

1 Introduction
Wood composites are a class of materials that generally consist of solid fragments of wood that are reconstituted in a form fit for a designated end-use by some sort of adhesion process. Plywood, fiber boards, particleboard, oriented strand board (OSB), and wood plastic composites are all members of this class of composites [1]. Over time, wood composites have been reconstituted from ever-smaller wood fragments, progressing from plies to strands to fibers to fine ‘flour’ measuring only microns in size. The latter represent the dimensions of wood fragments typically used for wood plastic composites produced by thermal extrusion processes [2]. Wood composites are capturing ever-larger markets, partially in response to reductions in the supply of solid, large dimension timber [3,4].
Composites in general are materials that combine the high strength and stiffness characteristics of a fiber (or particle) with the ductility of a (continuous) matrix [5]. In many man-made composites the fiber-matrix interface is the weakest point resulting in ‘fiber pull-out’ and failure before the fiber reaches its true strength potential [6]. In wood plastic composites, the dimensions of the wood fibers (if they existed in fiber form in the first place) often suffer a significant loss of aspect ratio caused by the high shear forces of the extruder at high temperatures [7]. Many wood composites also suffer from the high density that is the result of thermal processing under high-pressure conditions [8,9]. The ideal wood-like composite would combine the features of a high-strength and high-stiffness (hollow) fiber embedded in a continuous matrix from which it never (under any condition of moisture or temperature) separates interfacially, and with which it produces a lightweight material [10,11]. That is to say, wood in its native state is an ideal composite, a material worth mimicking! This conclusion has been recognized by a recent report of the National Academy of Sciences of the USA, which advocates the use of the “hierarchical structures in biology as a guide for new materials technology” [12].
In order to adopt the principles of biomimetics to the process of composite formation from wood fibers, two crucial elements must be understood. First, how can a matrix (i.e., lignin)-rich layer be deposited on the surface of a cellulose fiber? Second, how can this layer of matrix material be consolidated to form an adhesive bond between fibers? Solutions to these problems may potentially be accomplished thermally or biologically, i.e., by biomimetics.
Thermoplastic cellulose composites may potentially be formed when cellulose fibers are surface-coated with melt-deformable copolymers. Such copolymers may consist of saccharidic amphiphiles containing waxy substituents. Cellulose and/or cellulose derivatives have served as both adsorbing surface substrates and adsorbable amphiphiles [13–24]. Employing this established method of surface modification using amphiphilic copolymers with olefinic character, a thermoplastic coating may be produced on cellulose fiber surfaces. Cellulose mono- and di-esters with long chain fatty acids have recently been shown to represent thermoplastic entities with melting/softening points that decline in accordance with methylene group content (Fig. 1) [25,26]. Thus, melt-processable cellulose esters with low degrees of substitution (DS) may potentially open a route towards thermoplastic fiber composites by adsorption processes.

Glass transition temperatures of long-chain cellulose mono-, di-, and tri-esters (LCCE) with fatty acid substituents as a function of the methylene content (in weight-%) of the ester substituent. The methylene content of acetate groups is thereby considered nil, and that of a propionate group is 1. The number of carbon atoms in the substituent, the acyl group size, is given by n. Only mono- and di-esters follow the Fox equation, triesters have a constant glass transition at around 70–80 °C (according to data by Vaca-Garcia et al. [51]).
A biology-mimicking approach may involve the surface adsorption of molecules amenable to enzymatic crosslinking. During secondary wall formation in wood cells, cellulose is spun from rosette structures into an aqueous sol-like hemicellulose solution [27]. There is a body of work describing how hemicelluloses regulate (bacterial) cellulose fibril diameter [28–33]. These studies demonstrate that adsorption of hetero-polysaccharides plays a key role in establishing an interfacial region that prevents delamination and fiber pullout in wood. This adsorption process is governed by self-assembly, a process driven by thermodynamics that results in the aggregation of bipolar molecules on the fiber surface. Self-assembly behavior has been observed with many other natural amphiphilic polymers including oligosaccharide-protein block copolymers [34], hydroxyethyl cellulose [35,36], fluorine-containing cellulose diblock structures, xylan-rich heteropolysaccharides and their derivatives [37], and lignin–carbohydrate complexes [38,39].
