

1 Introduction
The prion protein (PrP) is a cell-surface glycoprotein that has a half-life of ∼6 hours in the body [1]. PrP is found in mammalian tissues, concentrated primarily in neuronal cells and it is believed to play a role in copper metabolism [2] and/or signal transduction [3]. Under certain conditions, the innocuous cellular form of the protein, PrPC, can convert to the lethal scrapie prion protein isoform, PrPSc, which can aggregate with other PrP molecules, resist degradation and destroy neuronal and glial cells. While PrPC is believed to play a positive role in cellular functions, PrPSc is responsible for transmissible spongiform encephalopathies (TSE), which include bovine spongiform encephalopathy in cattle, fatal familial insomnia, Creutzfeldt–Jakob disease (CJD) and Kuru in humans and the namesake of PrPSc, scrapie in sheep and goats.
PrPSc, while sharing the same covalent structure as PrPC [4], can be distinguished from PrPC based on secondary, tertiary and quaternary structure. PrPSc is rich in β-structure with some α-helical structure (43–45% extended structure, 17–30% α-helix) [5–7], whereas PrPC has little β-structure and is largely α-helical (3% β-structure, 47% α-helix) [5]. Unlike the monomeric PrPC, PrPSc can form oligomeric structures that can be cytotoxic and/or infectious (reviewed in [8]). PrPSc does not arise from misfolding during the synthesis of PrP, but directly from PrPC. Newly synthesized 253 amino acid PrP polypeptide (human numbering) is targeted to the rough endoplasmic reticulum (ER), after cleavage of the N-terminal signal sequence (residues 1–22), where an initial core glycan is attached to one or both possible glycosylation sites (Asn-181 and Asn-197), the disulfide bond is formed (between residues 179 and 214) and after the cleavage of the C-terminal signal sequence (residues 231–253) a glycosylphosphatidylinisotol (GPI) anchor is attached (Fig. 1A). On its way to the plasma membrane, PrPC transits through the Golgi where the glycans are further modified. In its final form, PrPC is comprised of a large unstructured domain and a globular domain that contains three α-helices and two short β-strands (Fig. 1B) [9,10]. At a point after PrPC reaches the cell surface, PrPC can be converted to PrPSc [11–13]. Factors that have been linked to the conversion of PrP include mutations (genetic or spontaneous), low pH environments, and exogenous PrPSc.
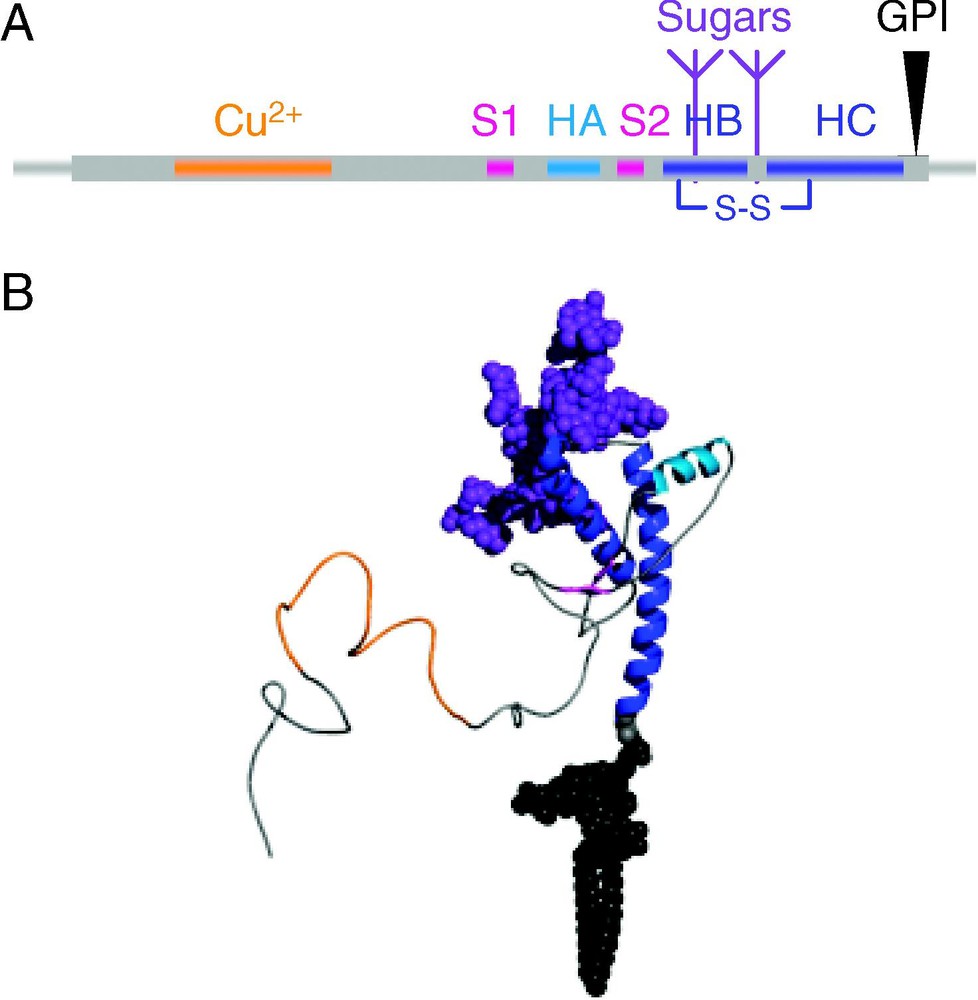
Sequence and structure of the prion protein. (A) The human PrP sequence contain two signal sequences (thin gray lines), at the N-terminus (residues 1–22) and at the C-terminus (residues 231–253), an octarepeat region that binds copper (residues 51–91, colored orange), two short β-strands (magenta), three helices, HA (light blue), and HB and HC (blue) which are bound by a disulfide bond. PrP can be glycosylated (purple) at Asn-181 and Asn-197 and has a GPI anchor (black) attached to the C-terminal Ser-230. (B) Structure of PrPC residues 23–230, colored as in (A). Structure of residues 125–228 were taken from the NMR structure of human PrP (1QLX, [9]), with the remaining residues, sugar and lipid groups modeled in.
