

1 Introduction
Kraft pulping is by far the most important industrial method in the world for the production of chemical pulps. The process has the advantage of being rather insensitive to the type of wood species and the inorganic process chemicals can be efficiently recovered and reused. The resulting cellulosic fibres are strong and can be used in a large variety of paper and board products. A major drawback is, however, the comparatively high amount of residual lignin that is still attached to the fibres when the cook, for reasons of fibre quality, must be interrupted. As a result, a rather large amount of lignin must be removed in the subsequent bleaching operation rendering this part of the total process expensive in terms of costs of bleaching chemicals and operation equipment. Consequently, if a prolonged delignification in the cook could be achieved without any detrimental effects on the pulp quality, a much-improved economy and a decreased environmental load might be obtained in the kraft pulp mill.
In kinetic pulping experiments, it has been demonstrated that the final part of the delignification, amounting to approximately 10% of the total, proceeds at a much lower rate as compared to the previous part [1]. The exact transition point between the two rates can be affected by the conditions in the cook and e.g. a higher alkali concentration has been shown to increase the amount of rapidly removed lignin. Kinetic pulping experiments do not, however, give any indication of a different type of lignin, merely that different chemical environments can affect the reaction rate. Thus, in such experiments any differences in the chemical structure of lignin are difficult to identify since e.g. no sharp drop of alkalinity takes place at the transition point to the final delignification phase. Spectroscopic and chemical analyses of isolated residual lignin from unbleached kraft pulps indicate, however, that the structure is much more complex than that of native lignin (MWL) but also of lignin isolated from pulp after a kraft cook followed by bleaching [2].
Much work on kraft pulping chemistry has been focused on the reasons for the low reactivity of the final portion of the lignin attached to the fibres. As a major reason, condensation reactions taking place in the lignin during the kraft cook has been suggested [3–7]. Among other alternative explanations, the formation of reduced and thus unreactive side-chain structures can be found [2,8].
It has been known since a long time that the γ-hydroxymethyl group present in virtually all of the lignin side-chains is, in part, released as formaldehyde during the course of a kraft cook. Based on NMR data, the extent of release of formaldehyde has been found to be comprehensive and less than half of the original hydroxymethyl groups are still present at the end of the cook [9]. Experiments with kraft pulping of wood meal containing radioactive lignin have shown that one portion of the released formaldehyde is found in a low molecular weight fraction, whereas another seems to be linked to the high molecular weight part of the dissolved lignin [10]. These results have been further confirmed by kraft cook of wood meal in the presence of radioactive formaldehyde [4]. Addition of formaldehyde has also been done to kraft cook of isolated native lignin (MWL) followed by analysis of the lignin with 31P NMR [7]. In the spectrum, signals were found supporting the view that a
Quantitative 13C NMR analysis of dissolved and isolated residual fibre lignin also supports the view that structures of the diguaiacylmethane type are present since the spectra contain a sharp signal at around 29 ppm attributable to the methylene group [5,8,11]. On analysis using the 2D HSQC NMR sequence, however, no signal above the noise level could be detected at the right place, i.e. at ∼29 and 3.8 ppm [12]. Obviously, the 1D NMR techniques are not able to provide an unambiguous assignment due to the extremely high degree of structural complexity of the lignin. It can, however, be concluded that formaldehyde, indeed, is incorporated into the polymeric lignin during pulping. Many individual reaction types are possible but, obviously, the coupling between two aromatic rings in polymeric structures is not a favoured mode of reaction.
The formation of quinone methides in phenolic lignin units as a result of elimination of an α-hydroxyl or α-ether group is a well-known reaction in alkaline pulping. In model experiments, it has been clearly demonstrated that an available phenolic compound will add to the α-carbon atom, thus giving rise to a substituted diarylmethane structure [3]. Despite the fact that this type of condensation occurs readily in alkaline conditions, the existence of such structures in the polymeric lignin is difficult to prove. In 1D NMR analysis, using the DEPT sequence, of various dissolved kraft lignins an increased number of methine carbons in the range of 0–55 ppm in comparison to MWL has been observed and interpreted as being due to condensation reactions [5]. Results which are contradictory to this interpretation have, however, been published [13]. Thus, a polymer-supported quinone methide was found to be unreactive when treated with alkali at high temperature in the presence of a reactive phenol:syringol. A corresponding experiment with a low molecular weight quinone methide, on the other hand, gave, as expected, a high yield of the condensation product. Therefore, it can be concluded that the difference between reactions in low molecular weight model systems and in polymers can be large.