Our studies were motivated by a vision of biomimetics. They presume that a cellulose surface enriched with lignin or lignin precursors becomes susceptible to the generation of phenoxy radicals by enzyme catalysis. Free radicals then form network polymers (thermoset adhesives) by coupling [40]. Felby et al. [41–45] and Huettermann et al. [46–53] have shown that wood fibers can be enzymatically activated in vitro with phenoloxidase and/or laccase, and this treatment can be used to produce wood composites with enhanced auto-adhesion between components [47,51]. The lignin-coated fibers that are formed when a lignin–carbohydrate complex is adsorbed to a cellulose surface may possess the potential for enzymatic activation and the development of adhesive bonds that are similar to those in wood.
This report describes some basic experiments to demonstrate how a model cellulose surface (which is devoid of the complexities that porosity, microfibril orientation, fiber dimension, etc. entail) and a model lignin–carbohydrate copolymer complex (which is devoid of the complexities of molecular non-uniformity, size, and structure) interact under conditions that often produce self-assembly.
2 Materials and methods
2.1 Cellulose model film preparation
Smooth, uniform films of regenerated cellulose were prepared on 12 mm × 12 mm SPR sensor slides using a protocol adapted from Schaub et al. [54] and previously described in conjunction with a study on the adsorption of lignin-containing hemicellulose derivatives [37]. Each sensor slide (Reichert, Inc.) consisted of a glass slide covered with 1 nm of chromium and 50 nm of gold. Each slide was cleaned by immersion in a 7:3 solution of concentrated sulfuric acid:hydrogen peroxide (30%) for 30 min. The glass portion of the sensor slide was hydrophobized by exposure to 1,1,1,3,3,3-hexamethyldisilazane in an 80 °C oven for 6 h. Upon cooling, the slide was placed into a 1 mM solution of 1-dodcanethiol (Aldrich) in absolute ethanol for 2 h according to a procedure described by Laibinis et al. [55]. Trimethylsilyl (TMS) cellulose (Jena Biosciences, Ltd.) was spread from a solution of 5 mg of polymer in 10 ml of chloroform onto the water surface of a Langmuir–Blodgett (LB) trough (KSV 2000) and transferred accordingly to prepare films 20 layers in thickness. TMS cellulose was removed from the glass side of the sensor slide using chloroform. Cellulose was regenerated by exposure of the film to gaseous wet HCl for 30 s [54].
2.2 Lignin–carbohydrate copolymer synthesis
The lignin–carbohydrate complex was modeled using a high-molecular-weight pullulan (water-soluble polysaccharide) derivatized with abietic acid (Fig. 2). The derivatization involved reacting pullulan with the acyl chloride of abietic acid in the presence of pyridine. The degree of substitution (DS) was approximately 0.027, or one abietic (non-polar) substituent per every 37 anhydroglucose units. The protocol was adopted from the work of Sunamoto et al. [18].

Structures of pullulan and abietic acid. The average degree of substitution used in this study is one abietic acid ester per 37 anhydroglucose linkages.
2.3 Surface-tension measurements
Surface-tension measurements of aqueous pullulan and pullulan abietate solutions were conducted using the tensiometer from a Langmuir–Blodgett trough (KSV 2000). The surface tension was determined by the Wilhelmy plate technique using a sandblasted platinum plate. Each solution was placed in a specially designed glass jar that consisted of an inner cup containing the solution and an outer jacket that allowed for insulation with 22.5 °C water flowing from a circulating thermostated bath.
