

1 Introduction
Recent progress in the field of microbial genome research has revealed that type-III secretion systems (TTSSs) are found in many Gram-negative bacteria in vertebrate and invertebrate animals, as well as in plants [1]. TTSSs function as systems for the translocation of bacterial proteins into host cells, whereby the physiological functions of host cells are then altered by translocated bacterial proteins [1]. TTSSs are known to play an important role in establishing disease processes, since the virulence of pathogens has been shown to be greatly reduced in TTSS-deficient strains. TTSSs are highly conserved in many mammalian pathogens such as Bordetella bronchiseptica, enteropathogenic Escherichia coli (EPEC), enterohemorrhagic E. coli (EHEC) O157:H7, Salmonella enterica serovar Typhimurium (S. typhimurium), Shigella flexneri, Pseudomonas aeruginosa, Chlamydia trachomatis, Yersinia spp., and some plant pathogens such as Erwinia amylovora, P. syringae, Ralstonia solanacearum, and Xanthomonas campestris [1]. Unexpectedly, TTSSs have also been found in symbiotic bacteria, e.g., Photorhabdus luminescens, Sodalis glossinidius, plant symbiotic rhizobia, and certain ‘non-pathogenic’ bacteria such as P. fluorescens [2]. Although the precise function of TTSSs in symbiotic and non-pathogenic bacteria is not well understood, it has been suggested that TTSSs may be required for adaptation to a host. It is generally accepted that TTSSs carry out one or both of the following functions: (i) they deliver virulence factors in order to occupy host functions and/or (ii) they deliver to the host cell certain modulators in order to establish symbiotic environments without inducing host perturbation and/or death. Molecules delivered by TTSSs perform a number of physiological functions, and are generally referred to as ‘effectors’.
To date, effectors have been identified in various types of bacteria with TTSSs. The host cell perturbation resulting from interactions involving effector-host factors has provided an increased understanding of various disease processes. However, the pathological features triggered by bacteria possessing TTSSs have remained difficult to elucidate, due to the fact that pathogens exploit multiple effectors, i.e., synergistic effects induced by multiple effectors contribute to the disease process. Furthermore, the spatiotemporal virulence properties of effectors in host cells remain to be considered in further detail. Comparative infection studies using wild-type strains and effector mutants will be necessary in order to determine whether or not an effector identified by in vitro experimental study is indeed a bona fide virulence factor.
2 Supermolecular structure of the TTSS
The TTSSs of pathogenic and non-pathogenic bacteria generally share the following features: (i) TTSSs function as an injection machinery that translocates effectors into a host cell; (ii) the expression of TTSSs is induced when bacteria attach closely to a host cell, or when bacteria are grown in vitro under appropriate conditions, i.e., those that mimic the environmental conditions of the host; (iii) TTSSs build pores on the host membrane via the translocation of a pore formation complex into the host membrane. Salmonella, Yersinia, and EHEC are known to possess two types of TTSS, and the timing and localization of effector delivery are precisely regulated by two different machineries in these pathogens.
Analysis using transmission electron microscopy has revealed the supermolecular structures of the TTSSs in human pathogens such as Salmonella [3], Shigella [4,5], and EPEC [6] (Fig. 1A). Such studies have shown that the supermolecular structure of TTSSs is quite similar to that of flagellar hook-basal body complexes, and the TTSS structure is generally referred to as a ‘needle complex’ [3] (Fig. 1B). The needle complex is composed of two distinct parts, with (i) a hollow extracellular ‘needle’ structure, and (ii) a cylindrical basal body similar to a flagellar basal body, which builds a channel extending beyond the two bacterial inner and outer membranes. Effector delivery into the host cell from the bacterial cytosol requires a certain amount of energy, which is generated by ATPase; furthermore, mutation in the gene encoding ATPase has been shown to fail to secrete all TTSS-dependent secreted proteins. ATPase and other components are located directly beneath the basal body, and these components have been observed to form a ‘bulb’ structure in Shigella [5]. Moreover, functional similarities between such bulbs and flagellar C-rings have been reported.
