

1 Introduction
The origin and evolution of modern mitochondria are matters of considerable biological interest. According to the endosymbiotic theory, mitochondria have a unique origin, arising from a symbiont closely related to an ancestral alpha-proteobacteria. This endosymbiont is supposed to have lived in a nucleus-containing host cell – either an amitochondriate eukaryote or an Archea-related cell – between 1.5 and 2 billion years ago when the oxygen content of the atmosphere started to increase [1–7]. Indeed, the obligate intracellular symbiont Rickettsia prowazekii, which belongs to the alpha-proteobacteria group, was previously described as the closest known eubacterial relative of mitochondria identified by the phylogenetic analysis of mitochondrial DNA encoded genes [4]. During the course of evolution, most of the endosymbiotic genes may have been lost or transferred to the nucleus of the eukaryotic host cell [5–9]. Numerous mitochondrial pseudogenes currently present in the modern human nuclear genome attest to the massive and ongoing transfer process of mitochondrial genes to the nucleus in the course of evolution [10–12]. It is believed that because of these complex evolutionary processes, only 13 protein-encoding genes may have persisted in the modern human mitochondrial DNA.
The human mitochondrial proteome is believed to contain over a thousand proteins [13]. However, the size of the mitochondrial proteome differs markedly among species [14]. Recent studies have demonstrated the dual origin of the mitochondrial proteome in yeast; 50–60% of the mitochondrial proteins have homologues in prokaryotic species, whereas 40–50% do not [7,15,16]. The current hypothesis is that these proteins may have been recruited from pre-existing nuclear genes and targeted toward mitochondria [4,15]. The large number of eukaryote-derived genes suggests that numerous specific functions of modern mitochondria were absent in prokaryotic mitochondrial ancestors. Thus, the modern mitochondrial proteome is composed of proteins with a dual eukaryotic and prokaryotic origin, the eubacterial proteins being encoded by two genomes, nuclear, and mitochondrial [17].
We have analysed in silico all the 393 human mitochondrial proteins annotated in SwissProt in order to determine their prokaryotic or eukaryotic affiliation. We have compared the size, the mitochondrial localization, and the function of the proteins according to their origin. Finally, we have considered the implication of these proteins in mitochondrial diseases.
2 Material and methods
2.1 Data selection
A search of the SwissProt database (http://www.ebi.ac.uk/swissprot/) yielded data on 393 human mitochondrial proteins. We classified these proteins according to their mitochondrial function and localization. A search of the NCBI database covering 94 prokaryote species produced data on 256,953 proteins.
2.2 BLAST analysis
The sequences of each of the 393 human mitochondrial proteins were compared with those of the 256,953 prokaryotic proteins using the BLASTP program [18]. We used as the criterion to avoid false positives in establishing similarity.
2.3 Mitochondrial N-terminal targeting prediction
To test the proteins listed without a transit peptide in the SwissProt databank, we used two independent N-terminal mitochondrial-targeting predicting methods: Mitoprot (http://ihg.gsf.de/ihg/mitoprot.html) [19], and TargetP (http://www.cbs.dtu.dk/services/TargetP/) [20]. The putative existence of the N-terminal transit peptide was admitted only when the results of both tests were positive.
2.4 Statistical analysis
Statistical analysis was performed using the Mann–Whitney and Kolmogorov–Smirnov tests. Differences were considered significant at .
3 Results
The BLASTP comparison of the 393 human proteins annotated as mitochondrial proteins in the SwissProt database with 256,953 proteins from 94 prokaryotic species allowed us to identify two distinct groups of human mitochondrial proteins. The first group of proteins with prokaryotic homologues, noted PH+, comprising 253 out of the 393 proteins (64%), included human mitochondrial proteins displaying a high score of homology with one or more prokaryotic proteins (). A more drastic cut-off value () did not greatly affect this result, since the number of mitochondrial proteins with prokaryotic homologues dropped from 253 to 233. The second group of proteins without prokaryotic homologues, noted PH−, contained the remaining 140 proteins (36%) that were non-homologous with any of the known prokaryotic proteins.
