mode. The conjunction occurs through a set of underlying rules that define the degree of variation and stability allowed for basic mechanisms such as replication, transcription, splicing, translation, protein folding, etc. In this context, biological systems are considered as performing contextual computation of the digital information, and it is a central goal of systems biology to uncover the rules of this computation, and ultimately to relate them to the physical parameters of the Universe.
Some have suggested that systems biology is nothing more than a new name for the integrative physiology practiced for the past 50 years. Yet, the context of biology has profoundly changed over the past 10–15 years and these changes provide a powerful new framework for systems biology that moves it far beyond classical integrative physiology.
1.2.1 The Human Genome Project
The Human Genome Project is providing a genetics parts list of the human and model organism genomes [2,3]. We can identify most of the genes, and, accordingly, their corresponding proteins. This provides the scientific framework for global analyses – where the behavior of all (or most) of the elements can, in principle, be studied. The Human Genome Project is also the first example of discovery science in biology, where all of the elements in a biological system can be defined and placed in a database, e.g., the sequence of the 3 billion nucleotides in the 24 chromosomal strings of the human genome. Discovery science now encompasses a quantitative characterization of all the RNAs in particular cell types, their transcriptome; the quantitative characterizations of all the proteins in particular cell types, their proteomes; the quantitative characterization of the small molecule metabolites in a cell, its metabolome; the characterizations of the protein/protein interactions, the interactome; etc. Discovery science provides systems biology with systems parts lists that are essential to its hypothesis-driven iterative and integrative cycles (see below).
1.2.2 Cross-disciplinary biology
Centers and institutes have been established with cross-disciplinary environments. Biologists are slowly starting to realize the power of cross-disciplinary scientists working in close apposition with biologists. In particular, biologists recognize the critical role applied mathematics, statistics, and computer science is playing in generating the tools for computing, storing, analyzing, graphically displaying, modeling, and ultimately distributing biological information, as well as the role of engineers, chemists, and physicists to produce tools for the global capture and analyses of biological information.
1.2.3 Internet and the Worldwide Web
The Internet and the Worldwide Web have provided biologists the capacity to transmit large datasets to colleagues throughout the world. Systems biology will thrive to the extent to which global datasets are openly available to all biologists, and will therefore greatly benefit from the ongoing developments of grid computing, both for enabling real-time large-scale data exchange and collaboration, and for performing high performance distributed computing.
1.2.4 Biology is an informational science
The genome structure is based on a digital code encompassing the core information necessary to initiate development and physiological responses [4]. Since digital codes are ultimately completely knowable, biology is founded with a core of knowable information. The digital genome encodes two fundamental types of information: the genes encoding proteins – the molecular machines of life, and the gene regulatory networks, which specify the behavior of the genes. The gene regulatory networks include the control regions of genes with their DNA-binding sites and their cognate transcription factors [5]. The DNA-binding sites carry out two important roles: (1) they assemble the transcription factors and co-transcription actors for each gene and these collectively operate as a molecular machine to specify the behavior of the gene across developmental or physiologic time – the emergent properties of the gene include temporal and spatial control, as well as the amplitude of expression –; and (2) the DNA-binding sites determine the architecture of the gene regulatory network; that is, which genes are linked in a network by virtue of shared transcription factors. Thus, the control regions act in a manner analogous to integrating computer chips, always sensing changes in the concentrations of transcription factors across developmental or physiologic time.
There are two major types of biological information: the digital information typical of the genome, and the generally analog information of environmental signals. The environmental information falls into two categories: (1) deterministic, where a given signal usually specifies a particular outcome, and (2) stochastic or random, where signals may be quite noisy. Some biological systems, such as the immune system, may have the capacity to convert the stochastic events into information; for example, the stochastic diversification of somatic hypermutation and the assembly of the junctional regions of their B or T cell receptors can be converted by antigen-driven selection into information. Clearly, a major challenge in biology will be to separate signal from noise in large datasets.
Biological information operates across three distinct time spans: evolution – tens to billions of years –; development – hours to a significant fraction of the life span of the organism –; physiologic – milliseconds to weeks. Gene regulatory networks lie at the heart of understanding each of these processes, because the toolbox of protein machines is highly similar across the evolutionary tree of contemporary metazoans. These gene regulatory networks enable the distinct phenotypes of different organisms to emerge as systems properties [5].
Finally, as one moves from the digital genome to the environment, there is a vast hierarchy of types of biological information: DNA → RNA → protein → protein → interactions → biomodules (sets of interacting proteins executing particular phenotypic functions) → networks of biomodules within individual cells → networks of cells and organs → individuals → populations of species → ecologies. The important point is that information is added to the operation of biological systems at each one of these levels; hence, to do systems biology properly, one must acquire global sets of information from as many different levels as possible and integrate them into a coherent picture of the biological system.