2.4 Surface adsorption and desorption tests
The docking behavior of pullulan abietate, DS = 0.027, onto a regenerated cellulose surface was investigated using surface plasmon resonance (SPR) spectroscopy. The SPR sensor slide was refractive index-matched to the prism of the Leica AR 700 automatic refractometer using immersion oil . The flow cell body was equipped with a Viton gasket (Dupont Dow Elastomers LLC) and mounted on top of the sensor slide. Solutions were pumped into the flow cell at a flow rate of 0.35 ml min−1 via PEEK tubing (Upchurch Scientific) connected to a cartridge pump (Masterflex). Prior to data acquisition, the cellulose surface was allowed to reach equilibrium swelling by flowing only Milli-Q water through the system. Once a stable baseline was established, solution containing the amphiphile was pumped into the flow cell. Each solution was allowed to flow for 20 min before switching to water via a solvent selection valve. Water was allowed to flow until a new baseline was established, as the signal did not return to the original baseline due to irreversible adsorption of the pullulan abietate. Solutions of higher concentration were run through the flow cell under the same conditions described above. It should be noted that once surface saturation occurs, molecules can adsorb onto material already docked to the surface.
2.5 Surface analysis
Atomic Force Microscopy (AFM) images were obtained for regenerated cellulose, pullulan on cellulose, and pullulan abietate on cellulose. Pullulan and pullulan abietate were adsorbed onto their respective substrates by submersion into solutions at the critical aggregation concentration of each material. Substrates were rinsed with Milli-Q water and dried with nitrogen prior to analysis. Measurements were conducted in tapping mode on a Digital Instruments Dimension 3000 Scope with a Nanoscope IIIa controller using silicon cantilevers and tips.
2.6 Other characterizations
Ultraviolet-visible spectroscopy was used to quantify the degree of substitution of pendant abietate groups on pullulan abietate. Solutions were prepared in a mixed solvent system composed of 44% water and 56% methanol by volume and analyzed using a Varian Cary 50 Bio UV-Visible Spectrophotometer.
3 Results and discussion
The lignin–carbohydrate complex consists of a family of copolymers that varies widely in composition. Discernible gradients exist with regard to the chemical (unit type and concentration) as well as physical (size, molecular weight, degree of crosslinking) structure of this complex across the space between fibers (middle lamella). There is no single copolymer structure that can represent this complex. In order to examine the behavior of hemicellulose-like molecules carrying lignin-like hydrophobic moieties in terms of their self-assembly behavior at cellulose surfaces, a well-characterized pullulan abietate (PA) was synthesized that represents a variant of the cholesteroylated pullulan used by Sunamoto et al. [13–20]. The PA prepared produced an NMR spectrum that was non-descriptive in terms of DS on account of the low abietate content. However, UV-spectroscopy provided clear evidence about the degree of substitution of the diterpene (Fig. 3).
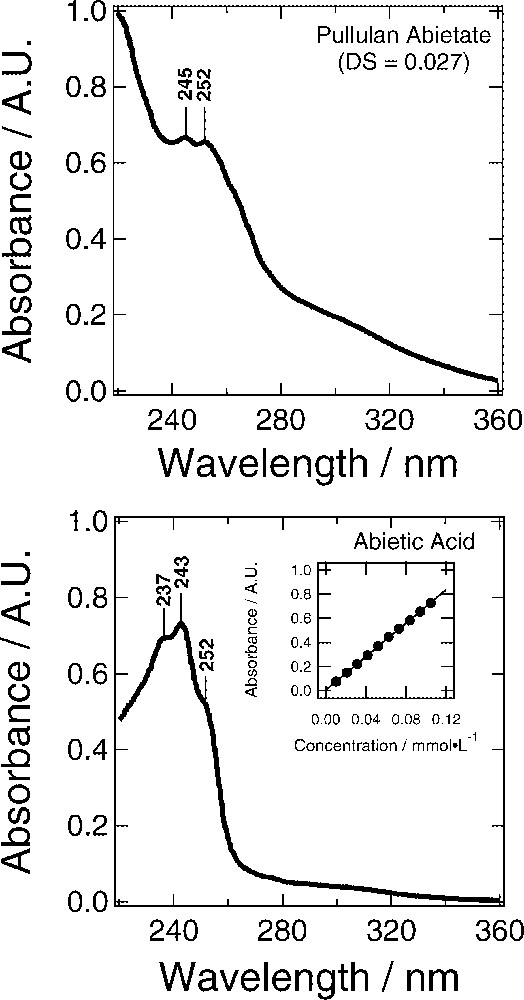
UV spectra of pullulan abietate (574 mg l−1, DS=0.027) and abietic acid (22.1 mg l−1) in a mixed solvent system composed of 44% water and 56% methanol by volume. Underivatized pullulan does not absorb in this region. The calibration curve of abietic acid (inset) provided an estimate for the molar absorptivity (ɛ) of PA at 252 nm, 6928 cm−1 mol−1, which was used to estimate the DS.