The presence of aliphatic methine carbons in the range of 0–55 ppm (13C NMR) in lignin polymers is to a large extent due to the presence of β-5, β-1 and β–β structures. In part, these structures survive the kraft cook and, consequently, the corresponding methine groups can be found in the NMR spectrum of dissolved as well as of isolated residual lignin. The observed increase in the number of such carbon atoms as given by DEPT analysis of kraft lignins [5] demonstrates, however, that also other structures must contribute. Recently, by NMR analysis using the HSQC sequence, one ‘new’ lignin structure has been found and identified as lignin-bound secoisolariciresinol. The two identical β-CH groups in this structure were found at 41.0 and 2.20 ppm (Cβ/Hβ) and the amount was calculated to be around 2% in softwood kraft lignins [14].
The presence of β-1 structures in native lignin has been the subject of some controversy and largely diverging values for the amount have been published. The reason for this is probably the fact that a substantial amount of the precursor, the spiro-dienone, can be present together with the β-1 structure itself [15]. During a kraft cook, all types of β-1 structures seem to be degraded early in the cook, however, and no contribution from such structures can be found in either the dissolved or the isolated residual lignin. Thus, the contribution to methine groups (0–55 ppm) in residual kraft lignins seems to be restricted to β–β structures of the secoisolariciresinol (∼2%) and pinoresinol (∼2%) types and to remaining β-5 structures (∼5%) [12]. Other types of structures, if present, are of very low abundance.
DEPT spectra of kraft lignins have also revealed a large quantity of methylene groups in the range of 0–55 ppm [5]. In analogy to the results obtained from lignin model experiments with soda/AHQ pulping liquor [16], the presence of such methylene groups in kraft lignins has in previous work been interpreted as being due to a one-electron reduction of quinone methide intermediates [2]. The large variety of this type of NMR signals indicates, however, that other explanations might prevail.
In the present work, the shortcomings of the results obtained from model experiments and from 1D NMR spectra for explaining the chemical structure of kraft lignins and the hope of finding a reasonable explanation for the unreactivity of the final portion of the fibre lignin have been addressed. For these reasons, kraft-pulping experiments have been combined with isolation and analyses of lignins using thioacidolysis, GPC and 2D NMR techniques as predominant analytical tools. Based on such analytical data, a new hypothesis on the origin of lignin condensation in kraft cooking has emerged. In order to gain further support for the presented view, some complementary data have been collected through selected analyses and model experiments.
2 Materials and methods
2.1 Kraft cooking experiments
All pulping experiments were carried out on industrial spruce wood chips (Picea abies). These were manually sorted prior to cooking to remove bark, knots and oversize chips. Each cooking experiment was done with 300 g of chips (dry basis) in stainless steel autoclaves at a liquor-to-wood ratio of 4:1. The cooking liquor (white liquor) contained 1.32 mol l−1 of sodium hydroxide and 0.34 mol l−1 of sodium sulphide. The liquor was introduced to the chips after 30 min of evacuation of the autoclave at a temperature of 70 °C followed by a temperature increase of 1 °C min−1 up to 170 °C. This temperature was kept until the desired time-temperature relationship, the H-factor, was reached. One series of cooks was carried out in the presence of 2,6-dimethylphenol (2,6-xylenol) which was added to the cooking liquor as its sodium salt (Na-2,6-DMP). All cooking data are collected in Table 1. After cooling of the autoclave in cold water, the black liquor was collected and the pulp sample washed overnight with distilled water and disintegrated. Residual alkali in the black liquor and the amount of oxidizable groups, the kappa number, in the pulp were determined according to the standard methods SCAN-N 33:92 and SCAN-C 1:77, respectively.