Conversion of PrPC to PrPSc in vitro is most commonly identified by a change in secondary structure and quaternary structure (i.e., aggregation) and an acquired resistance to enzymatic digestion. It is important to note that these characteristics are not sufficient to claim an infectious, and thus biologically relevant, PrPSc species. The nomenclature, PrPSc, was originally used to denote an infectious species, while it is used here in a broader sense to denote a misfolded species with structural and chemical features consistent with brain-derived and infectious PrPSc. Instead of the term PrPSc, PrPres is often used as it denotes a species resistant to enzymatic digestion [6]. The term PrPres more appropriately describes the aggregate state observed both in vitro and in vivo, for which the infectivity of the aggregate has either not been tested or observed. Nevertheless, we adhere to the prevailing convention here and use PrPSc throughout this text. The reader must keep in mind, particularly with in vitro and computational studies, that it remains uncertain, that besides the structural and chemical resemblances, whether the misfolding observed bears the infectivity and transmissibility properties of in vivo misfolded PrP.
Since TSEs arise from the unfolding and misfolding of PrPC, any environmental factors that can affect the structure, and thus the conversion, of PrP are of interest. For instance, PrPC is constitutively cycled between the cell-surface and endocytic compartments. At the cell surface PrP experiences a predominantly neutral pH environment whereas once endocytosed it enters a low pH environment. How does the structure of PrPC at the cell-surface differ from that of PrPC once it enters an endocytic compartment? Any pH-dependent conformational transition could help determine where conversion is taking place. Also, there exist NMR and X-ray crystallographic structures of soluble, non-glycosylated, recombinant (rec) PrP for several mammalian, a reptilian and an avian species, but how do these structures compare with the structure of glycosylated, GPI-anchored PrPC found in vivo? In this review, we will explore small molecules, covalent modifications and environments that may influence PrP structure including: pH, carbohydrate moieties, lipid membranes, metal ions and small molecule therapeutics.
2 pH
The presence of PrPSc deposits in the endosome-like compartments of scrapie-infected mouse brains [14] and the lack of PrPSc deposits in cell cultures where endosome formation was inhibited [15] led investigators to label endocytic organelles as plausible locations for conversion. Inside an endosome, the average pH is 5 and can be as low as pH 4.3 [16]. It is also possible that conversion takes place at the cell surface in caveolae domains as these domains may create locally acidic environments similar to those of endocytic compartments [17].