1.3 High-throughput platforms make it possible to acquire global sets of data
The past twenty years or so have seen the development of high-throughput platforms for generating genomic, proteomic, metabolomic, and cellular assays. For example, the prototype of the DNA sequencer was developed in 1986. From that time until today, there has been a more than 3000-fold increase in DNA sequencing throughput accompanied by a high degree of automation of the process. Corresponding increases in throughputs have been realized for the global analyses of mRNAs (DNA and oligonucleotide chips), genetic markers (oligonucleotide arrays and mass spectrometry), protein quantitation (capillary separations and mass spectrometry), protein interactions (clever biology and mass spectrometry), and metabolomics (NMR, mass spectrometry). Likewise, high-throughput platforms for a variety of cellular assays have emerged. All of these assays are moving toward the miniaturization, integration of multiple procedures, parallelization and automation that will be possible with microfluidics and, ultimately, nanotechnology. However, tools are already available for the global analyses of many different molecules in the information hierarchy mentioned earlier.
In high throughput data collection, it is essential to distinguish signal from noise, and to develop reliability indexes on the global datasets generated. Until this is achieved, the comparison of the results of different studies will remain limited in scope and depth. This is well illustrated by the history and present stage of development of the experimental tools and databases on the genome, transcriptome and proteome.
1.3.1 The genome is complex, not simple
Mapping and sequencing of the human genome, and of the genome of model bacterial, plant and animal genomes has been the major goal of the Human Genome Program. At present, the human genome has been announced as ‘completed’, the mouse genome is nearing completion, and those model organisms such as baker's yeast, nematode, fruit fly and the plant Arabidopsis have been essentially completed, as well as those of about a hundred bacteria, and many more are in the pipeline.
These advances have relied heavily on the progress made since the initial description of DNA sequencing chemistries in 1977. Initial attempts at automation of the Maxam–Gilbert and Sanger method were unsuccessful. Attachment of fluorescent dyes to nucleotides was described in 1985, enabling implementation of a first version of the DNA sequencer in 1986 [6]. However, scaling up of sequencing to the massive scale required to generate an accurate human genome sequence was possible, more than twelve years after the introduction of the first automated sequencer, only after significant improvements in data quality generation were made available through advances such as the use of dye terminators and fluorescence energy transfer for enhancement of signals, and when assessment of data quality across sequencing platforms and laboratories became a standard practice with software packages such as Phred/Phrap. In the process, a 3000-fold throughput increase has been achieved, and another 3000-fold increase is anticipated during the next decade, with the prospect of nanoscale instruments operating at the single molecule level.
1.3.2 Computer science tools have transformed global data handling and analysis
The content and accessibility of public electronic repositories have increased dramatically during the same period, benefiting largely of advances in computer technology and the development of the World Wide Web after 1992. As a matter of fact, the size of the DNA sequence databases is increasing so rapidly that its growth exceeds Moore's law predicting a doubling of computer chip performance every 18 months to two years [4]. It is therefore anticipated that smarter ways of dealing with the avalanche of DNA sequencing data will have to be invented and implemented, while maintaining a high level of accuracy, since we cannot rely merely on the projected increase in computing power. Some of the most powerful computers, which were commonly used so far in fields such as forecasting or telecommunications, are now used in the field of genomics.
As simple as it might look, completion of the sequencing of large genomes is not an easy task, even if one considers only its euchromatin part, leaving the heterochromatin aside. In the initial ‘complete’ descriptions, the human genome sequence was split in over 100 000 fragments of various sizes, with significant uncertainties as to the order and orientation of many of them, as illustrated by a comparison of the genome assemblies of the publicly and privately generated versions (Tajashi Gojobori et al., personal communication). This might be due to the existence of regions of the genome that are intrinsically unstable, and therefore difficult to clone and sequence; they might contain important genes and regulatory DNA sequences, as suggested by the observation that disease-related genes tend to be found around the regions of discordance between the physical, genetic, radiation hybrid and cytogenetic maps [7].
As the sequencing of the human and mouse genomes was proceeding to completion, it could be anticipated that the number of genes would be narrowed down to a consensus number. The current guess is that there are 30 000–35 000 genes in these species. More may be identified if small regulatory RNAs are defined as genes and if many small protein-coding genes are identified.
Given that the number of potential genomes made of 3 billion base pairs of DNA is approximately equal to 10 to the power of 1 billion, a number so large that a computer made of all particles in the Universe (1070) would have a hard time to browse through all of them, the debate as to whether a particular eukaryotic genome (ours) contains 104 or 105 genes seems to lack relevance. Emphasis should be rather placed on understanding the combinatorial rules that make use of a similar pool of genes by different species in different contexts, taking into account the other dimensions of the genome, including the various types of DNA polymorphisms, the effects of chromatin structure and methylation, etc.