The proportion of PH+ and PH− mitochondrial proteins involved in the main mitochondrial functions varied considerably (Fig. 1). Remarkably, 160 out the 166 proteins involved in metabolism belonged to the PH+ group, whereas nine out the 10 mitochondrial proteins involved in apoptosis belonged to the PH− group. The other functional classes were associated with both the PH+ and the PH− groups of proteins, attesting to their dual evolutionary origin. The metabolite carriers were evenly divided in the two groups: 12 were PH+ and 12 were PH−, whereas 12 out of the 13 ion carriers and 15 out of the 20 protein carriers belonged to the PH− group. As expected, a majority of proteins involved in mitochondrial DNA maintenance and expression, i.e. 34 out of the 52 proteins, or 65%, were of prokaryotic origin. Most of the matrix proteins, i.e. 175 out of the 200 proteins, or 88%, were PH+ (Fig. 2). The majority of the inner membrane proteins, i.e. 92 out of the 153 proteins, or 60%, was PH−, as was the majority of the outer membrane proteins, i.e. 17 out of the 27 proteins, or 63%. Out of the seven proteins in the intermembrane space, six were PH+ and only one was PH−.
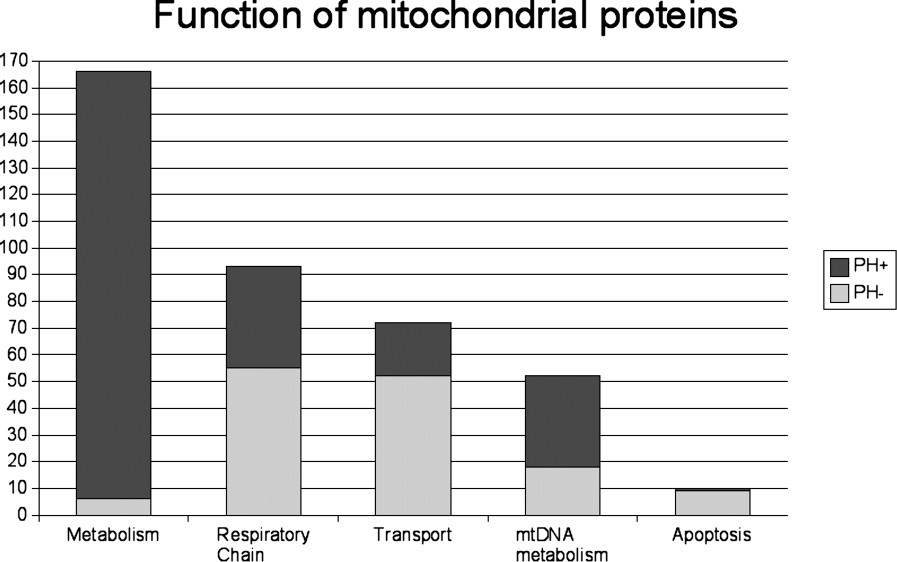
Functional categories of the 393 known human mitochondrial proteins and functions of the proteins according to their eukaryotic (PH−) or prokaryotic (PH+) origin.
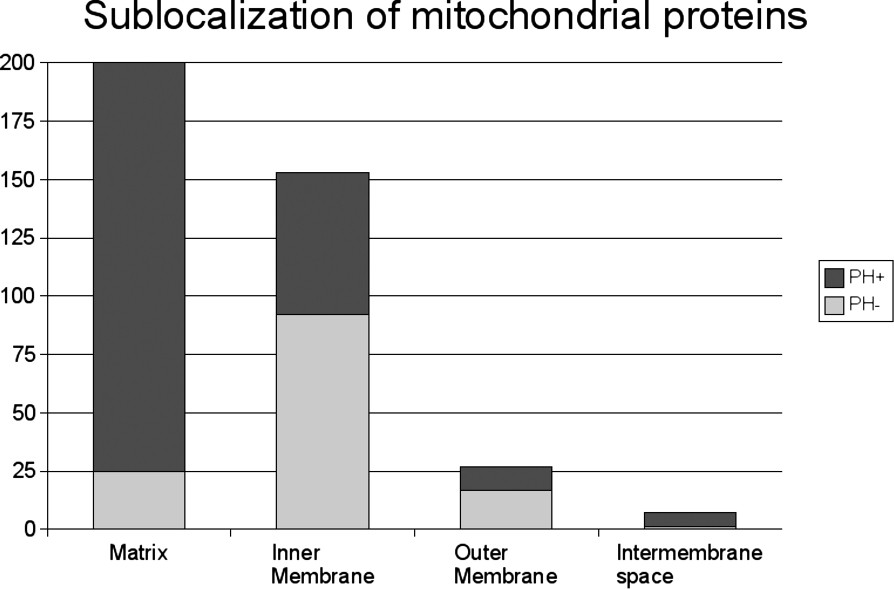
Distribution of the 393 known human mitochondrial proteins according to their localization and sublocalization of the proteins according to their eukaryotic or prokaryotic origin.