Using endosomal-like conditions, synthetic PrPC can be converted to PrPSc in vitro [18]. At low pH (usually in the presence of mild denaturants), PrPC gains β-like extended structure and tends to aggregate. Again, the PrPSc product is usually deemed to be similar to that observed in vivo based on structural characteristics alone. There have recently been claims of infectivity of synthetic PrPSc aggregates inoculated intracerebrally into transgenic mice overexpressing PrP [19], however, questions regarding the lack of a control group and possible contamination are, as of yet, unanswered. Synthetic models of PrPSc aggregates seem to resemble natural PrPSc in structure and possibly infectivity, and while the latter property requires further consideration and experimentation, rec-PrP conversion systems seem to be suitable models of the misfolding process.
2.1 Recombinant PrP
The dependence of PrP structure on pH has been studied for a variety of rec-PrP species including, human (Hu) [18], mouse (Mo) [20], and Syrian hamster (SHa) [21]. To characterize the behavior of rec-HuPrP (residues 90–231) at pHs ranging from neutral to highly acidic, equilibrium unfolding, far-UV CD, and 1,1′-bi(4-anilino)naphthalene-5,5′-disulfonic acid (bis-ANS) binding and fluorescence experiments were undertaken [18]. To assess the conformations of PrPC and what was identified as a folding intermediate, the ellipticity of PrP in the presence of 1 M guanidine hydrochloride (GdnHCl) at various pHs was measured [18]. Between pH 5 and 7.2 (with or without 1 M GdnHCl) the CD spectra indicate an isoform with predominantly α-helical structure. At a lower pH range, pH 3.6 to 4, and with 1 M GdnHCl, the CD signal indicative of α-structure (double minimum at 222 and 208 nm) is replaced by a signal characteristic of β-structure (a single minimum at ). In support of these results, similar experiments, on the structured domain of rec-MoPrP (residues 121–231) and 3.5 M urea instead of 1 M GdnHCl, demonstrate a conformational transition at low pH [20]. The conformational conversion is characterized by a loss of the α-helical signal and an increase in the signal at 215 nm [20]. Bis-ANS binding experiments also demonstrate a conformational conversion of rec-HuPrP in the lower pH range in the absence of denaturants [18]. Bis-ANS can be used to monitor conformational changes because binding of bis-ANS to a protein (typically to solvent exposed hydrophobic patches) results in increased fluorescence intensities. The results of the small-molecule binding experiments reveal a conformational transition between pH 6 and 4.4 that involves a significant increase in exposure of hydrophobic surfaces.
To determine the regions where pH-dependent structural conversion were occurring, Matsunaga et al. used antibodies to probe the structure of rec-SHaPrP (residues 90–231) over a pH range of 2 to 12 [21]. The results of this anti-PrP antibody binding study indicate that the conformation of epitopes localized in the C-terminus are insensitive to pH, whereas the conformation of N-terminal residues are pH sensitive. Below pH 4, there is a significant decrease in binding of antibodies that target N-terminal epitopes (Fig. 2). The pH dependence of the conformational change was also studied in the absence of denaturants using far-UV CD. Rec-SHaPrP folded, after dissolution of the inclusion bodies, at pH 5.5 is primarily α-helical and, when the pH is lowered to approximately pH 4, there is a minor shift towards β-structure, and at pH 2, a shift to random coil. Below pH 4.7, PrP undergoes conformation changes that likely involve a loosening of the native tertiary structure and a gain of β-like secondary structure that, based on antibody mapping studies, likely occurs at the N-terminus and includes residues 94–112 (no data available for residues 113–132, which are likely involved) [21].