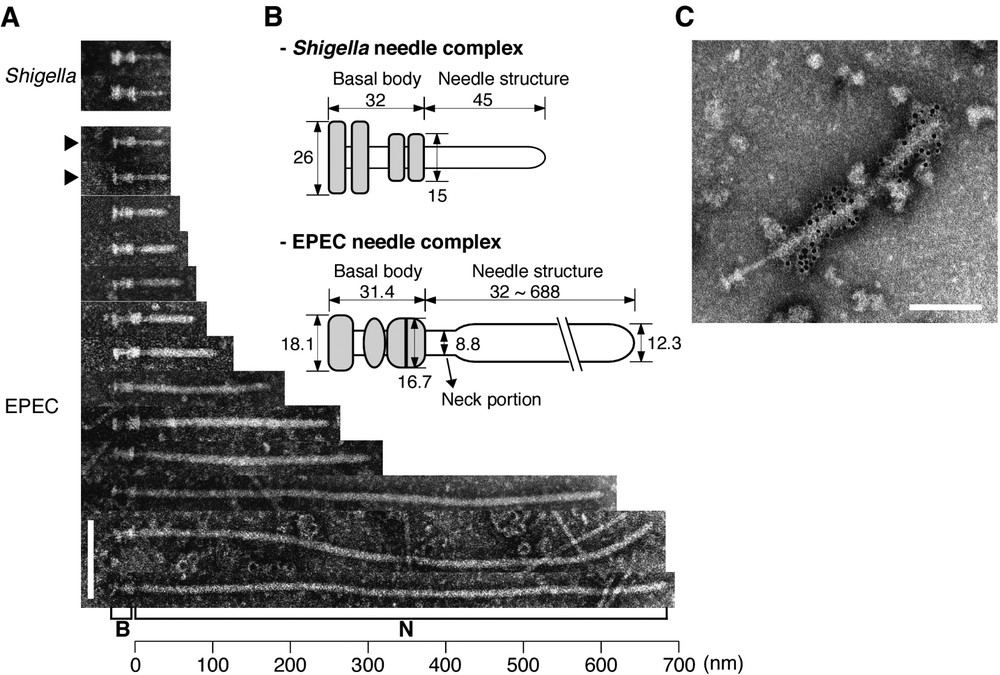
The supermolecular structures of the TTSSs of EPEC and Shigella. (A) Electron micrographs of negatively stained needle complexes from EPEC and Shigella [6]. B and N indicate the basal body and needle structure, respectively. Black arrowheads indicate immature needle complexes without the sheath-like structure. (B) Putative models of the EPEC [6] and Shigella [4] needle complexes are shown, and the original figures are modified and illustrated here. Grey areas indicate basal bodies. Size is represented in nanometers. (C) Direct interaction of the EPEC needle complex with EspA. EspA was detected by immuno-gold-labeled anti-EspA antibodies. The sheath-like structure, but not the needle complex, was covered with 6-nm gold particles. Electron micrographs of Fig. 1A and C were taken from [Proc. Natl Acad. Sci. USA 25 (2001) 11638]. (Bars=100 nm.)
3 Control of needle length
Needle complexes are highly conserved among the pathogens listed above, with the exception of the acquisition of sheath-like structures in both EPEC [6] (Fig. 1A) and EHEC O157:H7 (Sekiya and Abe, unpublished data). Such a sheath-like structures is located at the tip of the EscF needle; this structure is expandable and exhibits variable length, ranging between (average length: 93 nm) [6]. Electron micrograph study using anti-EspA antibody has shown that the sheath structure is composed of EspA, which is secreted via the EPEC/EHEC TTSS (Fig. 1C). The length of the sheath appears to be regulated by the amount of EspA, and sheath length has been shown to increase markedly in association with the overexpression of EspA in EPEC [6]. In contrast, no such sheath-like structures are found in either Salmonella (SPI-1) [3] or Shigella [4] needle complexes, and in these bacteria, needle length appears to be fixed in the wild-type strains of both types of bacteria under in vitro culture conditions, i.e., 80 or 45 nm. In the case of Shigella, the needle portion is composed of MxiH. When the needle complex was prepared from Shigella that overexpresses MxiH, needle elongation from 200 to 1000 nm was observed [4]. Moreover, similar results have been obtained using the Spa32 defective mutant strain of Shigella [7]. Spa32 is known to be secreted into the culture supernatant via the Shigella TTSS [7], and its homologue, InvJ, has also been identified in Salmonella; in addition, needle elongation has been observed in an InvJ-deficient strain [8]. Although the mechanisms affecting needle length remain unclear, Journet et al. have proposed the fascinating hypothesis [9] that the needle length in Yersinia depends on the molecular size of the YscP protein, and the authors proposed that YscP regulates needle length by acting as a ‘molecular ruler’ during the stepwise assembly of the needle structure of the TTSS (Fig. 2). Deletions affecting both the N- and C-termini of YscP were shown to result in a loss of length control, suggesting that the two ends of the YscP protein may function as anchors for the TTSS. One end of YscP apparently binds to the basal body, whereas the other end binds to the growing tip of the needle. When the needle reaches its mature length, YscP is fully stretched, and the secretion apparatus responds to a signal from the YscP internal anchor, thus resulting in a termination of the export of needle components; this signal might also trigger the secretion of both translocators and effectors. Interestingly, a strict linear relationship has been observed between needle length and the number of amino acids in YscP, with 1.9 Å per amino acid residue of YscP [9]. Although the YscP protein shares no substantial sequence identity with either Spa32 or InvJ, their encoding genes are located in similar positions in the respective TTSS gene clusters; moreover, YscP is a secreted protein, as are both Spa32 and InvJ.

Assembly and regulation of the TTSS. A model of the regulation of needle length is shown [9], and the original figure is modified and illustrated here. The type-III secretion system is composed of a needle structure and a cylindrical basal body. YscP in Yersinia acts as a molecular ruler during the stepwise assembly of the needle structure of TTSS. The N- and C-terminal domains of YscP protein may function as anchors for the TTSS. When the needle reaches its mature length, YscP is fully stretched, and the secretion apparatus responds to a signal from the YscP internal anchor, resulting in the termination of the export of needle components. OM, outer membrane; PP, bacterial periplasm; IM, inner membrane.