1.3.3 Expression profiling, a technology coming of age but still in its infancy
RNA expression profiling to monitor the transcriptome has also a long history starting with RNA complexity measurements performed by kinetic reasssociation in the 1960s and early 1970s, the famous Rot curves. The first cDNA array experiments were performed soon after the initial description of cDNA cloning in 1975 [8]. The technology was based on bacterial colonies spotted by hand on nitrocellulose filters using toothpicks, then hybridized with radioactively labeled cDNA and revealed on X-ray films, providing semi-quantitative measurements. After the invention of PCR in 1985, cDNA inserts could be amplified in microtiter plates, spotted onto Nylon membranes; exposure to phosphor plates introduced around 1990 made it possible to obtain quantitative measures on a dynamic range spanning three orders of magnitude. Automation of the full process was made possible with the introduction of commercial picking and spotting robots after 1992, around the time when in situ photolithography oligonucleotide synthesis was first described [9]. The now popular two-color fluorescence cDNA arrays were introduced in 1995 [10], and deposition and in situ oligonucleotide synthesis by ink-jet technology in 1997 [11].
Although this technology has made important progress, particularly during the past decade, it has not yet reached the level of maturity and robustness of DNA sequencing. This is well illustrated by the fact that public repositories of microarray data have been introduced only very recently, and discussions on initial formats and standards are still ongoing. As a result of the availability of a myriad of instruments, reagents and tools for capturing and analyzing the data, quality assessment and standards have yet to come. This imposes severe restrictions on the possibility of comparing datasets generated using similar but somewhat different procedures, and on the depth of analysis possible when multiple samples are compared. In spite of early efforts to introduce statistical assessment of the data, it is only recently that mathematicians and statisticians have started to tackle the problems associated with microarray experiments, such as signal identification and measurement, data filtering and normalization, and data analysis and mining. Large amounts of data have been generated, most often with limited or no replication, and with little attention paid to the basics of experimental design. As a result, the complex experimental and biological variations associated with microarray data are rarely documented, preventing a thorough analysis and seriously limiting the power of integration with other types of data.
In the frame of systems biology, it is essential to perform transcriptome measurements based on a robust experimental design, including a precise description of the biological problem, and following standard operating procedures under quality assurance. The current technologies provide reasonable access to significant variations of relatively intense signals, which has been largely documented by a variety of other analytical techniques during the past decades. Reproducing established results, and generating some novel ones, such as identification of biomodules operating in galactose metabolism of yeast discussed below, is reassuring on the value of the technology in its present state.
However the challenge ahead is to access the small variations of the weak signals, corresponding to the vast majority of genes, since they potentially convey collectively more biologically relevant information than the limited number of those associated with strong signals and variations. Novel technologies under development, enabling massively parallel and controlled measurements down to the single molecule level, will help to overcome the current hurdles. Indeed, the single molecule approaches will move the analysis of transcriptomes from an analog mode (DNA and oligonucleotide arrays) to a digital mode where it will be possible to analyze transcriptomes down to the single mRNA per cell. This ability to visualize mRNAs expressed at very low levels is critical because much interesting biology operates at this low level of mRNAs (e.g., gene regulatory networks and some signal transduction pathways). The ability to measure digitally in cells down to the single RNA molecule level will also enable us to address other dimensions of the roles of RNA such as alternative splicing, editing, folding, stability and degradation, each of which adds to the complexity of the transcriptome.
1.3.4 The proteome world of molecular machines
An analysis of the proteome begins with a listing and quantitative enumeration of all the molecular species of proteins in individual cells. Proteomic analyses represent an ever-greater challenge than genomic or transcriptomic analyses for one simple reason: the dynamic range of protein expression in a cell vary from one to 106 copies. There is no equivalent of PCR for proteins – hence the limits of detection for proteins are limited by the sensitivity of the analytic tools. A widely used tool for protein analysis – the mass spectrometer – may have at best down to attomole sensitivity (e.g., requires 105 molecules). This challenge can be overcome, of course, by analyzing the proteins from large number of cells.
Proteomic analyses also pose two other challenges – shared in part by genomic analyses. First, cells change across physiologic and developmental time dimensions. One wants to capture the changing snapshots of the changing patterns of protein expression across these time dimensions – for they reflect the changing biology. Physiologic responses may occur in a fraction of a second. How can these real time snapshots be obtained with global analyses (e.g., all protein elements)?
Finally, proteins exhibit a hierarchy of different informational states and locations that inform the biology they execute. We need to be able to identify and quantitate all of the molecules species of proteins in a cell. We need to characterize all their chemical modifications and correlate them, where possible, with changes in biological function. We need to measure accurately the protein/protein and protein/DNA interactions for these data constitute the foundations for building protein and gene regulatory networks. We need to measure protein half-lives and activations. Where proteins are localized in cells is key. We also need ab initio and better experimental methods for determining dynamically changing three-dimensional structures and correlating these with protein function. A wide range of platforms will be necessary for all these measurements.