Kraft cooking conditions
| Cook | Reference | 2,6-xylenol |
| White liquor | [OH−] = 1.32 M | [OH−] = 1.32 M |
| [SH−] = 0.34 M | [SH−] = 0.34 M | |
| [Na-2,6-DMP] = 0.2 M | ||
| Liquor:wood ratio | 4:1 | 4:1 |
| Initial temp. (°C) | 70 | 70 |
| Temperature (°C) | 170 | 170 |
| Time (min) | 81, 103, 132, 140 | 79, 104, 131, 148 |
| H factor | 752, 1007, 1324, 1403 | 735, 1020, 1314, 1498 |
| Residual alkali (M) | 0.37, 0.35, 0.33, 0.31 | 0.45, 0.41, 0.39, 0.36 |
| Kappa number | 40.1, 34.3, 26.4, 24.7 | 40.4, 31.3, 26.1, 21.4 |
2.2 Analysis of industrial black liquors
A series of black liquors covering the whole cooking cycle were taken out from a Swedish pulp mill producing softwood kraft pulp in the batchwise mode. Each sample was analysed for the presence of elemental sulphur and polysulphide sulphur using a gas chromatographic method based on derivatization of sulphur with triphenylphosphine at two different pH values [17]. At a pH of 11.5, only the originally present sulphur can be derivatized, whereas at a pH of 5.5 both the originally present sulphur and the sulphur originating from the decomposition of polysulphide are included in the analysis.
2.3 Model experiment with 2,6-xylenol
In a 100-ml stainless steel autoclave, 2 g of 2,6-xylenol (0.0164 mol), 0.0164 mol of sodium hydroxide and 0.246 moles of polysulphide solution, prepared according to [18], were added under pure nitrogen giving a total volume of 75 ml. The autoclave was heated at 135 °C during 60 min and, after a temperature increase of 1 °C/min to 165 °C, a further heating at this temperature was done for another 30 min. After cooling, the resulting mixture was neutralised, extracted with methylene chloride and subjected to analysis by GC-MS using a DB 5MS J&W Scientific column (30 m length, 0.32 μm I.D., 0.25 μm film thickness) with the program: 100 °C/5 min, 4 °C/min, 280 °C/10 min. Split injection was used.
2.4 Isolation of lignin from the black liquor
To each of the black liquors produced in the laboratory cooks, disodium-EDTA (approximately 1 g/100 ml) was added to chelate any metal ions present in the sample. The pH was adjusted to pH = 5 with sulphuric acid while continuously adding nitrogen to the liquor in order to remove all malodorous gases liberated in the pH-adjustment. The combined precipitate and solution was put in a freezer over-night and subsequently allowed to tow to ∼0°C. At this point, the precipitated lignin was filtered on a glass filter G4 and the lignin further washed with icy water. After suspension of the lignin in water and freeze-drying, the lignin was extracted with pentane for 6 h in a continuous extractor to remove all extractives. Further purification of the lignin was done by dissolving the lignin in dioxane-water 9:1, whereby carbohydrate-containing material is left as a residue. After repeated washing of the residue with dioxane-water 9:1, the combined solution was evaporated and the lignin again suspended in water and freeze-dried.
2.5 Analytical methods
Thioacidolysis was carried out as described in [19]. Samples of kraft pulp and wood (milled in a Wiley mill using a 40 mesh screen) were first extracted with acetone in a Soxhlet extractor for 24 h prior to analysis in order to remove extractives and any remaining excess of 2,6-xylenol (if added in the cook). All samples were also pre-swollen in water as described in [20] and solvent-exchanged to dioxane before addition of the thioacidolysis reagents in order to achieve a complete dissolution of the lignin.
Analysis by gel permeation chromatography (GPC) was done with a Waters system on all thioacidolysis mixtures obtained from wood and pulp samples as described in [20]. Each analysis was run on a series of three Ultrastyragel columns having pore sizes of 100, 500 and 1000 Å, respectively. A mobile phase of dioxane-water 9:1 and a flow rate of 0.6 ml min−1 were used and UV detection was done at 280 nm.
13C NMR spectroscopy was done on a previously isolated kraft residual lignin and a dissolved black liquor lignin obtained from a cook in the presence of 2,6-xylenol [21]. Both 1D and 2D (HSQC) sequences were employed in order to find out the possible coupling mode between lignin and 2,6-xylenol. On similar lignins, isolated from normal kraft cooking, several 2D sequences, such as HSQC, HMBC and HSQC-TOCSY, were used in order to get detailed information about the origin of the aliphatic methylene carbons present in the range of ∼15–45 ppm. The data obtained are collected in Table 2 with the structural assignments shown in Fig. 1. The following experimental conditions were used: instrument Bruker Avance 400 MHz, sample amount ∼100 mg, solvent acetone(d6)/D2O 5:1, ambient temperature, 1D runs (quantitative) with a 5-mm broadband probe using an inverse gated proton decoupling with 10 s between pulses and a 90 degree pulse angle, 2D HSQC, HMBC and HSQC-TOCSY spectra acquired with a 5-mm inverse probe equipped with a gradient coil. For the HSQC and HSQC-TOCSY spectra, the data acquisition was done with a delay of 2 s between pulses, a coupling constant of 150 Hz, an acquisition time of 0.2 s, a spectral window of 12.8 ppm in F2 and 150 ppm in F1 and with 2 K× 1 K increments. In the HSQC-TOCSY experiment, a mixing time of 60 ms was used. The 2D HMBC spectrum was acquired with a spectral window of 12.8 ppm in F2 and 200 ppm in F1 with increments of 2 K × 0.5 K, giving an acquisition time of 0.2 s in F2. A delay time of 60 ms was used for the evolution of the long-range couplings.