4 Delivery of translocators into the host membrane
There are two types of secreted proteins, effectors and pore-forming factors, both of which are delivered into the host cell via TTSSs (Fig. 3). The delivery of the pore-forming factors into the host membrane is a prerequisite for the translocation of effectors into the host cell. The pore-forming factor, which is referred to as a ‘translocator’, is delivered and translocated into the host membrane, and then builds a physical pore on the host membrane. The bacterial effectors are then able to translocate into host cells via the TTSS and the translocator-mediated pore. TTSS-deficient strains result in the abolishment of effector and translocator secretion. However, mutant strains of translocator proteins have been shown to retain the ability to secrete both effectors and other translocators into the bacterial culture supernatant, although such mutations have not demonstrated the ability to translocate effectors into host cells. BopB–BopD in Bordetella, SipB–SipC in Salmonella, IpaB–IpaC in Shigella, EspB–EspD in EPEC/EHEC, and YopB–YopD, LcrV in Yersinia are involved in the translocator complex in these strains. Approximately 40% sequence similarity between SipB/IpaB/EspD/YopB and most of the translocator proteins containing one or two putative trans-membrane domains; these components appear to build a continuous channel that connects with the TTSS, since EspA protein for the sheath-like structure is associated with EspB protein for the translocator, as determined by binding assay [10]. The ability to form pores on the host membrane can be evaluated based on hemolytic activity. When red blood cells (RBCs) are infected with bacteria possessing a TTSS, hemolysis is induced by the translocator-mediated membrane pore on RBCs [6,11–13]. This hemolytic activity is often used in the process of characterization in order to determine whether or not a newly identified secreted protein is a translocator.

Effector translocation into host cells via a TTSS. Pathogenic bacteria build physical bridges between themselves and host cells by using a TTSS. First, the type-III secretion machinery is expressed in response to environmental stimuli. Second, translocators such as pore-forming factors are translocated into the host cell; these factors then localize on the host cell membrane, resulting in the formation of a pore complex. Finally, effectors are able to translocate into the host cell via the bacterial-mediated bridge and pore.
5 Secretion signal of effectors and TTSS-specific chaperones
Proteins that are regulated by a type-II secretion system are secreted through the bacterial inner and outer membranes to the extracellular space by following mechanism. (i) The secreted protein contains an N-terminal signal sequence that is recognized by the Sec system localized in the bacterial inner membrane and is cleaved during translocation into the periplasm from the bacterial cytosol. The signal sequence consists of ∼30 hydrophobic residues and can be predicted by computer analysis due to its highly conserved region. (ii) The mature (shortened) protein translocated into the periplasm is secreted into the bacterial culture via the outer membrane channel of the type-II secretion system [14]. A twin arginine translocation (Tat) system is also involved in the translocation of bacterial proteins into the periplasm [15,16]. Thus, Tat as well as Sec is now known to be coupled with the type-II secretion system.
Secretion signals of type-III effectors are thought to be contained in (i) the 5′-region of the mRNA [17,18], (ii) the N-terminus of the secreted protein [19,20], and/or (iii) they may possess the ability to carry out chaperone binding to the secreted protein prior to secretion [19,21]. Deletion analysis has revealed that the ∼15 residues of N-termini of Yersinia Yops (YopE, YopN, and YopQ) are required for secretion into a bacterial culture supernatant. However, the lack of a consensus sequence among effectors in this region remains unexplained. Frame-shift mutations that alter the amino acid sequence of the ∼15 residues of N-termini Yop (YopE, YopN, and YopQ) have not been shown to affect secretion, a finding that has led to the mRNA signal hypothesis [17,22]. However, this hypothesis remains controversial, as common features among the N-terminus of the YopE effector have been reported. The replacement from 2 to 8 amino acid residues in the YopE N-terminus with all possible permutations of synthetic serine/isoleucine sequences (resulting in 128 mutants) demonstrated that an amphipathic N-terminal sequence, including four or five serine residues, had a particular secretion phenotype. In contrast, YopE secretion was either greatly reduced or completely abolished with the replacement of 2 to 8 N-terminal residues with a hydrophobic or a hydrophilic amino acid sequence [20]. Although the precise secretion signal for TTSSs remains unknown, it does appear to exert a universal function; for example, the translocated intimin receptor (Tir), an EPEC effector, was shown to be secreted in Shigella by use of its TTSS [23]. Although the secretion signal is thought to be limited to the N-terminal residues, the translocation signal, which is required for the translocation of an effector into a host cell, is located in the 50–75 N-terminal residues [24,25]. Unlike type-II secreted proteins, neither the secretion nor the translocation signal sequences of TTSSs are cleaved upon secretion.
Molecular chaperones interact transiently with one or more partners to prevent degradation and/or incorrect molecular interactions in bacterial cytosol. Most chaperones for the type-III effectors have only one or two cognate effector(s), and such chaperones are not secreted and released upon secretion from those cognate effectors. The TTSS chaperones possess certain conserved features in common with small (), acidic proteins (pI<5), and they are thought to have an amphiphilic α helix in the C-terminus. To date, the TTSS chaperones have be divided into two classes as follows: (i) class-I chaperones interact with one (class IA; e.g., the SycE chaperone interacts with YopE in Yernsinia) or several (class IB; e.g., Spa15 interacts with IpaA, IpgB1, OspC3, and OspB in Shigella) effectors; and (ii) class-II chaperones interact with translocators (e.g., SycD (LcrH) interacts with both YopB and YopD translocators in Yersinia).