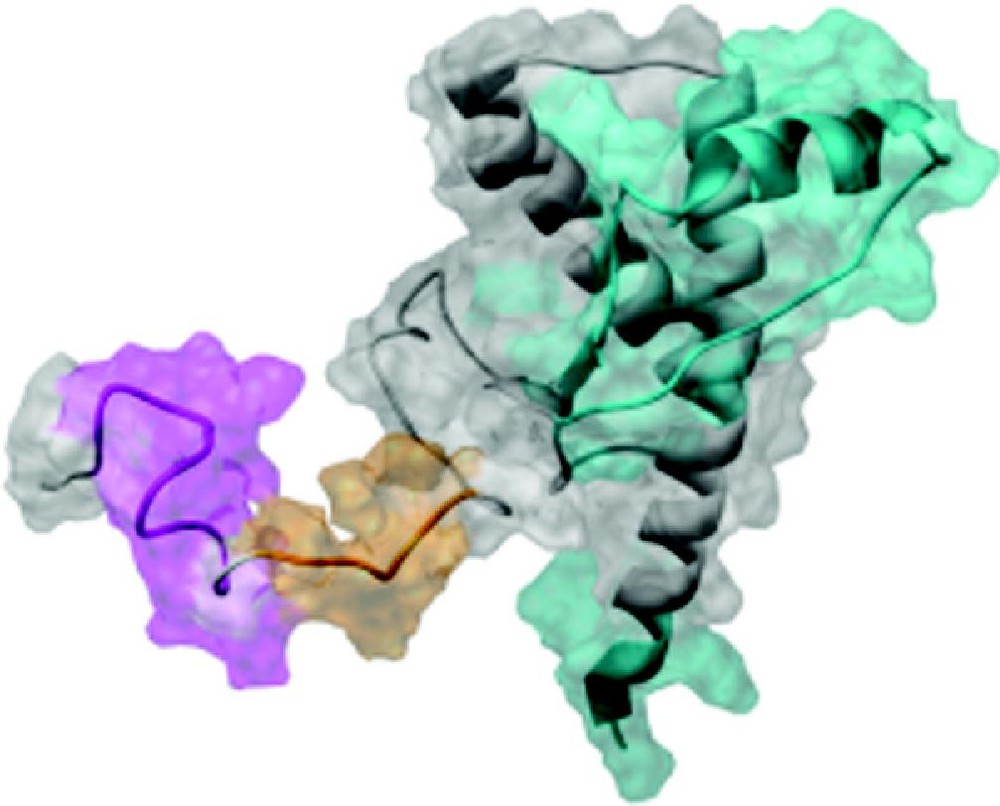
At low pH, PrPC (residues 90–231, shown as a ribbon diagram with transparent space-filling view) undergoes a conformational transition that can be observed by antibody mapping. All epitopes shown (colored regions of the protein) are exposed in PrPC. Under low pH conditions, the conformation of PrPC changes and binding to N-terminal epitopes mapped to residues 94–105 (magenta) and 107–112 (orange) significantly decreases. There are no observed changes in binding to C-terminal epitopes mapped to residues 133–157, 138–142, 144–150, 146–154, 152–163, and 226–231 (cyan).
From the above studies we can draw several conclusions: (1) the structure of PrP is dependent on pH; (2) the pH-dependent structural changes are linked primarily to N-terminal residues, while the C-terminal structured domain undergoes only minor conformational changes; and (3) mild denaturants coupled with a low pH environment stimulate the conversion of PrPC to a species with secondary structure consistent with that observed in natural PrPSc.
2.2 Brain-derived PrP
Conversion experiments using brain-derived PrPC and PrPSc echo some of the findings of conversion studies using rec-PrP. In particular, using conditions similar to those employed by Matsunaga et al. [21], 1.5 M GdnHCl and pH 3.5, conversion assays ofPrPC isolated from healthy human brains were performed [22]. These conditions cause HuPrPC to change quaternary structure, from soluble monomers to a detergent-insoluble species (aggregate). Insolubility of PrP is deemed to be pH dependent in non-denaturing conditions (low GdnHCl concentrations), with HuPrPC forming detergent-insoluble fractions at a pH ⩽3.5 and detergent soluble fractions at pH >4.5. While sharing the detergent insolubility of PrPSc, the aggregated PrP species does not share other properties of PrPSc with respect to proteinase K resistance and epitope inaccessibility. Despite the mix of PrPC and PrPSc properties, the acidic and GdnHCl treated HuPrP more readily converts to proteinase K resistant aggregates upon exposure to brain-derived PrPSc from CJD patients, as compared to untreated HuPrPC. Zou and Cashman [22] hypothesize that HuPrPC in low pH conditions with mild denaturant initiates an initial conformational transition that allows for aggregation. In a second step, PrP can then undergo a conformational transition in the presence of a PrPSc template to a detergent insoluble, proteinase K-resistant species – PrPSc [22].