Two-dimensional gel electrophoresis (2D-GE), introduced in 1975, was probably the first analytical technique available to analyze the proteome, but it could not provide directly access to the protein sequence information. The introduction of the automated protein sequencer proved invaluable to collect such information on specific proteins, including some isolated by 2D-GE [12]. The automated peptide synthesizer made it possible to produce proteins or protein segments [13], and to generate specific reagents such as polyclonal and monoclonal antibodies to probe their structure and function as individual elements, or as part of molecular complexes. Although these methods could not manage the diversity of proteins, they helped revealing the multidomain structure of proteins, and their assembly in multimeric and supramolecular molecular machines performing simple and complex biochemical functions. A global approach to the diversity of protein–protein interactions (the interactome) was made possible by the yeast two-hybrid system in its various forms, although it remains to be seen how many of the interactions revealed by such techniques are relevant to the real context of the original biological system. In fact, comparison of independently derived datasets has indicated limited overlaps, indicating that many of the interactions identified may be yeast-context specific, or simply occur by chance. Recently, a number of novel technologies have started to provide the means by which to interrogate proteomes on a more global scale, combined with the ability to provide non-ambiguous identification of their elements. These include specific labeling of subsets of proteins, based on their content of specific residues or protein modifications (ICAT), and the use of liquid chromatography coupled to tandem mass spectrometry [14]. A variety of array-based technologies are currently under development to probe proteomes with antibodies, or to reveal protein-DNA interaction.
There are many more aspects of protein structure and function that require similar technological advances before global analyses become practical. Mass production of recombinant proteins will help decipher their structures by X-ray crystallography and Nuclear Magnetic Resonance. The generation of large collections of antibodies to all human proteins is under way to provide the reagents to probe tissue and cell arrays and identify the spatial and sub-cellular localization of these proteins.
2 What is systems biology?
Systems biology is the global analysis, ideally, of all of the elements in a biological system in response to hypothesis-driven perturbations that are necessary and sufficient for elucidating systems functions [15]. It is not merely discovery science, which is itself not hypothesis-driven. Our approach to systems biology can be formulated in the following algorithmic manner. (1) Gather all available information on the biological system in a particular model organism and formulate a preliminary model of how it functions. This model may be descriptive, graphical, or mathematical. (2) Define all of the elements in the system with the tools of discovery science. This begins with the genome sequence of the organism defining all (or most) of its genes and can proceed to transcriptomes, proteomes, metabolomes, interactomes, etc. (3) Perturb genetically the central elements in the system (knock-outs, knock-ins, silencing, etc.) under various environmental cues. Gather global sets of data from as many informational levels as possible. These are steady-state experiments. One may also sample systems across their developmental or physiological time spans – these are kinetic experiments. Both steady-state and kinetic experiments provide powerful information for deciphering the functioning of biological systems. (4) Integrate the various global datasets and compare them against the model. There will be discrepancies. Explain the discrepancies by hypothesis-driven formulations and use these to design additional perturbations to discriminate among alternative explanations for the disparity between the model and the data. With each new set of perturbations, gather global datasets, thus iteratively repeating steps (3) and (4). With each set of perturbations, the model can be recast in light of new experimental findings. Thus, the systems biology approach is iterative, integrative, and hypothesis-driven. The process will be repeated until theory and data are brought into apposition with one another. It is important to stress how important it is to integrate for each system the two types of digital information – the protein (gene) components, and the corresponding gene regulatory networks.
The global integrative process will initially be graphical. Ultimately, one would like to cast the model in mathematical terms. One fascinating question is whether we have an appropriate mathematics for this iterative, integrative process.
The ultimate objective of the mathematical (and even graphical) model of a biological system is two-fold: (1) to be able to predict how the system will behave – given any genome or environmental perturbation; (2) to be able to redesign the system to produce new (and eventually predictable) emergent properties.
Several general comments can be made about this view of systems biology. First, the computational, physical, and biological scientists must work closely together on this hypothesis-driven iterative and integrative approach. Physical proximity enormously facilitates this integration of cross-disciplinary talents. The key point is that data space is infinite and that hypothesis-driven formulations are necessary to shed light on that portion of data space that will inform us about the system.
Second, there is a question about the granularity at which systems biology should be explored. Some argue that systems biology demands precise quantitative measurements for the on/off constants of all important interactions. Obviously, with current methodologies this will be difficult to achieve, particularly in the context of the cells or organisms environment within which these systems operate. An alternative is to take a more granular approach measuring the global networks of physical interactions (protein/protein, protein/DNA, protein/metabolite, etc.), the changing concentrations of RNAs and proteins, the modifications of proteins, etc. This more granular ‘information’ level will clearly lead to significant insights into how systems work (see the galactose utilization example in yeast described below).
Finally, it is important that phenotypic assays can be taken all the way back to the digital code of the genome. Failing this, there can never be an integration of the genes and gene regulatory networks associated with the functioning of the biological system and, accordingly, one cannot predict the system's behavior given any perturbation, nor can one redesign the system to create new emergent properties. One will be frozen in phenomenology, albeit sophisticate phenomenology in some cases, and biology would remain mostly a descriptive science.