Class IA chaperones bind to effectors by recognizing the chaperone binding domain that is located among the 50–150 N-terminus residues of effectors. Crystallographic structure analyses of SicP and SigE in Salmonella, SycE in Yersinia, and CesT (which has also been categorized under class IB due to its interaction with Map effector as well as Tir [26]) in EPEC have confirmed that the class IA chaperones function as dimers [27–30]. Interestingly, YopE, SigD, and Tir exhibit similar enzymatic or physical properties in the presence or absence of their cognate chaperones. The complex of the YopE effector with its cognate SycE chaperone has been found to be catalytically active, and the Rho GAP activity of SycE–YopE is similar to that of uncomplexed YopE [21]. Similar property have also been obtained with EPEC CesT–Tir and Salmonella SigE–SigD TTSS chaperone–effector complexes [31]. Such findings suggest that the binding of chaperones to a certain region of their cognate effectors may not affect the global anti-folding activity on the entire effector. Recent crystallographic studies of two effectors, Salmonella SptP and Yersinia YopE, in complex with their respective chaperones, have revealed that these effectors are quite similar to each other, although the overall sequence similarity among the chaperone binding domains is poorly conserved [21]. Such findings have provided evidence for the generality of TTSS targeting by chaperones; moreover, such results have indicated that the targeting signal is encoded in the three-dimensional structure of the chaperone–effector complex.
6 Regulation and switching of effector/translocator secretion
In EPEC, the expression of both flagella and TTSS structures is precisely regulated by environmental signals. We have revealed that when the environmental stimulus is shifted to an in vivo conditional medium such as Dulbecco's modified Eagle's medium (DMEM) from a rich in vitro medium such as LB medium, the supermolecular structures on the bacterial cell surface of EPEC change drastically [6]. These observations indicate that EPEC undergoes two different phases: (i) a planktonic phase, in which flagella are used to aid in bacterial movement; and (ii) a virulent phase, in which the production of flagella is repressed, and a TTSS is produced instead, to be used for the translocation of the effector into a host cell. Thus, EPEC is able to assess environmental conditions; host-body temperature is possibly one type of signaling in this regard. Indeed, the secretion of translocators is finely tuned to the host-body temperature, and the maximal secretion of translocators of human EPEC has been shown to occur when the temperature of the culture media was adjusted to simulate human body temperature () [32]. It is noteworthy that the maximal expression of translocators of rabbit EPEC (REPEC) occurred when the REPEC culture conditions were adjusted to rabbit body temperature (). Thus, pathogens can respond to host-body temperature in order to exert full virulence, and this may provide an explanation for host tropisms.
A recent study revealed that switching between effector/translocator secretion is regulated by certain bacterial factors. Mutations of both SepL and Rorf6 in EPEC have been shown to inhibit the secretion of detectable translocators such as EspA, EspD, and EspB, even though the translocators were produced in the bacterial cytosol, as is the case in wild-type EPEC [33]. In contrast, both SepL and Rorf6 defective mutants exhibited a markedly enhanced secretion of the Tir effector. These results suggest that SepL and Rorf6 may act as ‘molecular switches’ that control the secretion of both translocators and effectors, in response to the environmental conditions. When a physical bacterial bridge (TTSSs and pore formation on the host membrane) is constructed between bacteria and host cells, the secretion of translocators may be down-regulated by these molecular switches, and instead, effector secretion may increase due to this signaling process.
The expression of effectors is not exclusively regulated by pathogen-specific regulators. The EPEC and EHEC TTSSs are regulated by integrative host factor (IHF) [34], nucleoid-associated protein, Fis [35], and the quorum sensing system (QSS) [36,37]. The QSS is a bacterial communication system that utilizes secreted chemical signaling molecules that are referred to as autoinducers (AIs) [38]. The QSS collectively governs a population of bacteria in order to control gene expression; bacterial dynamics are thus shifted into a mode of synchronized behavior as a bacterial community, starting from the planktonic phase. By using the QSS, bacteria can assess population density by detecting a particular AI, the concentration of which is correlated with bacterial density. This ‘cross-talk’ in turn permits the bacterial group to control the expression of specific genes only at particular population densities. An AI is produced in many species of bacteria, and AIs are able to activate the expression of a subset of genes when the microbial population reaches a crucial level. The QSS regulates the expression of the EPEC TTSS, as well as the expression of the flagella [37]. Normally, EHEC infects humans at very low infection doses (10–100 organisms). Although it is difficult to use own AIs such low density of EHEC, a hypothetical model has been proposed, in which EHEC detects AI signals produced by the intestinal bacterial flora such as commensal E. coli and other enterobacteria [36]. In response to an AI signal, the expression of major virulence factors, including type-III effectors and Shiga toxin, is induced [39]; such synergistic effects of virulence factors induced by the QSS are required in order to exert full virulence with such a small number of EHEC cells. Furthermore, mammalian hormones, such as adrenaline and noradrenaline, appear to function as AI molecules, resulting in activation of the EHEC QSS [36]. Thus, EHEC can assess the in vivo environmental condition by the QSS in response to the eukaryotic hormone.
7 Contribution of type-III effectors to bacterial pathogenesis
To date, various effectors have been discovered among numerous pathogenic bacteria (see Table 1). Some bacteria have been found to exploit and/or alter host factors and cytoskeletal components in order to establish proliferation, invasion, modulation of vesicular traffic, and escape from autophagy (Fig. 4). Salmonella has the ability to invade intestinal epithelial cells that do not engulf bacteria in a normal manner. Salmonella-induced macropinocytosis in epithelial cells is dependent on the TTSS. S. typhimurium possesses two TTSSs, each of which is used at a different stage of infection. One of these TTSSs is encoded in Salmonella pathogenicity island 1 (SPI-1), and it governs the first event of contact between Salmonella and an epithelial cell, leading to bacterial internalization. The other type of TTSS, encoded in Salmonella pathogenicity island 2 (SPI-2), is required to elicit systemic infection. Salmonella can internalize into non-phagocytic epithelial cells by temporal regulation of the Rho family GTPases and these events are governed by a subset of SPI-1 effectors.