Most studies focus on the effect of pH on PrPC structure as it relates to the conversion process, however, it happens that PrPSc conformation is also pH dependent. Strains of PrPSc (isolated from the brains of patients diagnosed with sporadic CJD) treated with proteinase K under acidic, neutral and basic conditions revealed a conformation dependence on pH for certain strain-types [23]. Full-length PrPSc produces significantly larger proteinase K resistant fragments at pH 4 and 6 than at pH 7.4, suggesting that low pH triggers a conformational change that blocks digestion sites. N-terminally truncated forms of PrPSc do not display the same resistance at low pH, as they produce indistinguishable digestion fragments at neutral and at low pH. It appears that the N-terminal residues, present in full-length PrPSc and absent or partially absent in truncated forms of PrPSc, are conformationally sensitive to pH, similar to what was observed for brain-derived PrPC [22]. Low pH may cause N-terminal residues to take on a conformation that protects them from enzymatic digestion, whereas the conformation of the C-terminus of PrPSc is not altered by pH, a finding that is supported by antibody binding studies to PrP 27–30 rods [21].
2.3 Computational studies of PrP
Conformational conversions that PrPC may undergo as a consequence of pH can be studied in atomic detail using molecular dynamics (MD) simulations. Using all-atom, explicit solvent simulations, fragments of PrPC have been explored at both low and neutral pH. In Fig. 3A, the misfolding pathway, in silico, of PrPC to a PrPSc-like species is shown [24]. Several different PrP sequences and their misfolding pathways have been investigated including wild-type and mutated SHaPrP [24,25], mutated bovine PrP and wild-type HuPrP [26]. PrP fragments comprised of residues 109–219 (SHa numbering) based on the corresponding NMR structure for hamster (1B10, residues 109–228 (structure provided by T.L. James and S. Farr-Jones [10]), bovine (1DWY, residues 124–227 [27]), and human (1QLX, residues 125–227 [9]). For both the bovine and human forms, residues 109–124 of the hamster structure are grafted onto the structures. To simulate at different pH levels the protonation state of Asp, Glu and His residues are adjusted; at low pH (pH < 4) these residues are protonated and at neutral pH (6 < pH < 10) Asp and Glu are charged, while His is neutral.

Conversion of PrPC to PrPSc. (A) Snapshots from a 20-ns MD simulation of the conversion of D147N SHaPrPC that occurred under amyloidogenic (low pH) conditions. The wild-type protein undergoes a similar conversion [25]. A misfolded intermediate rich in β-like structure formed from 6 to 20 ns (represented by the 8-ns structure). A trimeric representation of a PrPSc protofibril, modeled by docking newly formed hydrophobic strands of the 8-ns structure, is shown. (B) Representative structures from the misfolding ensembles found in low pH MD simulations of hamster, bovine and human PrP fragments (residues 109–219, hamster numbering). The largely α-helical PrPC converts to a species with more extended structure (colored magenta).
For the wild-type PrPC fragments, neutral pH simulations are typified by low root-mean-square deviations from the native structure, maintenance of native secondary and tertiary structure and mobility of the ∼15-residue N-terminal sequence. At low pH, all simulations show a slight decrease in α-helix and an increase in extended structure. The new extended structure nucleates at the existing S1–S2 sheet, as shown by structures representative of the misfolded ensemble in each of the simulations in Fig. 3B. Along with the commonalities between the PrPSc-like species, there are local differences in the location of the extended structure. This suggests that the sequence can introduce a level of heterogeneity between PrPSc conformations, a property that may account for the barrier of transmission observed between several species.
Based on MD simulations, the effect of low pH on PrPC fragments is generally characterized by a loosening of tertiary structure and a gain of extended secondary structure in N-terminal regions of the protein. In addition to characterizing the early steps in the misfolding pathway, structures from MD simulations, in particular, those representative of PrPSc-like species, are valuable for exploring the oligomerization process of PrPSc. Using a misfolded species from an MD simulation, a model of a PrPSc protofibril, in agreement with experimental observations of PrPSc protofibrils and fibrils, was built [24]. By docking extended strands, that present new stable hydrophobic surfaces, intra-molecular extended sheets form to connect subunits (Fig. 3A).
2.4 Summary
These results serve to highlight the influence of pH on the structure of the N-terminal sequence of PrPSc versus the lack of influence on more C-terminal regions of PrPSc. As for PrPC, the N-terminal region appears to undergo the greatest structural change as a result of variations in pH, although the C-terminal structured domain also undergoes more modest changes in response to lower pH.