3 Experimental approaches to systems biology
3.1 Galactose utilization in yeast
The galactose utilization system in yeast has been well studied for the past 30 years or more. A fascinating question is whether new insights can be gained by systems approaches to this well-studied functional biomodule [16]. This system converts galactose to glucose-6-phosphate and a model of this system as determined by experiments over the past 30 years is presented in Fig. 1. The system has at least nine genes (proteins). Four are the enzymes that catalyze the enzymatic conversions. One is a galactose transporter that brings galactose into the yeast cell and, in so doing, sets the biological state of the system. In the presence of galactose, the system is turned on; in its absence, the system is shut down. The four remaining proteins are the transcription factors and co-transcription factors that turn the system on and off.
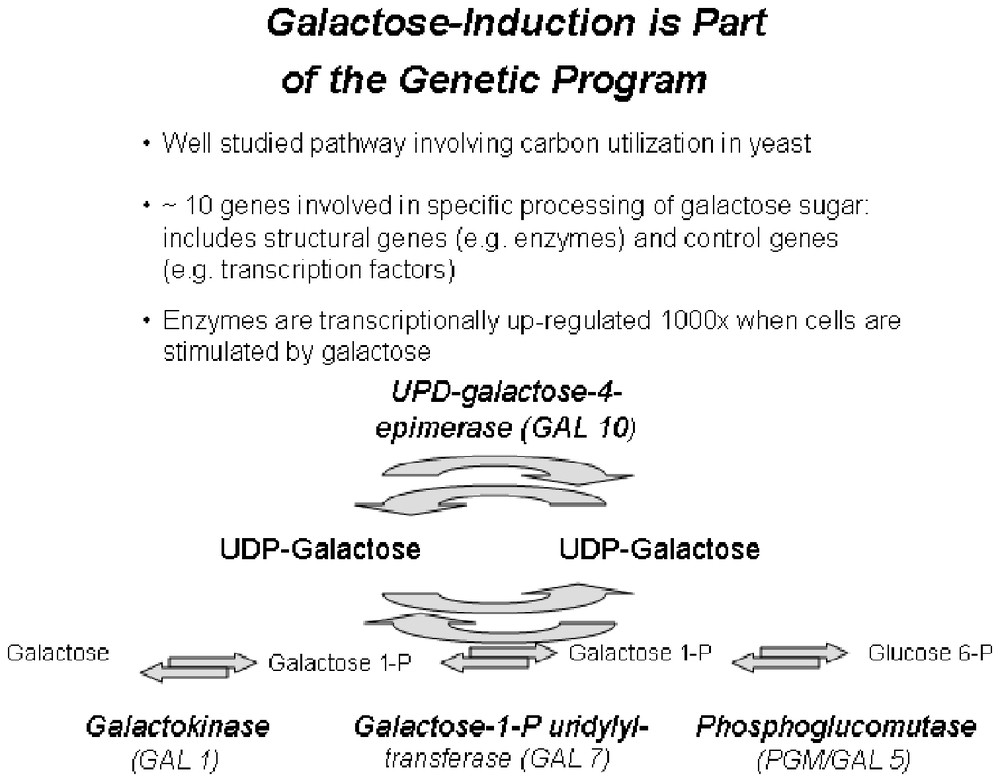
A diagram of the functioning of the galactose biomodule drawn from more than 30 years experiments (adapted from [15]).
Four distinct types of global datasets were generated and analyzed. First, genetic perturbations were executed leading to nine mutant strains of yeast, each with one of the nine genes in the system knocked out. These nine mutants and the wild-type strain were analyzed in the presence and absence of galactose (with the system on and off) by DNA array analyses that monitored the expression levels of most of the ∼6200 yeast genes. These data revealed two types of insights. First, when the behavior of the nine systems genes were examined under the 20 different perturbations (10 genetic states operating in each of two biological states), in most cases the model quite accurately predicted the behaviors. However, in a few cases, unexpected results were obtained. In two cases, hypotheses were formulated to explain these discrepancies, and a second round of perturbations (double knock-outs) and global analyses were carried out and further insight into the operation and control of the galactose biomodule was obtained. These new insights could be added to the model. Second, 997 out of the ∼6200 mRNAs changed in a statistically significant manner across the 20 perturbations. These could be clustered into 16 groups where the genes within a group behaved in a similar manner across the 20 perturbations. Each group contained one or more functional biomodules for the yeast cell (e.g., cell cycle, amino acid synthesis, synthesis of other carbohydrates, etc.). Hence, it was postulated that the galactose biomodule was directly or indirectly connected to each of these other biomodules and that these connections led to the perturbations.