Effectors delivered via TTSSs
| Bacterium | Effector | Natural ligand ⁎a | Activity and function | Reference |
| Salmonella | SopE, SopE2 | Rac1, Cdc42 | GEF for Rac1 and/or Cdc42. Induction of membrane ruffling. | [41,43] |
| SptP | Rac1, Cdc42 | RhoGAP for Rac1 and Cdc42. Host cell recovery. | [42] | |
| SopB/SigD⁎b | Phosphatidylinositol phosphate | Inositol phosphatase activity. Induction of membrane ruffling and macropinocytosis. Modulation of vacuole traffic. Anti-apoptotic activity. | [47,48,62] | |
| SipA | Actin | Inhibition of depolymerization of actin filaments. Induction of membrane ruffling. | [63] | |
| SipB | Caspase-1 (ICE) | Autophagy-mediated programmed cell death in macrophages. Pore-forming factor. | [64–66] | |
| SipC | Actin | Actin bundling activity at the N-terminus. Actin nucleation activity at the C-terminus. Induction of membrane ruffling. Pore-forming factor. | [67] | |
| SpiC | TassC (NIPSNAPhomologue) | SPI-2 effector. Inhibition of phagosome–lysosome fusion. | [68,69] | |
| SifA | ? | SPI-2 effector. Survival and replication of Salmonella in macrophages. | [70–72] | |
| Shigella | VirA | Tubulin | Promotion of Rac1 activity by microtubule destabilization. Induction of membrane ruffling. | [58] |
| IcsB | VirG (bacteria) | Escape from autophagy. | [49] | |
| IpaA | Vinculin | Induction of F-actin depolymerization. Induction of membrane ruffling. | [73] | |
| IpaB | Caspase-1 (ICE),CD44, IpaBCDcomplex binds to-integrin | Macrophage death by induction of apoptosis. Induction of actin polymerization. Pore-forming factor. | [74–78] | |
| IpaC | ? | Promotion of Rac1 and Cdc42 activities. Induction of membrane ruffling. Pore-forming factor. | [74,78,79] | |
| IpgD | PI-4,5-P2 | Inositol 4-phosphatase activity. Induction of membrane ruffling. | [80] | |
| enteropathogenic E. coli(EPEC)enterohemorrhagicE. coli (EHEC) | Tir | Nck (for EPEC) | Receptor for bacterial outer membrane protein, intimin. Activation of actin nucleation beneath the adherent bacteria. | [81] |
| EspG, Orf3⁎c | Tubulin | Promotion of RhoA activity by disruption of microtubule network. | [50] | |
| EspF | Cytokeratin 18 | Induction of disruption of intermediate filaments. Tight junction disruption. | [82] | |
| EspFU (TccP)(for EHEC) | N-WASP | Adapter protein between Tir and N-WASP. Nck-independent actin assembly. | [83,84] | |
| Map | ? | Tight junction disruption. | [85] | |
| Cif | ? | Inhibition of cell cycle G2/M transition. Formation of actin stress fibers | [86] | |
| Yersinia | YopE | RhoA, Rac1, Cdc42 | RhoGAP for multiple Rho GTPases. Disruption of actin filaments and inhibition of bacterial uptake into host cells. | [87] |
| YopH | p130Cas, FAK | Protein tyrosine phosphatase. Inhibition of bacterial uptake into host cells. | [88] | |
| YopO/YpkA⁎d | RhoA, Actin | Serine/threonine kinase. Down-regulation of RhoA activity. | [89] | |
| YopP/YopJ⁎e | ? | Cysteine protease. Inhibition of MAPK and NF-κB. | [90] | |
| YopT | RhoA, Rac1, Cdc42 | Cysteine protease that cleaves RhoA GTPases. Disruption of actin cytoskeletons. | [91] | |
| Pseudomonasaeruginosa | ExoS, ExoT⁎f | RhoA, Rac1, Cdc42 | RhoGAP domain at the N-terminus, resulting in disruption of the actin cytoskeleton. ADP-ribosyltransferase domain at the C-terminus. ExoS induces apoptosis. | [92,93] |
| ExoU | ? | Phospholipase. Induction of lung injury. | [94] | |
| ExoY | ATP | Adenylate cyclase. Induction of cytotoxicity. | [95] | |
| Chlamydia | Tarp | ? | Recruitment of actin to the site of attachment. Formation of pedestal-like structure. | [96] |
⁎a : Host factors are described;
⁎b : SopB and SigD are found in S. dublin and S. typhimurium, respectively, and the functions of both effectors are identical;
⁎c : EPEC possesses both EspG and Orf3, which share functional similarities, and EHEC possesses only EspG;
⁎d : YopO is found in Y. enterocolitica, YpkA is found in both Y. pseudotuberculosis and Y. pestis, and the functions of both effectors are identical;
⁎e : YopP is found in Y. enterocolitica, YopJ is found in both Y. pestis and Y. pseudotuberculosis, and the functions of both effectors are identical;
⁎f : although ExoS and ExoT are functionally similar effectors, their precise functions differ to some extent [93].