The self-assembly behavior of the amphiphilic PA in water was examined by surface tension measurements. It is unlikely that PA forms traditional ‘micelles’ due to the low degree of substitution. This is the reason why the term ‘aggregate’ will be used to describe these self-assembled structures. In Fig. 4, the critical aggregation concentration (cac)-value of PA is compared to un-derivatized pullulan (P). Although the transition from non-aggregated to aggregated solution occurs over a range of concentrations, it is evident that (a) the presence of PA has a more dramatic effect on the surface tension of water, and (b) PA's cac occurs at a significantly lower concentration than that of the corresponding P-control. These observations indicate that the copolymer aggregates more readily than its parent polysaccharide due to the presence of the diterpene substituents.
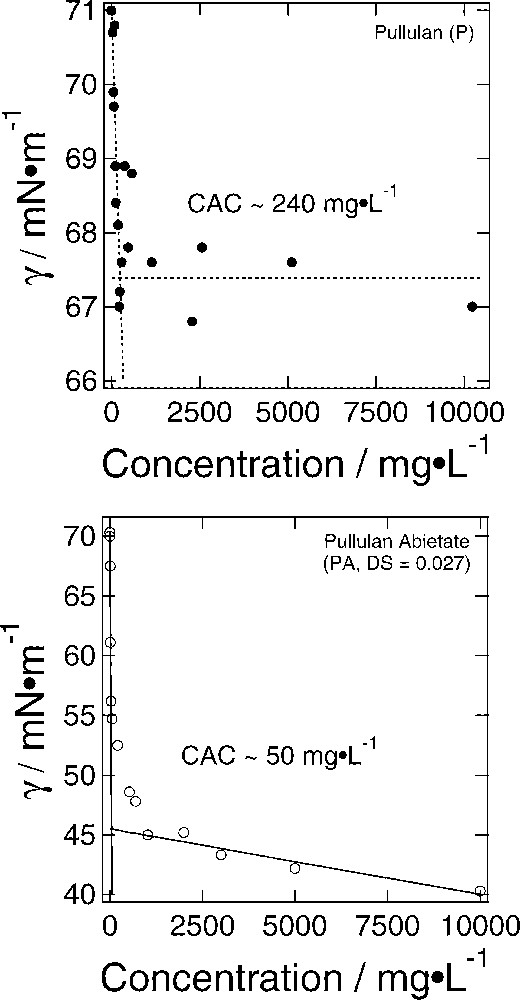
Determination of the critical aggregation concentration (cac) of P and PA using the Wilhelmy plate technique. The copolymer has a lower cac value, 50±5 vs. , indicating a greater tendency to self-aggregate. Error estimates represent ± one standard deviation. In addition, the copolymer impacts the maximum surface tension change of the solution more significantly than the homopolysaccharide.
The aggregation behavior of the PA in the presence of a solid cellulose surface was examined by surface plasmon resonance (SPR) spectroscopy. SPR is capable of detecting the docking event by a single molecule at thin film surfaces [56–60], and it has previously been employed for measuring the relative rate of adsorption of water-soluble oligo- and polysaccharides at cellulose surfaces [37]. The data recorded illustrate that PA adsorbs to cellulose LB film surfaces rapidly, in significant quantities, and remains mostly desorption-resistant (Fig. 5). To test the final feature, flow-through experiments with PA solutions at concentrations below the cac are alternated with those of pure (deionized) water for time periods of . The adsorption/desorption process reveals the build-up of a layer of copolymer at the surface that can not be removed (within the time period examined) by washing with water.