Identification of methylene groups in kraft lignin based on HSQC, HMBC and HSQC-TOCSY NMR spectra with the 1H/13C chemical shift values listed. Chemical structures are shown in Fig. 1. It should be noted that the fatty acid methylene group c may contain more than one methylene group
| Structure | a | b | c | d | e | f a |
| Dihydroconiferyl alcohol | 2.54/32.1 | 1.74/34.9 | ||||
| Fatty acid | 2.22/34.2 | 1.51/25.2 | 1.21/29.9 | 1.19/32.1 | 1.20/22.9 | 0.80/14.0 |
a Methyl group.
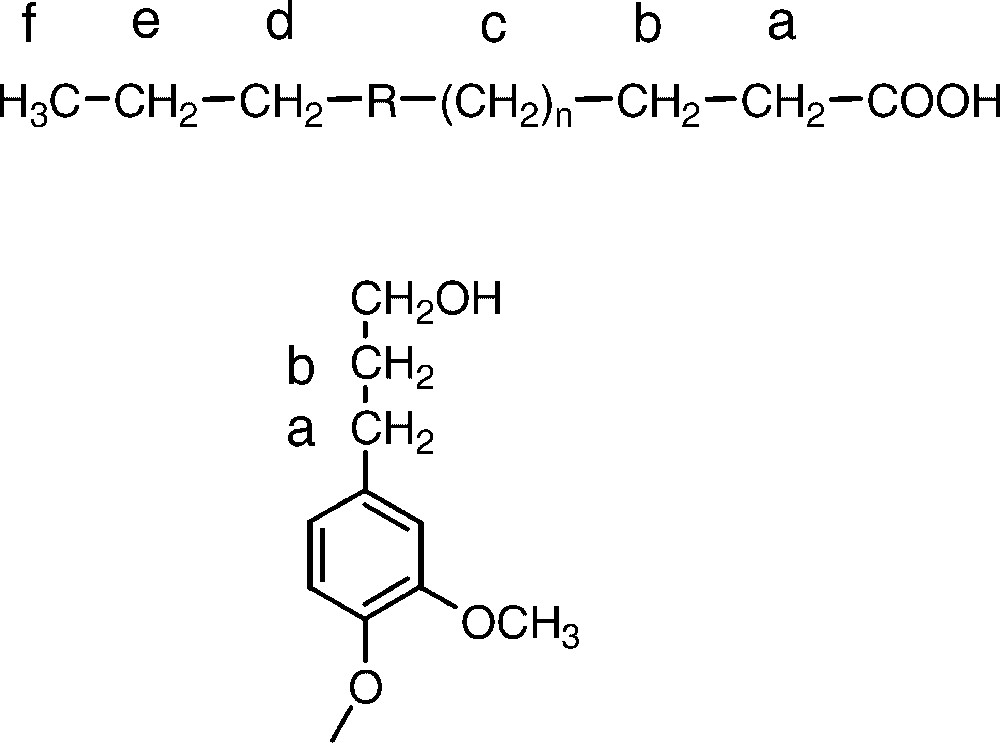
The fatty acid and dihydroconiferyl alcohol structures suggested as origin of aliphatic methylene groups present in the 13C NMR range of ∼15–45 ppm. Shift values for the groups a–f are given in Table 2. R denotes the unidentified portion of the fatty acid.