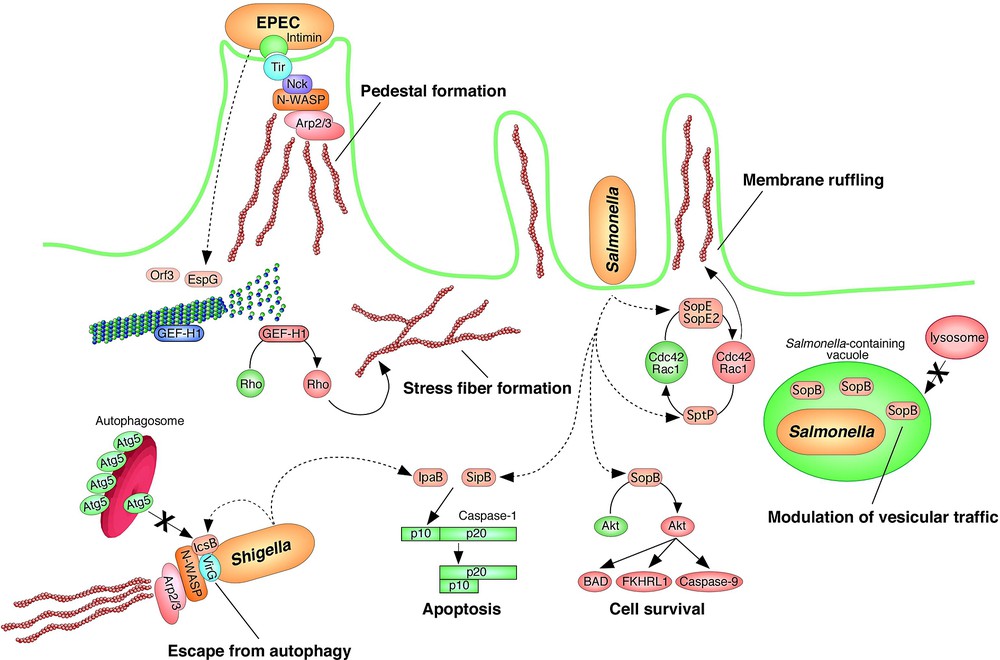
Perturbation of host homeostasis by effector translocation. Effectors that are translocated into host cells exert various physiological effects on host cell signaling, including the perturbation of members of the Rho protein family, the reorganization and/or disruption of the host cytoskeleton, the modulation of vesicular traffic and/or autophagy, and the induction of programmed cell death and/or anti apoptotic activity.
The Rho family of guanine nucleotide binding proteins consists of the RhoA, Rac1, and Cdc42 subfamilies [40]. The Rho family proteins alternate between a GDP-bound (inactive) state and a GTP-bound (active) state. In the active (GTP) state, these GTPases relay signals from growth factors, cytokines, and adhesion molecules in order to control the host signaling involved in cell growth, morphogenesis, cell motility, cytokinesis, and the trafficking and organization of cell cytoskeleton components. The Rho family proteins are exquisitely regulated by three different regulatory proteins: (i) guanine nucleotide exchange factors (GEFs); (ii) GTPase activating proteins (GAPs); and (iii) guanine nucleotide dissociation inhibitors (GDIs). GEFs catalyze the exchange of GDP for the GTP-bound form by promoting the release of GDP. GAPs promote the intrinsic GTP hydrolyzing activity of Rho proteins, resulting in an enhancement of their conversion to the GDP-bound (inactive) form. GDIs preferentially associate with the GDP-bound form and they prevent the spontaneous and GEF-catalyzed release of nucleotides, thereby maintaining the Rho family proteins in the inactive state. The active forms of any of the three subfamilies of RhoA, Rac1, and Cdc42 will induce actin cytoskeletal rearrangement, including the formation of (i) actin stress fibers and focal adhesions (RhoA), (ii) membrane ruffles and lamellipodia (Rac1), and (iii) spikes and filopodia (Cdc42).
7.1 Unwelcome guests: bacterial internalization
The two known Salmonella effectors, SopE [41] and SptP [42], possess the ability to modulate the cytoskeletal organization of the host cell by exploiting regulators of the Rho family proteins (Fig. 4). Contact between Salmonella and a host epithelial cell induces the upregulation of Cdc42 and Rac1 by two highly related bacterial GEFs, SopE (for both Cdc42 and Rac1) and SopE2 (for Cdc42), resulting in bacterial uptake into the host cell as a consequence of actin cytoskeletal rearrangements and profuse membrane ruffling [41,43]. Membrane ruffling is required for the invasion of Salmonella into the non-phagocytic epithelial cell, a step known as bacterial-mediated endocytosis (BME). After bacterial internalization into the epithelial cell is complete, Salmonella triggers a reversal of the cellular morphological changes induced by the SopE and SopE2 effectors, and the host cells recover their normal cellular morphology. In other words, Salmonella possesses the ability to restore normal cellular morphology and architecture by the translocation of an additional effector, SptP [42], which antagonizes the activities of SopE and SopE2. When cells are infected with wild-type Salmonella, profuse membrane ruffling is induced within 10–30 min. Interestingly, such morphological changes are only temporary, and the host cell appears to once again exhibit the normal actin cytoskeletal structure 2–3 h after infection, although a large number of bacteria remains in the host cell. In contrast, cells infected with an SptP mutant strain do not recover normal morphology, even 3 h after infection. Analysis of SptP has revealed that this molecule acts as a bacterial GAP that inactivates Cdc42 and Rac1 by promoting the hydrolysis of GTP to GDP. Indeed, SptP shows sequence similarity with the consensus sequence of the Rho family GAP, and mutation in a critical arginine residue in this motif results in the loss of in vitro SptP GAP activity [42]. Thus, SptP exhibits the ability to reverse the effects of both SopE and SopE2 effectors.