Second, this network hypothesis was tested by examining global datasets for protein/protein and protein/DNA interactions in yeast. A graphical platform termed Cytoscape was developed to integrate global mRNA concentrations, protein concentrations (see below), protein/protein and protein/DNA interactions. The global datasets of the 997 perturbed mRNAs were then joined to the global datasets of protein/protein and protein/DNA interactions (Fig. 2). This graphical display does confirm the interactions, direct and indirect, among many of the biomodules delineated in the expression profiling data from the 20 perturbations. It provides one of the first glimpses of the myriad of interactions among biomodules in the yeast cell and opens doors for further investigation of the nature and control of these fundamental molecular machines. A more detailed examination of the galactose biomodule shows the central role galactose 4, its major transcription factor, plays.
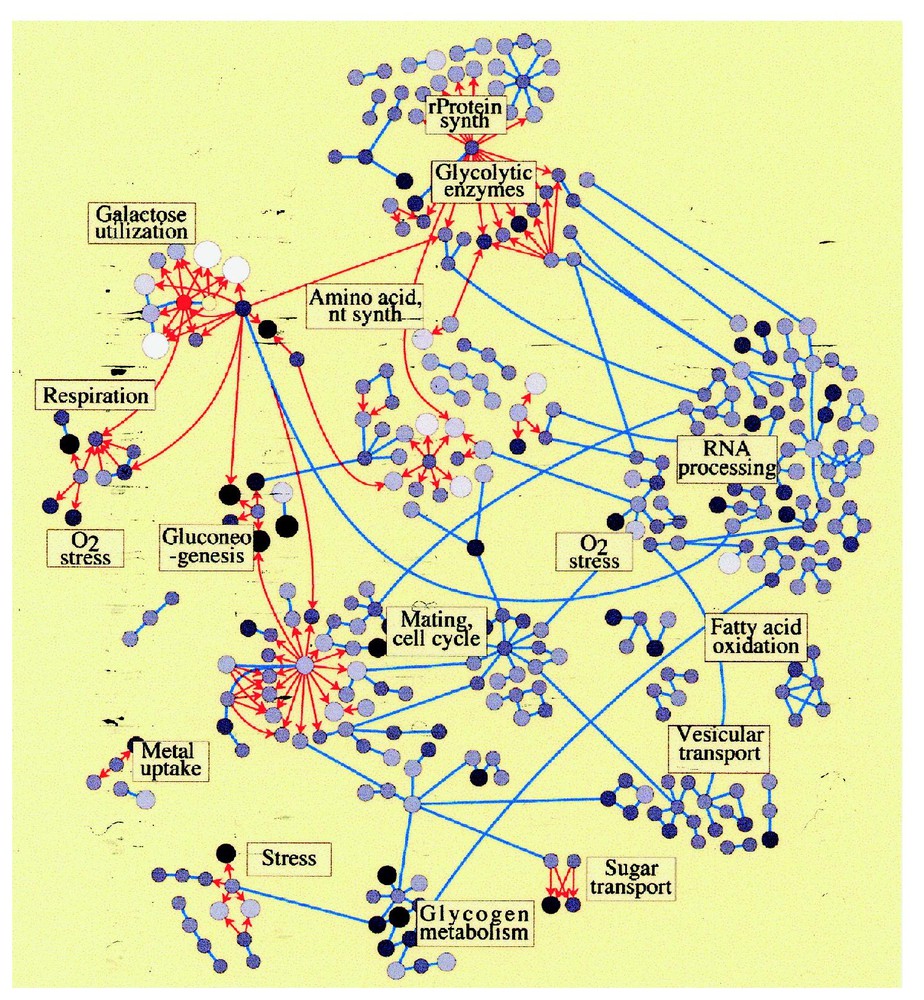
A network diagram of the yeast galactose utilization biomodule (red center circle) and its interrelationships to other biomodules in the yeast cell. The circles represent genes (or proteins). Arrows indicate transcription factor DNA binding site interactions. Bars represent protein/protein interactions. The mRNA concentrations are indicated by a gray-scale (black: high expression; white: low expression). The size of the circle indicates a change from the wild-type expression (big circles up and small circles down). (Adapted from [16].)
Third, we used quantitative proteomics (ICAT technology) to analyze 300 proteins in wild-type yeast with the system turned on and off. Thirty of these proteins changed in the transition between these two biological states. What was striking was that the mRNA and protein changes went in different directions for 15 of these examples. Hence, post-transcription control mechanisms must regulate protein synthesis in half of the examples. This is a beautifully explicit example of why multiple levels of biological information must be analyzed and integrated to understand how biological systems function.
Fourth, kinetic data on global mRNA concentrations change across the physiological time span of activation of the galactose biomodule has been generated. Kinetic data provide powerful new approaches to understanding the temporal operation of the galactose biomodule and its temporal connections to other biomodules in the yeast cell (A. Weston, personal communication).