3 Results and discussion
3.1 GPC analysis of kraft lignins
On thioacidolysis of wood, the lignin can be completely degraded into a mixture of monomeric, dimeric and trimeric products with only traces of higher oligomers. This can be clearly shown by GPC of the product mixture and from wood the monomeric and dimeric thioacidolysis products constitute the predominant peaks in the chromatogram with only a weak shoulder containing the higher oligomeric material [20]. A similar analysis of kraft pulp reveals, however, that a considerable amount of material having a higher molecular weight is now present. Thus, lignin reactions in the kraft cook, resulting in the formation of non-degradable structures, take place. In order to further verify the formation of new structures, not degradable by thioacidolysis, in the lignin during kraft cooking, a series of four kraft cooks was carried out covering a range of kappa numbers of ∼40–20 (Table 1). All pulps were subjected to thioacidolysis and the products subsequently analysed by GPC. As shown in Fig. 2, the same pattern was obtained from all samples. By normalizing the chromatograms using the ‘polymeric’ peak as standard, a trend towards decreasing relative amounts of monomers and dimers could be clearly seen as the cooking time was increased.
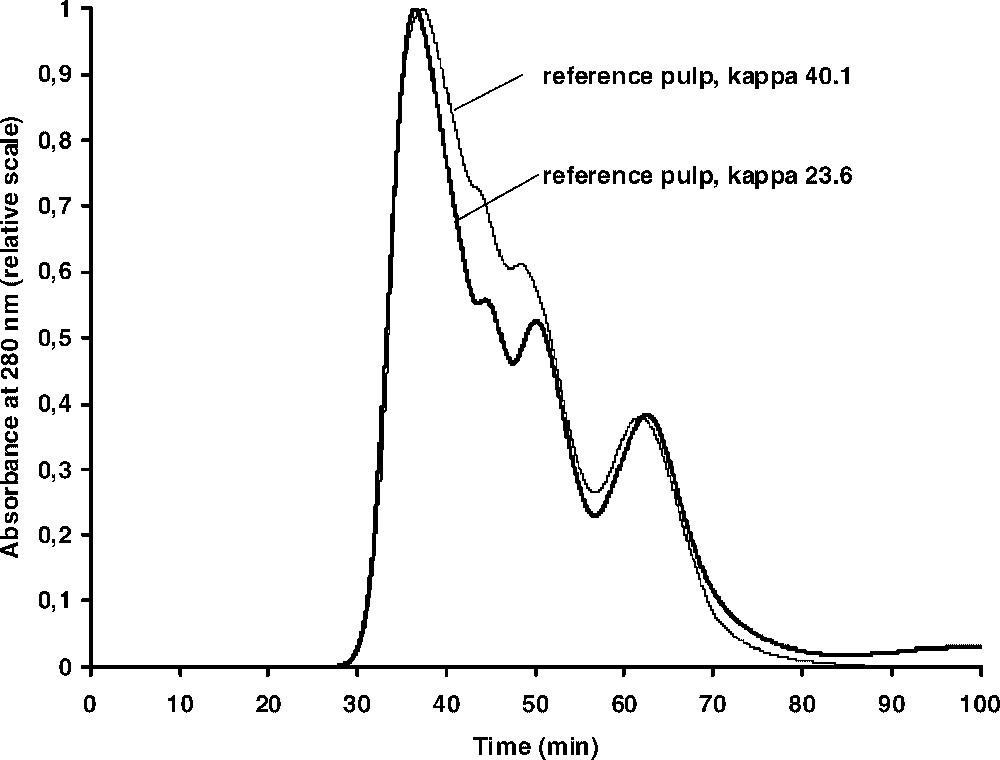
GPC curves of thioacidolysis mixtures obtained from two of the reference kraft pulps shown in Table 1 (the other two pulps gave similar chromatograms). In each chromatogram, the prominent peak at around 60 min is assumed to be derived from carbohydrates, whereas the thioacidolysis monomers and dimers elute at around 50 and 45 min, respectively (cf. [19]).
A second series of kraft cooks was also done covering approximately the same kappa number range (Table 1) but now in the presence of a large amount of a reactive phenol, 2,6-xylenol, since, in previous work, it was shown that this phenol can be readily incorporated into the polymeric fibre lignin [21]. Again, thioacidolysis was done and the resulting mixtures analysed by GPC. The resulting chromatographic curves were found to be very similar to those shown in Fig. 2. In order to get a more detailed comparison between the GPC-curves obtained in the absence and presence of 2,6-xylenol, the thioacidolysis mixtures from the pulps at around kappa number 30 were compared in the same chromatogram. The result, shown in Fig. 3, demonstrates that at similar kappa numbers and H-factors, the cook containing 2,6-xylenol seems to contain a higher amount of the ‘polymer’ fraction, assuming similar amounts of monomers and dimers in both cooks.