Temporal regulation and/or the sequential delivery of a reversal function of effector may be required to establish the internalization of Salmonella, but the exact mechanism of this process has long remained unclear. However, Kubori and Galan have shed light on this issue [44]. In their study, comparable levels of translocated SopE and SptP were observed shortly after infection. Although the levels of the bacterial GEF, SopE, were markedly reduced with the passage of time and the SopE levels were found to be below the level of detection ∼30 min following infection, the level of SptP decreased very slowly after infection. This difference in SopE and SptP levels is now known to be due to differences in the respective degradation patterns, achieved by a proteasome-dependent pathway. These findings are supported by the following results: (i) proteasome inhibitors inhibit SopE degradation, resulting in sustained levels of this effector; (ii) ubiquitination of translocated SopE is immediately detected after infection; and (iii) although SptP is also degraded by proteasome, the kinetics of this process are much slower than those of the process involving SopE. These results therefore indicate that differences in the stability of these two effectors that perform opposed functions are responsible for the regulation of the Salmonella-programmed internalization into host cells.
7.2 Molecular mimicry: hiding and perturbation of host organs
In mammalian cells, phagocytosis is essential for homeostasis, including such functions as tissue remodeling/clearance and the host defense against invasive bacteria and viruses. Phagocytosis consists of several continuous steps, namely, (i) an initial step of binding to and recognition of a particle (pathogen) by numerous mammalian cell surface receptors; (ii) the induction of phagocytic receptors, which triggers the subsequent host signal-transduction events by the formation of an actin-rich membrane protrusion around the pathogen; (iii) the enclosure of the pathogen as a consequence of membrane fusion, which results in phagosome formation, and which in turn is the prerequisite for the maturation of a phagolysosome. Pathogens inside the phagolysosome are killed by low acidity and hydrolysis, and as a result of attacks by the following agents: radicals, lysozyme, and defensin. As a result of phagocytosis, pathogens are degraded, and the resulting pathogen-derived molecules are translocated as antigens to the host cell surface for antigen presentation. In addition to phagocytosis, autophagy is involved in the elimination of bacteria such as group A Streptococcus (GAS) [45] and Mycobacterium tuberculosis [46]. Autophagy involves the formation of double-membrane structures referred to as autophagic vacuoles, and their contents are eventually degraded in lysosomes. Both phagocytosis and autophagy are involved in the elimination of pathogens. However, certain intracellular pathogens inhibit phagosome formation and/or are able to escape from autophagy. Thus, pathogens can modify their environmental conditions by calling upon a number of different strategies of escape from the innate host defense system, and these pathogens are thus able to exploit certain type-III effectors using the mechanisms outlined below.
By modulation of the Rho family proteins, Salmonella can gain entry into epithelial cells. After internalization, this pathogen is able to replicate and escape from the host defense system. Interestingly, the intracellular replication of Salmonella at a certain niche is regulated by an SPI1 effector, SopB [47]. SopB is a phosphoinositide phosphatase and contributes to maintaining high levels of phosphatidylinositol-3-phosphate in the membranes of Salmonella-containing vacuoles (SCVs). However, a SopB mutant strain is known to be unable to elicit the maturation of SCVs, resulting in the failure of Salmonella to replicate in epithelial cells [47]. Furthermore, SopB is also required for the activation of the serine-threonine kinase Akt, also known as protein kinase B (PKB) [48]. Akt is a regulator of apoptosis in normal intestinal epithelial cells. SopB has been shown to possess anti-apoptotic activity by the sustained activation of Akt and inhibition of caspase-3 cleavage [48]. In contrast, it is known that both SipB and SpvB have pro-apoptotic activities. Thus, Salmonella can selectively and temporally regulate macrophage or epithelial cell events by the modulation of programmed cell death. In general, SopB is known to be involved in the maintenance of SCVs and anti-apoptotic activity.
Like Salmonella, Shigella is an invasive bacteria that multiplies in host cells. Shigella has the ability to escape from the autophagic response by exploiting a type-III effector, IcsB [49]. An IcsB mutant strain has been shown to be trapped by autophagy during the process of multiplication in a host. Shigella VirG, which is required for the intracellular actin-based motility of this organism, was found to be a target for the autophagy protein, Atg5. IcsB competitively inhibits the association of Atg5 to VirG, thus contributing to avoidance of the host autophagy response.