We come away from these integrated studies with several striking points. First, the systems biology approach to this biological system did give fundamental new insights into both how the galactose biomodule functions and how it is connected to other biomodules in the cell. Second, the power of integrating different global sets of biological data became obvious. Third, perturbations in just a single element can have widespread consequences in the system as a whole. Thus, even apparently subtle changes (perturbations) can be/have far reaching consequences in many biological systems (we presume that the galactose biomodule is representative). This has important consequences for systems approaches to diagnostic markers, therapeutic targets, and even preventive measures in disease. Finally, the integration of scientists skilled in technology, computation, and biology was essential.
These systems approaches are now being extended to multi-cellular organisms and more complex biological systems.
3.2 Endomesodermal development in sea urchin
The sea urchin is a fascinating model system for studying development [17]. Its developmental program is rather simple, moving from fertilization to the development of a mobile larva in 72 h, followed by implantation of the larva in the sea floor and emergence of the sessile adult sea urchin thereafter. One can readily obtain 30 billion eggs in a single summer, and large aliquots can be synchronously fertilized and development terminated at will at any particular developmental stage. This permits the purification and identification of many different transcription factors, which often are expressed at very low levels. It is possible to generate thousands of transgenic sea urchins per hour, and these can be used to assess developmental patterns of gene expression with the promoters of specific genes attached to reporter genes such as green fluorescent protein. Finally, development can be perturbed with n-morpholino oligonucleotide derivatives specific for individual genes (which act as antisense), various environmental agents such as lithium (which blocks development at a certain stage), and dominant negative transcription factors (which block development at certain stages) [17]. These perturbations can be used to assess mRNA populations at differing stages of development (e.g., to establish which transcription factors are active). Together, these features make the sea urchin a powerful developmental model. Larval development has been studied exclusively during the first 72 hours of endomesodermal development. Developmental gene regulatory networks involve three types of components: transcription (and co-transcription) factors, their cognate DNA binding sites on cis-regulatory regions, and the environmental signals that impinge on the gene regulatory networks [5]. These latter may be mediated by ion gradients, changes in cell-surface receptors responding to their cognate ligands, and all trigger various signal transduction pathways which end up mediating the effect of specific genes and gene regulatory networks. The architecture of a gene regulatory network is specified by the DNA binding sites, for these establish the linkages of the transcription factors that coordinate the behaviors of genes throughout the gene regulatory networks. The gene regulatory networks contribute to determine the behavior of the peripheral (structural) genes in the network. The peripheral genes ultimately execute the specific development functions that underlie particular aspects of development. Because creatures spanning the evolutionary spectrum from sea urchins to humans have similar toolboxes of transcription factors (obviously apart from gene expansions and the like), it is the representation and organization of DNA binding sites in the cis-regulatory regions that specifies the species-unique features of developmental gene regulatory networks and their corresponding manifestations as very different body plans. Accordingly, gene regulatory networks are fundamentally digital in nature [4]. Let us now consider the representation, organization, and function of DNA binding sites in what is to date the best-studied cis-regulatory system, the endo16 gene of the sea urchin.
The endo16 gene is expressed early in endomesodermal development and exhibits a complex pattern of gene expression [17]. It is expressed initially in the early endodermal cells; later throughout the entire gut as the gut anlage evaginates; and finally, it comes to be expressed only in the mid-gut of the mature larva. The entire cis-control region is encompassed in a 2.3 kb DNA sequence 5′ to the endo16 coding region. There are 34 DNA binding sites that are cognate sites for 13 transcription factors. The cis-regulatory region behaves in a manner analogous to a computer chip with six modular regions, each sensing a changing development environment reflected by changing concentrations of transcription factors. The G module is a general booster of endo16 transcriptional activity; the E/F modules are spatial inhibitors; the C/D modules are spatial inhibitors early in development; the B module runs endo16 in the mid-gut of the mature larva and the A module is a grand integrator for the activities of all of the other modules. Logic diagrams can be constructed for the behavior of each of the modules of the endo16 gene (Fig. 3). By the hypothesis-driven iterative and integrative approach of systems biology, data gathered on the expression patterns of the endo16 gene for the first 72 hours of development permit an accurate mathematical description of its behavior over this time span [18]. This is one of the first examples of where the systems approach has been carried to a conclusion formulated in mathematical terms. The endo16 gene is just one peripheral gene in the endomesodermal gene regulatory network.

A portion of the gene regulatory network for endomesodermal development in the sea urchin. Upper section. The green (upper) panel depicts primarily transcription factors and their interactions with the control regions of other transcription factors. Genes are indicated by horizontal lines. Arrowheads indicate activation. I indicate repression. The yellow (lower) panel indicates peripheral genes that carry out the functions of endodermal development. Middle section. The 2.1-kb promoter region of the endo 16 gene is enlarged here and depicts 34 DNA binding sites (rectangles) and 13 different transcription factors and cofactors (rectangles or lollipops connected by lines to the DNA binding sites). Experiments indicate that there are six modules (A–G) that carry out discrete functions for the developmental regulation of endo 16. The ultimate objective is to convert this logic diagram into a mathematical formulation that accurately represents the subtle complexities of this developmental circuit. Lower section. A logical diagram depicting the functions of the A and B modules throughout endomesodermal development is provided in the lower panel. This logic then embodies a mathematical relationship between the control segments. (Adapted from [4].)