Fig. 3 shows the composition of the respiratory chain. Out of the 73 respiratory chain proteins annotated in the SwissProt database, only 25 were PH+. This analysis confirmed the dual origin of complex I subunits that contain 18 proteins known to originate from eubacterial ancestors. These include 7 mitochondrial DNA-encoded proteins and 11 nuclear DNA-encoded proteins. The other 23 proteins were contributed by eukaryotic ancestors. Furthermore, the study of complex IV (cytochrome c oxidase, COX) revealed that all the subunits originated from eukaryotic ancestors, except for the three mitochondrial DNA-encoded subunits known to originate from prokaryotic ancestors.
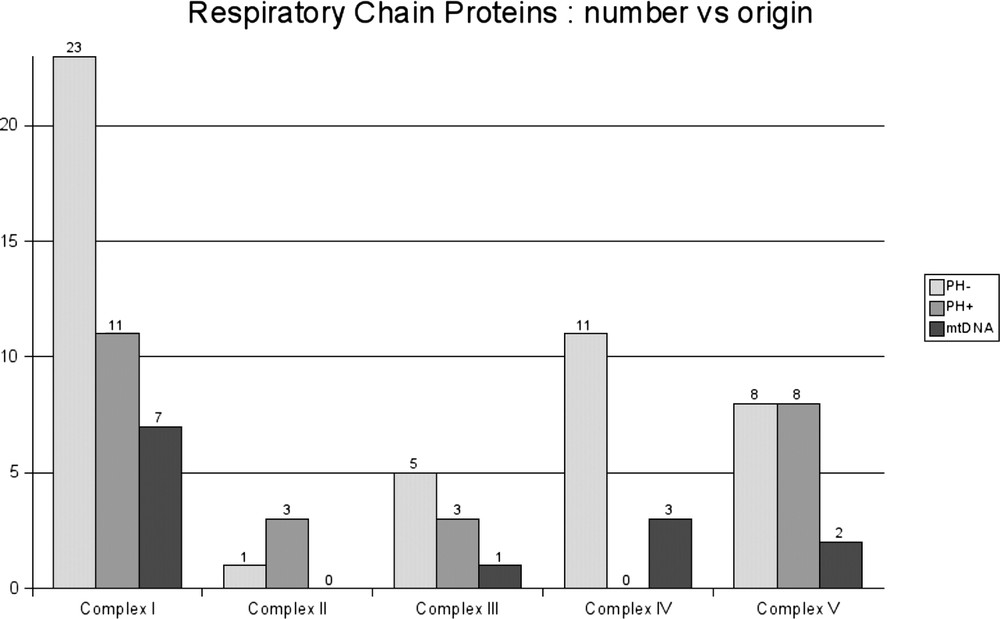
Seventy-three nuclear-encoded human mitochondrial proteins of the respiratory chain according to their eukaryotic or prokaryotic origin. The 13 proteins of prokaryote origin encoded by mitochondrial DNA (mtDNA) are also shown.
The comparison of the size of the mitochondrial proteins according to their origin revealed that the proteins of prokaryotic origin were significantly larger (). The PH+ proteins were made up on average of 435 amino acids (range: 69–1500), whereas the PH− proteins had an average size of 238 amino acids (range: 50–1816). As Fig. 4 shows, this difference was mainly due to the larger size of the PH+ respiratory chain components and transport proteins.