7.3 Molecular mimicry: exploitation of host factor
EPEC regulates host MT networks by using certain effectors [50]. EPEC remains a major cause of infantile diarrhea worldwide, primarily in the developing world. Related pathogens are known to cause disease by similar mechanisms, include EHEC, REPEC, and Citrobacter rodentium. These pathogens induce a histopathological lesion, referred to as an attaching/effacing (A/E) lesion, which is defined by the intimate attachment of a bacterium to an intestinal epithelial cell and the subsequent effacement of the host cell microvilli [51]. These processes are directly involved in the development of diarrhea [52]. Factors responsible for A/E lesion formation are encoded by the locus of enterocyte effacement (LEE), which includes a TTSS, intimin, effectors including Tir, and translocators. A/E lesion formation is induced by interactions between the bacterial membrane protein, intimin, and Tir, which is translocated onto the host cell membrane via the TTSS [53]. The Tir–intimin interaction triggers the accumulation of actin cytoskeletal components beneath the adherent bacteria, and this phenotype is not affected by the addition of the RhoA-inhibitor. Tir is known to possess the ability to recruit the host factors N-WASP and the Arp2/3 complex via use of the host adapter protein, Nck, which results in the accumulation of actin [54,55].
Like EPEC, most pathogenic bacteria elicit the rearrangement of actin cytoskeletal components in order to enable internalization into epithelial cells. In addition to actin reorganization, the microtubule (MT) structure is also exploited by the entry or attachment of certain pathogenic bacteria into host cells. Indeed, the invasion of epithelial cells by Campylobacter, Listeria, and Neisseria is inhibited in the presence of an MT-destabilizing agent such as nocodazole [56]. MTs are composed of tubulin molecules, and MT networks are a major cytoskeletal component in most eukaryotic cells and they are responsible for the regulation of mitosis, organelle movement, and actin dynamics.
Recently, we revealed that the EPEC EspG/Orf3 effectors elicit the disruption of MT networks beneath the adherent bacteria by the direct association of EspG and Orf3 with tubulins [50]. Although it remains unclear why EPEC possesses two functionally identical effectors, the gene encoding EspG is located in the LEE, but the gene encoding Orf3 lies outside of the LEE. It is of note that both effectors are also required for the induction of actin stress fiber formation in a Tir-intimin interaction-independent manner. A guanine nucleotide exchange factor, GEF-H1, is known to be involved in EspG/Orf3-mediated signaling. GEF-H1 specifically promotes RhoA activation and is localized in MTs in its inactive form, but switches to active form when it is released from MTs. Indeed, both EspG/Orf3 effectors possess the ability to release GEF-H1 from the MT network to the cytosol as a result of MT destabilization [50]. These findings indicate that EPEC regulates the cross-talk between the formation of actin stress fibers and the MT network. A recent study has shown that GEF-H1 is associated with tight junctions and regulates size-selective paracellular permeability by the activation of the RhoA-ROCK signaling pathway [57]. Indeed, EspG- and Orf3-expressing MDCK cells have been shown to exhibit normal overall morphology and maintain fully assembled tight junctions. However, the paracellular permeability to 4-kDa dextran, but not that to 500-kDa dextran, was greatly increased (Matsuzawa, unpublished data). Thus, EPEC has the ability to regulate size-selective permeability via EspG/Orf3 effectors. A recent study demonstrated that EspG is required for Citrobacter rodentium, which does not possess Orf3, to achieve full virulence in vivo [33].
Although EspG and Orf3 in EPEC, as well as VirA in Shigella, can exert effects on microtubule networks, EspG/Orf3 induces the formation of actin stress fibers, and VirA induces membrane ruffling. The VirA effector induces MT instability, leading to Rac1 activation, which in turn results in the induction of membrane ruffling and the subsequent promotion of bacterial entry into the host cell [58]. VirA binds -tubulin dimers, but not MTs, suggesting that VirA specifically associates with tubulin heterodimers. The VirA domain that binds to tubulin heterodimers shares amino acid identity (∼21%) with a certain region of the EspG/Orf3 effector. The different mechanisms underlying the cytoskeletal rearrangement of host cells induced by these factors remain to be elucidated. Both the EspG and Orf3 effectors are involved in an increase in paracellular permeability by the perturbation of MT networks. A/E lesion formation due to Tir-intimin interactions [53], as well as the disruption of tight junctions by EspF [59,60], both exert effects on the overall morphology and physiology of host cells. The synergistic effects caused by multiple effectors are expected to contribute to triggering EPEC-mediated diarrhea.
8 Concluding remarks
Type-III effectors are known to be involved in disease processes. As a common effector delivery system, the TTSS is a good candidate that could serve as a target of anti-microbial agents. To date, several compounds have been reported as inhibitors of the Yersinia TTSS [61]. TTSS inhibitors are expected to specifically inhibit the delivery of type-III effectors into the host cell and yet they do not kill the normal intestinal bacterial flora, resulting in elimination of bacterial resistance to antibiotics. Although antibiotic-resistant bacteria are an emerging public-health threat, no drugs have been made available to date that eradicate only pathogenic bacteria, leaving the non-pathogenic bacteria intact. Thus, it is likely that TTSS inhibitors could contribute to the development of such a pathogen-specific drug.
Furthermore, an effector-deficient, non-pathogenic strain that still possesses a functioning TTSS might be applicable as a vaccine delivery system. The signals required for both secretion and translocation have been well characterized among the type-III effectors, and it has been demonstrated that the chimera between an effector and a foreign protein is effectively translocated into host cells via the TTSS under the appropriate conditions. Thus, investigations of bacterial type-III effectors contribute to gaining a better understanding of bacterial virulence; future studies in this area will provide new approaches to the development of therapeutic drugs and vaccine delivery systems.