3 Carbohydrates
Despite the focus on the protein portion of PrP, PrP can be glycosylated with very large and complex carbohydrate moieties that also play a role in disease. Glycoforms can attach to two sites on the protein, Asn-181 and Asn-197 (human numbering) [28,29]. N-linked glycosylation of the two PrP glycosylation sites, identified by the AsnXaaThr sequence, is both a co- and post-translational event occurring in the ER and the Golgi. In the first step of PrP glycosylation, the AsnXaaThr sequences are identified (in the ER) and high-mannose core glycans are linked to the polypeptide. Upon exiting the ER, PrP glycosylation sites contain a core glycan typical of N-glycosylated proteins: two N-acetylglucosamine, eight mannose and two glucose residues. This standard glycan is then further modified in the Golgi. Mature PrP glycoforms have in the range of 8–20 carbohydrate residues and there are greater than 60 different types of carbohydrates residues in hamster and mouse PrP-glycoforms including charged sialic acid, which is found in both PrPC [28,30,31] and PrPSc [32]. Analysis of in vivo PrP proteins has yielded the identification of over 400 different PrP glycoforms [28] and identification of un-, mono- and di-glycosylated forms of PrP in cell cultures [1].
No specific glycoform (or group of glycoforms) or glycosylation pattern has been linked to prion disease. However, glycosylation does play a role in the disease process. Using transgenic mice, and selectively deleting the first, second or both glycans, DeArmond et al. found that the glycans were involved in PrP expression, distribution (within the brain and among different types of neuronal cells) and deposition of PrPSc plaques [33]. Deletion of the first glycan results in low levels of PrPC and unusual distribution of PrPC (accumulation in nerve cell bodies and decreased deposition in dendritic trees). Deletion of the second glycoform results in expression levels of PrPC comparable to wt-PrP and partial mis-trafficking of PrPC to neuronal cells. The second glycoform deletion mutant alters the deposition pattern of PrPSc as compared to diglycosylated PrPSc. Since different regions of the brain express different PrP glycoforms [34], PrPSc deposits could be targeted to regions of the brain expressing similar glycosylation patterns. Supporting this hypothesis is the observation that glycoforms found in PrPSc deposits of patients with variant-CJD, a disease causally linked to an outbreak of BSE, are similar to those in cattle infected with BSE [35]. From these results it was hypothesized that glycosylation can modify the conformation of PrPC and/or affect the affinity of PrPC for a strain (particular conformation) of PrPSc.
3.1 Brain-derived glycosylated PrPC
All structures of PrPC, whether a product of NMR or X-ray crystallographic experiments, are of rec-PrP expressed in Escherichia coli (E. coli). Therefore, they lack post-translational modifications except for the disulfide bond. Then, one wonders, is rec-PrP is similar in structure to brain-derived PrP? To answer this question, Hornemann et al. compared PrPC from calf brains to that of rec-bovine PrPC expressed in E. coli [36]. To isolate ‘natural’ PrPC, neuronal cells were treated with phosphatidylinositol-specific phospholipase C (PIPLC) to release PrPC from the membrane and in the subsequent purification steps the use of detergents was avoided to maintain the non-protein portions of PrP, namely the remaining GPI anchor and the carbohydrate moieties. The secondary structure and stability of PrP were characterized using far-UV circular dichroism. The two types of PrPC, ‘natural’ (likely residues 23–230, as the isolated species contains the N-terminal sequence that follows the cleaved N-terminal signal sequence and a portion of the GPI anchor) and recombinant (residues 23–230), have nearly identical spectra, indicating little, if any, differences in secondary structure content. The thermal unfolding transitions, also monitored by far-UV CD, have similar sigmoidal shapes, indicative of cooperative unfolding. The melting temperatures of natural diglycosylated PrPC and unglycosylated recombinant PrPC are 60.6 and 61.0 °C, respectively. Tertiary structure was studied using 1D 1H-NMR spectroscopy, showing that the two PrP types have similar conformations within the structured domain of PrP. Despite the attractive proposition that glycosylation can affect PrPC structure or stability, it appears that PrP glycoforms do not alter the thermostability or structure of PrPC and that rec-PrPC is an adequate structural model for natural di-glycosylated PrPC [36].