SPR results illustrating the adsorption and desorption process of PA at regenerated cellulose LB surfaces. The SPR sensor is alternately exposed to PA solutions (rise in ) and water (drop in ). The PA solution concentrations are (A) 27, (B) 56, (C) 106, (D) 212, (E) 524, (F) 1024, (G) 2000, (H) 3000, (I) 5000, and (J) 10 000 mg l−1.
The results of this adsorption can be visualized by atomic force microscopy (AFM) (Fig. 6). Comparing the control LB-surface with one that had been in contact with P and one that was present during PA flow (at a concentration equal to the cac), the surface roughness-values revealed significant differences. Whereas surface roughness increased by 40% following contact with P, the roughness rose by 160% when the amphiphilic copolymer was present in solution. Noteworthy is that, with minimal substitution of the PA, the decrease in water's solvent quality caused by a few abietic acid groups is enough to promote the adsorption of the copolymer to a higher degree than the unsubstituted pullulan. This effect suggests that the degree of roughness can be controlled by the degree of substitution of the copolymer with hydrophobic moieties; and hence, the interphase thickness of a composite can be controlled over a significant range. Overall, these results indicate a propensity of cellulose surfaces to adhere spontaneously to carbohydrate-based amphiphiles in an aqueous environment similar to what is found during secondary cell wall synthesis in woody plants. This process is depicted in Fig. 7 with respect to the cac. During initial exposure of the amphiphile to the cellulose surface (a), chains adsorb with a loop-tail conformation. The presence of abietic acid groups drives the deposition of material from the bulk onto exposed abietic acid groups on the surface (b–c). Once a solution, whose concentration is above the cac, is introduced into the system, aggregates begin to form in the bulk (d). These aggregates are also capable of docking to material on the surface (e–f). Due to the inability to desorb pullulan abietate by rinsing with water, this material seems to be adaptable for convenient and practical surface modification of cellulose films and fibers.
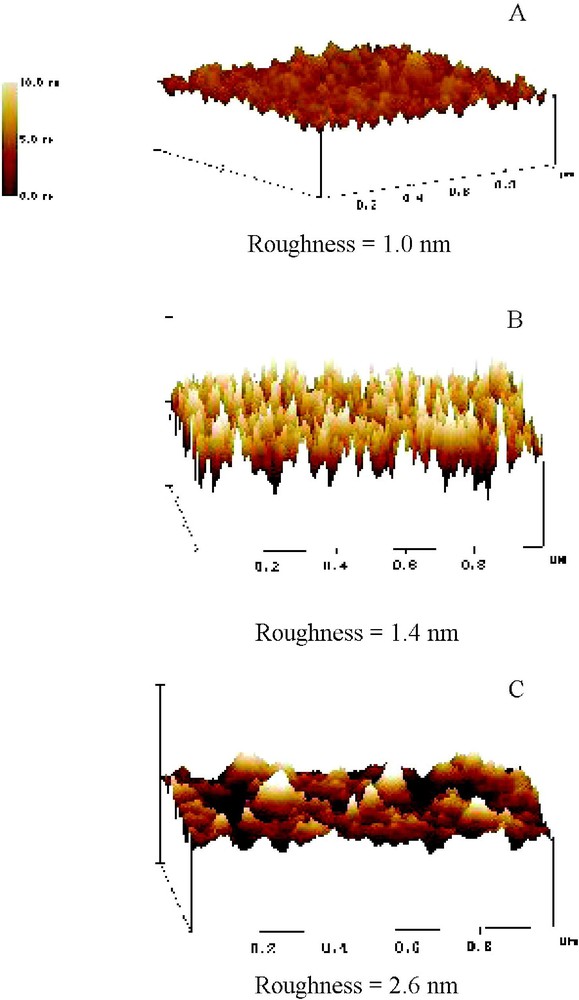
AFM images of cellulose LB-film surfaces before and after adsorption of P and PA: (A) regenerated cellulose before adsorption, (B) after adsorption of P, and (C) after adsorption of pullulan abietate. Note the change in the root mean square surface roughness, reflecting the modification of the surface by adsorption. All images are 1 μm×1 μm.