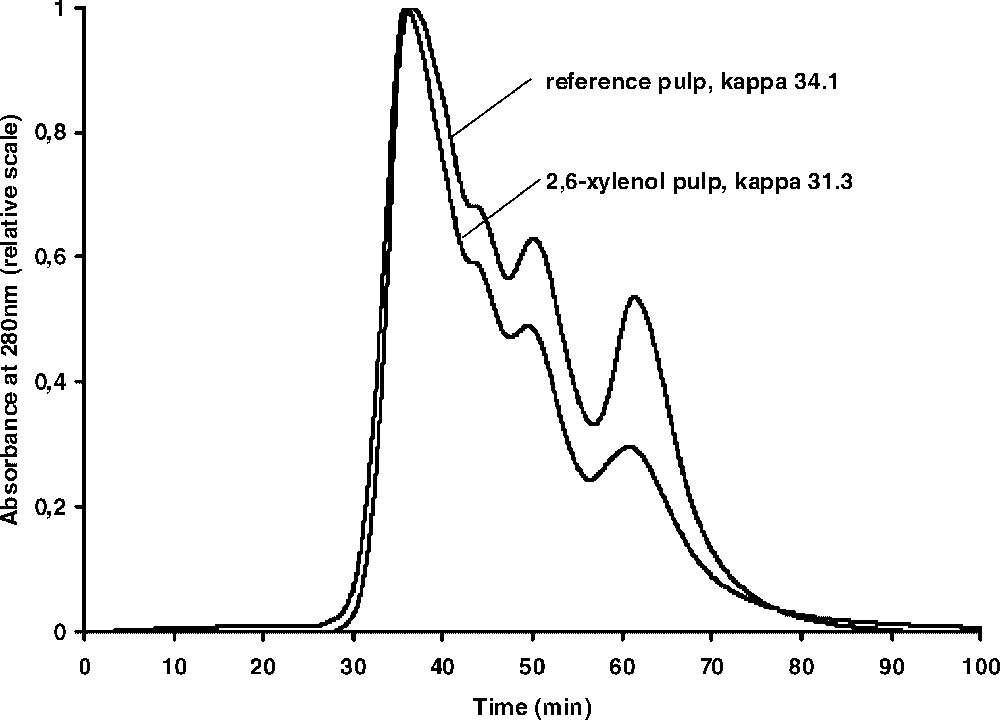
GPC curves of the thioacidolysis mixtures obtained from a kraft cook to kappa number 34.3 and a kraft cook in the presence of 2,6-xylenol to kappa number 31.3.
The presence of non-degradable material in the thioacidolysis reaction demonstrates (most likely) that new carbon–carbon linkages have been formed in the lignin during the kraft cook. Based on the discussion above, the presence of diarylmethane structures formed through condensation either with formaldehyde or involvement of a quinone methide seems, however, rather unlikely. Another alternative for the formation of higher molecular weight material would be radical coupling reactions.
Already in the 1960s, radical coupling reactions were suggested to occur in the soda pulping of wood [22]. A more thorough understanding of any possible electron transfer reactions in alkaline pulping was not obtained until some 10 years ago, however. By the use of lignin model compounds such as syringyl alcohol it could be demonstrated that electron transfer reactions indeed are possible during the conditions encountered in soda [23] as well as in kraft or soda/AQ pulping [24]. Thus, the treatment of syringyl alcohol with sodium hydroxide resulted in the formation 4-methylsyringol together with several other products resulting from one-electron transfer reactions. 4-methylsyringol was also found as a product in both kraft and soda/AQ pulping albeit in lower and much higher amounts, respectively.
3.2 Kraft cooking with 2,6-xylenol
From kraft cooks, carried out in the absence and presence of 2,6-xylenol, the corresponding fibre lignins were isolated [21] and subjected to NMR analysis. By recording 1D and 2D spectra of these lignins as well as the two corresponding difference spectra, it could be demonstrated that the only observable differences between the two samples were those related to the two methyl groups and to the aromatic ring carbons in 2,6-xylenol. From the integral values for the lignin methoxyl group and for the methyl groups in the 2,6-xylenol unit, respectively, an incorporation of approximately 14% 2,6-xylenol units per phenylpropane unit could be calculated. Furthermore, the aromatic methine groups from xylenol could be integrated giving a value of 2–3 such groups per xylenol, indicating that the xylenol had been incorporated in the residual lignin both as biphenyl and biphenyl ether structure.