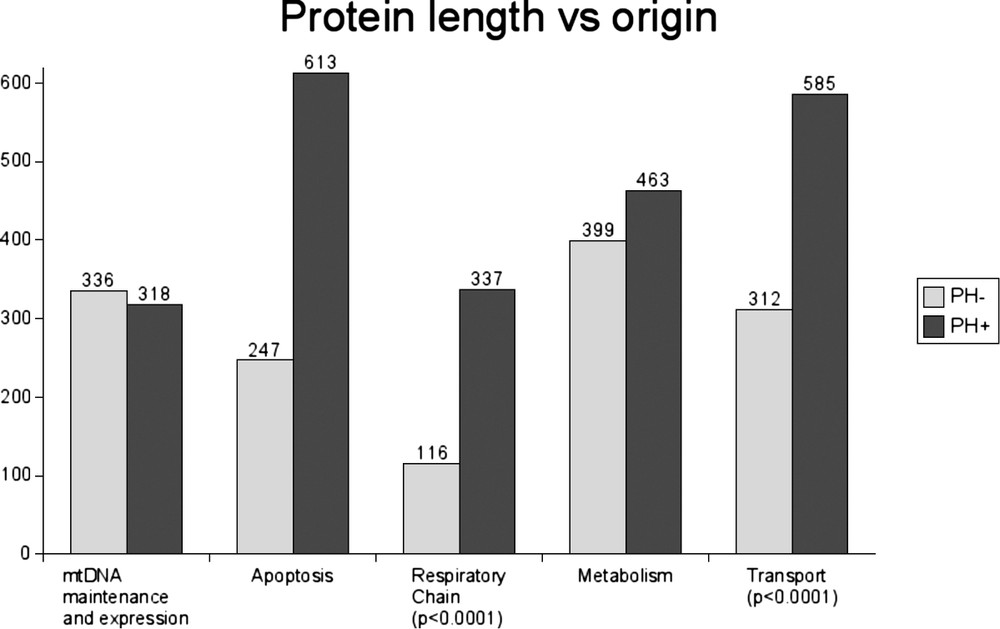
Average protein length of human mitochondrial proteins according to their function and origin.
We found that 223 out of the 253 human mitochondrial PH+ proteins (88%) were significantly larger (average: 435 amino acids, range: 69–1500) than their prokaryotic homologues (average: 400 amino acids, range: 38–1353) (). This difference was mainly due to the presence of a supplementary N-terminal sequence. These additional sequences displayed no homology with prokaryotic proteins and were probably of eukaryotic origin. Thus, most of the PH+ proteins are in fact probably prokaryotic-eukaryotic chimeral proteins. The use of TargetP and Mitoprot software showed that 213 out of the 253 mitochondrial PH+ proteins (84%) either had, or were predicted to have, a mitochondrial-specific N-terminal targeting sequence. In contrast, only 54 out of the 140 mitochondrial PH− proteins (39%) possessed this targeting sequence. A possible explanation is that proteins of eukaryotic origin may have mainly contributed to the membrane compartment in which proteins frequently lack the typical N-terminal targeting sequence.
Lastly, we examined the nuclear-encoded proteins that have been previously linked to human mitochondrial pathology in terms of their evolutionary origin (Table 1). Strikingly, 18 out of the 20 mitochondrial proteins known to be involved in diseases associated with the respiratory chain and the Krebs cycle belonged to the PH+ group, whereas only two belonged to the PH− group ().
Origin of nuclear genes associated with human mitochondrial diseases [29–32]
| Gene | OMIM | Prokaryotic homologue |
| Respiratory chain and Krebs cycle | ||
| NDUFS1 (complex I) | 157 655 | + |
| NDUFS2 (complex I) | 602 985 | + |
| NDUFS3 (complex I) | 603 846 | + |
| NDUFS4 (complex I) | 602 694 | + |
| NDUFS7 (complex I) | 601 825 | + |
| NDUFS8 (complex I) | 602 141 | + |
| NDUFV1 (complex I) | 161 015 | + |
| NDUFV2 (complex I) | 600 532 | + |
| SDHA (complex II and Krebs cycle) | 600 857 | + |
| SDHB (complex II and Krebs cycle) | 115 310 | + |
| SDHC (complex II and Krebs cycle) | 605 373 | + |
| SDHD (complex II and Krebs cycle) | 168 000 | − |
| HUMQPC (complex III) | 191 330 | − |
| BCS1L (complex III assembly factor) | 603 647 | + |
| SURF1 (complex IV assembly factor) | 185 620 | + |
| SCO1 (complex IV assembly factor) | 603 644 | + |
| SCO2 (complex IV assembly factor) | 604 377 | + |
| COX10 (complex IV assembly factor) | 602 125 | + |
| COX15 (complex IV assembly factor) | 603 646 | + |
| FH (Krebs cycle) | 150 800 | + |
| MtDNA maintenance and expression | ||
| TP (thymidine phosphorylase) | 603 041 | + |
| DGUOK (deoxyguanosine kinase) | 251 880 | + |
| TWINKLE (DNA helicase) | 157 640 | + |
| TK2 (thymidine kinase 2) | 251 880 | + |
| DNC (deoxynucleotide carrier) | 607 196 | − |
| ANT1 (adenine nucleotide translocator 1) | 157 640 | − |
| POLG1 (DNA polymerase γ) | 157 640 | − |
| Other mitochondrial functions | ||
| SPG7 (paraplegin) | 607 259 | + |
| FRDA (frataxin) | 229 300 | + |
| ABC7 (ABC transporter) | 301 310 | + |
| DDP1 (deafness-dystonia protein) | 304 700 | − |
| OPA1 (optic atrophy 1) | 165 500 | − |
4 Discussion
We found that 64% of the proteins of the human mitochondrial proteome had prokaryote homologues, whereas 36% of the proteins were non-homologous with existing prokaryotic proteins. Mitochondrial proteins involved in energetic metabolism, biosynthetic metabolism, and mitochondrial DNA maintenance and expression were mainly of prokaryotic origin, while those involved in the transport and control functions originated from eukaryotes. The majority of the matrix proteins originated from prokaryotic ancestors, whereas the proteins of the inner and outer membrane compartments were of eukaryotic origin. Most of the eukaryotic proteins were probably targeted to the endosymbiont in order to develop communication with the host cell (protein, ion and metabolite transport), to regulate ancestral mitochondrial functions (such as ATP production and regulation of membrane potential) and to establish the more recent biological functions (such as apoptosis or androgen synthesis).