Using the perturbations described above and others across the first 72 hours of sea-urchin development, it has been possible to begin the delineation of the endomesodermal gene regulatory network [19]. It is a work in progress that currently contains about 55 genes, most of them encoding transcription factors (Fig. 3). Several striking conclusions emerge from an analysis of this gene regulatory network. First, the network may be broken down into sub-circuits that have discrete functions (e.g., positive feedback, negative feedback, switches, etc.). This suggests that, in time, we may be able to create a lexicon of sub-circuits that are the building block components of all metazoan developmental gene regulatory networks. Second, development is inexorably driven forward. In general, development is not reversible; rather it is driven to an end point. This stands in stark contrast to the necessary reversibility of physiological networks. Third, as we come to understand the logic of gene regulatory networks, we will be able to reengineer them to create very different developmental outcomes (emergent properties). For example, by one simple manipulation of the endomesodermal gene regulatory network, it is possible to generate a sea urchin with two guts. As we learn more about how to engineer development, many useful opportunities will emerge in the fields of plant and animal development. Finally, the challenge of describing mathematically the endomesodermal gene regulatory network is striking, and raises the provocative question as to whether current mathematical approaches can handle the problem, or whether we will have to invent a new type of mathematics.
4 Future directions in systems biology: nanotechnology, data integration and modeling
In all instances discussed above, as well as in the many other areas of biology that are not discussed here, development of standards and quality assurance procedures will be instrumental to data validation and curation, a prerequisite for the types of data integration and mining that are essential to the success of any systems biology program. This represents an unprecedented challenge that will be addressed effectively only through partnership between academy and industry at all stages of technological development, data collection and analysis.
As illustrated above in the case of DNA, the combinatorial space of biological systems is virtually infinite, so that it is impractical to search this space for regularities in a systematic, comprehensive manner. Constant recycling of established building blocks, rather than systematic random testing has occurred during evolution as a tinker [20]. In this context, a central goal of systems biology is to identify the rules that underlie the emergence of biological structures and functions, keeping in mind that there might not be a simple relation between the complexity of these rules and that of the biological system. As a matter of fact, research in many different fields has provided evidence that simple rules can generate complex systems or behaviors, and complex rules can generate simple systems and behavior. If this can be achieved, then we should be in a better situation to reduce the infinite data space to that relevant to the biological problem under study.
The delineation of biomodules by unsupersived analysis of integrated data in the yeast system is a rather encouraging first step in this direction, pointing to limitations and possible avenues to extract such rules and use them. It also illustrates the fact that hypothesis generation and hypothesis-driven interrogation cannot be purely abstract, but must rely on graphical display and visualization tools that are essential to provide the biologists with a global view of the data. Matrix display of sequence comparisons, in which the sequences of two organisms are displayed on the x and y axis, immediately provides an overview of relatedness, of the presence of repeats and other features that would not be evident to the human mind by reading each of the sequences or even the printed output of the sequence comparison algorithm. It also makes it possible to fine-tune the parameters of the sequence comparison to highlight certain features. Similarly, image maps of microarray gene expression data [21,22], where samples are displayed on one axis, and genes on the other, coupled with color coding of the registered hybridization intensities, provide instant overview of large datasets; they allow human assessment of the results of various clustering methods, by revealing patterns of similarities between the samples (similar origin or cell type, similar drug sensitivity), between the genes (co-expression or regulation). These two examples also illustrate the iterative loop of modeling, testing and assessing which is the hallmark of systems biology.
The ultimate challenge of genomics, transcriptomics, proteomics, metabolomics, interactomics, etc., is to move to the characterization of single molecules and single cells. This requires rapid global analyses with high data quality and low cost per informational unit analyzed. Accordingly, we need to parallelize, miniaturize, integrate successive chemical and biological procedures and automate these analytic procedures. This automatically moves us to the realms of microfluidics and nanotechnology. The biological imperatives described in our paper must drive the design of microfluidics and nanotechnology machines. It is our prediction that these technologies will move us to the sophisticated analyses of single molecules and single cells in real time – thus bring to biology a revolution that will transform how we think about and practice biology and medicine. Nanotechnology and microfluidics will be the cornerstones of systems biology, provided that we develop and use them in a standardized mode in the context of appropriate formalisms. In time, these advances will be integrated with new molecular imaging technologies that will permit the visualization of discrete types of biological information in living cells and organisms in which the final stages of hypothesis-driven systems biology must be carried out.
Acknowledgements
We thank Tawny Biddulph, Odile Brasier and Patrick Zaborski for manuscript editing.