Based on these results, any major condensation reaction between e.g. the side chain
3.3 NMR analysis of kraft lignin
In a previous work, the presence of large quantities of methylene groups in kraft lignins had been interpreted as being due to a reduction of intermediate methylene quinones formed during the course of the kraft cook [2]. In addition, any diarylmethane structures formed through condensation between phenolic lignin units and released formaldehyde will result in such groups [7]. By careful NMR analysis of dissolved as well as of residual kraft lignins using 2D NMR sequences like HSQC, HMBC and HSQC-TOCSY it could be demonstrated, however, that the vast majority of the methylene groups seems to originate from dihydroconiferyl alcohol units and from fatty acid(s) (Fig. 1). Whereas the former of these is well known as an integral part of the lignin structure [26], the presence of chemically linked fatty acid structures has not been observed before. The estimated abundance for each one of these structures is around 2%, based on the intensity of one methylene group per aromatic ring in lignin. Despite the application of different 2D NMR sequences, the exact identity of the fatty acid(s) could not be revealed completely although several of the methylene groups were unambiguously identified. A possible structure is pinolenic or linoleic acid, both major unsaturated fatty acids present in softwood. It should be noted that the carbon shift for one of the methylene groups in the fatty acid coincides well with the shift for a
3.4 Sulphur as an electron transfer agent
The possibility of one-electron reactions playing a dominant role in the fragmentation reactions taking place in kraft pulping has been the subject of experiments with lignin model compounds. No conclusive evidence for such a mechanism could be obtained, however [27]. Thus, the predominant cleavage of phenolic β-O-4 structures is thought to proceed via addition of a hydrosulphide ion (or a polysulphide ion) to a quinone methide intermediate followed by an intramolecular attack and fragmentation [28,29]. The cleavage reaction is accompanied by a formation of elemental sulphur that, via reaction with hydrosulphide ions and formation of polysulphide, may disproportionate into hydrosulphide and thiosulphate ions. The latter reaction is, however, not complete and throughout a kraft cook, various amounts of sulphur and polysulphide can be found in the black liquor as shown in Fig. 4. Obviously, the amount of polysulphide + sulphur is rather low towards the end of the cook (∼2–3% of the charge of hydrosulphide ions in the white liquor). It is suggested, however, that the sulphur/polysulphide may participate in electron transfer reactions between the residual fibre lignin and dissolved low molecular weight phenols present in the cooking liquor. Thereby, new biphenyl and diaryl ether (thioether) structures can be created successively during the cook and, at a certain point, give rise to a complete blocking of any further lignin cleavage reactions (cf. [30]).
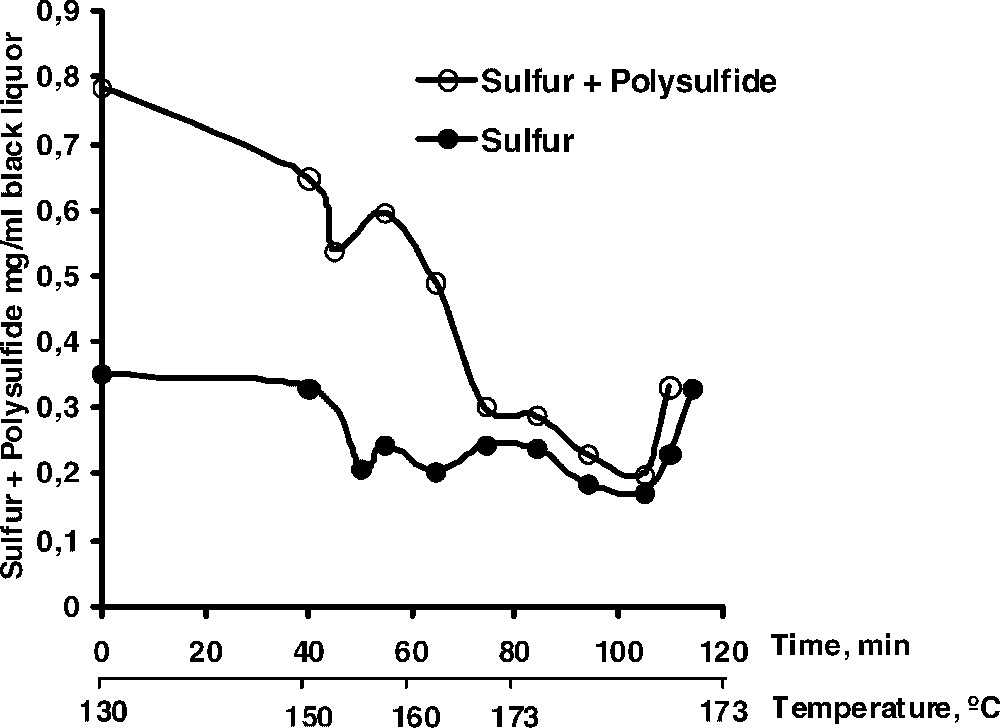
Content of elemental sulphur and polysulphide in the cooking liquor as a function of cooking time in an industrial kraft cook.