Mitochondrial proteins of eubacterial origin are significantly larger than proteins of eukaryotic origin. The difference is particularly striking in the case of proteins involved in membrane transport and the respiratory chain. We hypothesise that the endosymbiont contributed the genes of the large core enzymes of the respiratory chain complexes and that the genes of the more recently evolved accessory proteins of smaller size came from the nuclear genome of the eukaryotic host. This hypothesis is supported by the fact that the three mitochondrial-encoded subunits of complex IV (cytochrome c oxidase) are known to be the largest proteins of the complex. It has been shown in Paracoccus denitrificans that two of these subunits were sufficient to ensure oxygen reduction in water and proton transport [21]. Thus, the other nuclear-encoded subunits (originating from eukaryotic hosts), which may not be necessary to ensure the basic catalytic function of complex IV, might be implicated in the assembly of the complex or the modulation and stabilization of its activity.
Interestingly, we found that 89% of the proteins encoded by nuclear genes involved in respiratory chain deficiencies have prokaryotic homologues. This is an unexpected result, since 59% of the respiratory chain subunits and assembly factors are of eukaryotic origin (38 of the subunits belonging to the PH− group and 55 to the PH+ group). In addition to these nuclear-encoded proteins, two other main categories of proteins of eubacterial origin are involved in human mitochondrial pathologies. The first consists of mitochondrial DNA-encoded proteins, which are also derived from alpha-proteobacteria. The second includes proteins implicated in several mitochondrial diseases associated with matrix metabolic enzyme deficiencies. Our study shows that these proteins are of prokaryotic origin in 96% of the cases. Taken together, these data indicate that the majority of human mitochondrial pathologies may involve proteins of eubacterial origin.
For instance, respiratory complex IV deficiency fits in with this hypothesis. The disorder has been attributed to mitochondrial DNA mutations (3 subunits) or to mutations in the nuclear genes such as SURF1, Sco1, Sco2 and COX10, encoding mitochondrial assembly factors [22–26], all of which were found to have eubacterial homologues in the present study. However, several authors failed to detect any mutations [27,28] in the 10 nuclear-encoded structural subunits of complex IV in other respiratory chain complex deficiencies. Our observation that none of these nuclear-encoded proteins of complex IV has a prokaryotic homologue reinforces our hypothesis of the specific involvement of proteins of eubacterial origin in human mitochondrial pathologies.
Our study indicates that the investigation of the evolution of mitochondrial proteins should lead to a better understanding of mitochondrial diseases. It is in accordance with previous study that showed that 18/100 human mitochondrial proteins that were strongly conserved among eukaryotic species were directly associated with human disease [14]. It should be emphasized that our study bears on the 393 known human mitochondrial proteins, whereas the mitochondrial proteome is estimated to contain over a thousand proteins. Thus, several hundred other proteins remain to be identified and investigated. To date, the nuclear genetic origin of many mitochondrial diseases remains to be identified and numerous genes await analysis. Our results suggest that, considering the implication of mitochondrial proteins in various pathologies, the screening of mitochondrial proteins with eubacterial homologues should be given the attention it deserves.
Acknowledgments
We thank Kanaya Malkani for critical reading of the manuscript. This work was supported by grants from INSERM, the ‘CER Pays de la Loire’, ‘CHU d'Angers’, and the University of Angers (France).