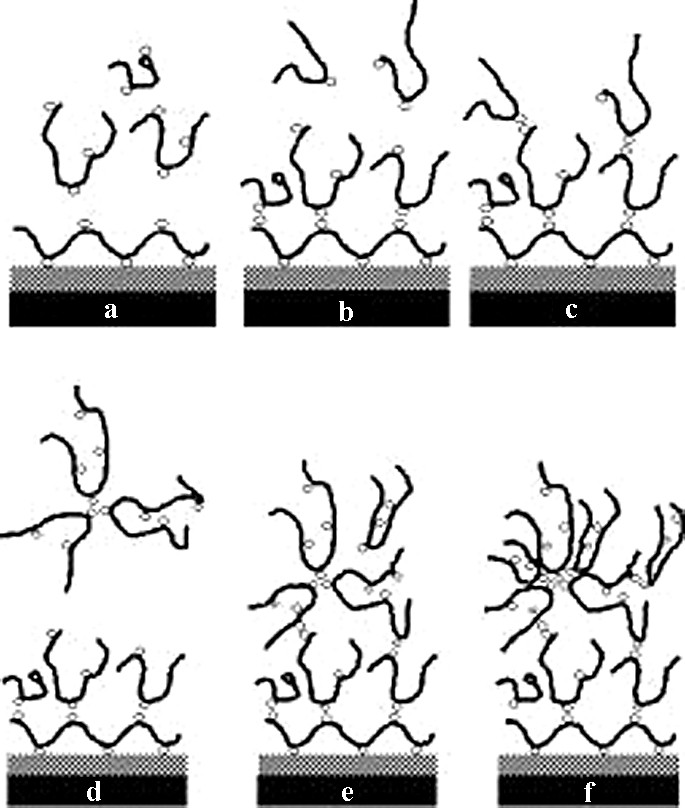
Illustration of the docking process of lignin–carbohydrate-like copolymers at cellulose surfaces. a–c demonstrates adsorption behavior below the cac, and d–f above the cac.
These results support the hypothesis that cellulose surfaces can be modified by the adsorption of self-assembled amphiphilic copolymers based on polysaccharides. If the hydrophobic character of the copolymers were to represent (a) a waxy (thermoplastic) substance, or (b) a phenolic (crosslinkable) entity (rather than a diterpenoid moiety), a fiber would result that would be amenable to thermal or enzymatic consolidation and composite preparation, respectively.
4 Conclusions
Cellulose (fiber) surface modification with cross-linkable, lignin-like copolymers, or thermoplastically deformable copolymers, on the basis of water-soluble carbohydrates, is envisioned as a critical step for the preparation of wood composites by biomimetics. A series of adsorption experiments were carried out that involved the self-deposition of water-soluble pullulan abietate derivatives (as models for a lignin–carbohydrate copolymer complex) on Langmuir–Blodgett films of regenerated cellulose (as models for cellulose fiber surfaces). The results indicated a significant, spontaneous, and apparently desorption-resistant modification of the cellulose surface by copolymer. This type of surface modification is seen as a critical biomimetic step for the creation of stronger composite interfaces involving cellulose fibers.
Acknowledgments
Financial support for this study was provided by the United States Department of Agriculture as part of its NRI program, contract #9902352; and by the National Science Foundation, #CHE-0239633, which allowed for the purchase of the SPR. The authors wish to thank Mr Muhammed Ajaz Hussain, University of Wuppertal, Germany for the preparation of the lignin-carbohydrate copolymer, Dr Ray Dessy and Miss Aysen Tulpar, Virginia Tech for guidance in the field of surface plasmon resonance, and Dr Stephen McCartney, Virginia Tech for assistance with atomic force microscopy.