Although this mode of reaction still is not unambiguously demonstrated, the incorporation of 2,6-xylenol into the residual lignin, described above, as well as the finding that a fatty acid is linked to kraft lignin are both in favour of a one-electron mechanism. A further support for such a mechanism was obtained by heating 2,6-xylenol with a polysulphide solution. Analysis of the reaction products revealed the formation of several radical coupling products as shown in Fig. 5 (cf. [31]).
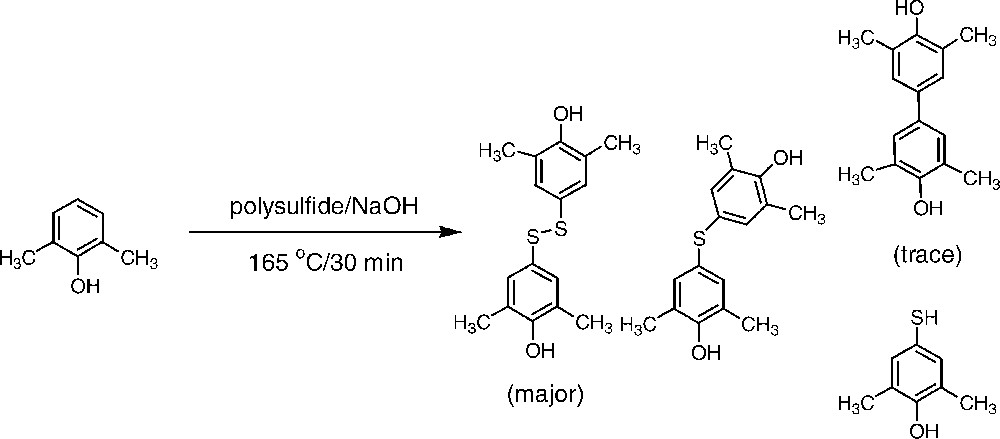
Products from the treatment of 2,6-xylenol with polysulphide solution at 165 °C.
The possibility of sulphur/polysulphide acting as an electron transfer agent during the course of a kraft cook needs further confirmation. The well-known fact that sulphur on heating is transformed from cyclooctasulphur into a chain-formed biradical [32] able to initiate e.g. cross-linking in rubber or to give coupling products with phenols are, however, observations in favour of such a view. In the kraft cooking liquor, formation of polysulphide proceeds according to Eq. (1), thus demonstrating the simultaneous presence of elemental sulphur and polysulphide [33].
| (1) |
4 Conclusions
The final portion of lignin is difficult to remove in a kraft cook. A major reason for this behaviour seems to be the occurrence of condensation reactions between fibre lignin and dissolved lignin fragments during the course of the cook. Several pieces of evidence for a condensation mechanism involving elemental sulphur as initiator in one-electron reactions have been collected. Although further work is necessary to finally confirm such reactions unambiguously, the data obtained are all in favour of such a mechanism. This will open up new possibilities for making process changes that can be used to extend the delignification in the kraft cook without detrimentally affecting the pulp quality.
Acknowledgments
Thanks are due for a grant to one of us (AM) from ‘Lars-Erik Thunholms Stiftelse för Främjande av Vetenskaplig Forskning’ and to the European Community, FAIR CT98 3460, for financial support. The skilful experimental help from Eva-Lisa Lindfors, STFI, and from Waleed Wafa Al-Dajani of this department (now with Paprican) is gratefully acknowledged.