3.2 Computational studies
Molecular dynamics simulations of di-glycosylated PrPC also support these experimental findings. Zuegg and Gready have simulated PrP, residues 90–231, the primary proteinase K-resistant fragment, with and without sugars for each [37]. In these simulations, native secondary and tertiary structure (NMR structure) are preserved in both diglycosylated and unglycosylated forms. Interactions between the protein and carbohydrate groups are limited and primarily involve the regions near the attachment sites. Also, solvent accessible surface area of the two PrP forms is similar. Preliminary analysis of our 15-ns simulations of di-glycosylated and un-glycosylated HuPrP (90–231), also indicate that secondary structure in both models is similar to that observed in the un-glycosylated HuPrP NMR structure 1QLX [9] (Fig. 4). As well as making transient short-range local interaction with the C-terminal helices, the glycoform at Asn-187 is capable of making long-range interactions with the N-terminal unstructured portion of the protein. In our di-glycosylated HuPrP simulations, the Asn-181 glycoform interacts with the loop preceding S1. These long-range interactions do not distort the native structure and in fact may serve to further stabilize the globular portion of PrPC (manuscript in preparation).
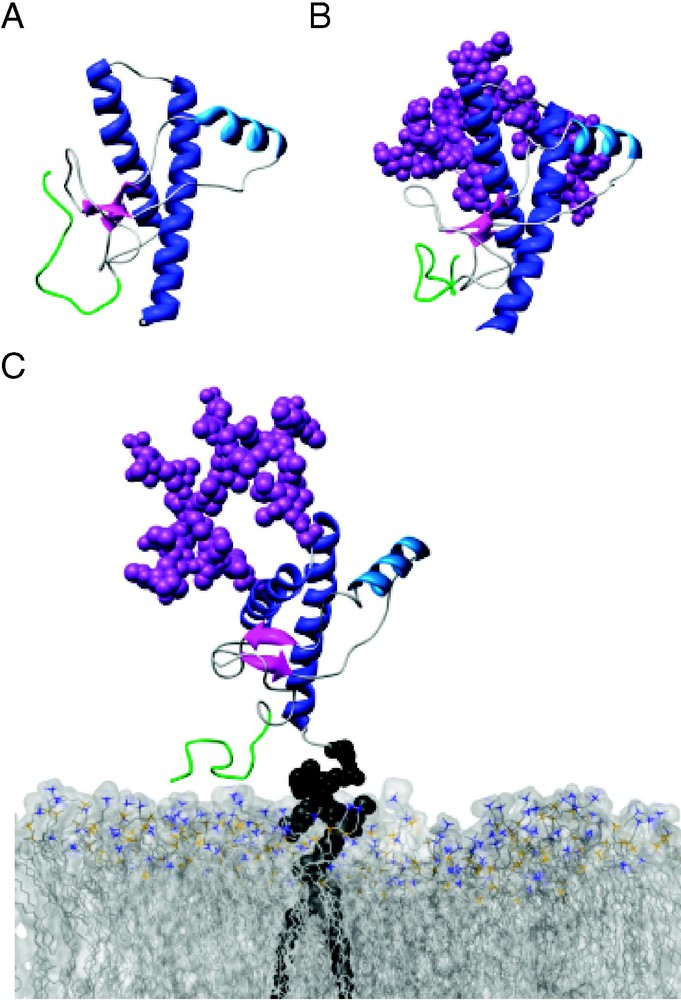
Glycosylation of PrPC has a minimal effect on its conformation. Snapshots from MD simulations of (A) unglycosylated (10 ns), (B) diglycosylated (10 ns), and (C) diglycosylated and membrane-bound (5 ns) HuPrPC (residues 89–230). Secondary structure colored as in Fig. 1, with residues 89–108 colored green. (C) HuPrPC is linked to a POPC bilayer (space-filling view, with nitrogens colored blue and phosphorous colored gold) via its GPI-anchor.
3.3 Summary
Both experimental and theoretical results are in agreement; glycosylation does not significantly affect the structure of glycosylated PrPC as compared to unglycosylated PrPC.
4 Lipid membranes
The availability of PrP structures of soluble rec-PrP fragments have been invaluable for the study of prion disease, unfortunately little is known about the structure of the membrane-bound form of PrP. The importance of the membrane-bound form has been made apparent by several studies that lead to the conclusion that conversion of PrPC to PrPSc occurs either at the cell-surface or in the endosomal/lysosomal pathway [12,15]. Typically, PrPC is bound to the extra-cellular side of the plasma membrane and cycles between the cell surface and the early endosomal pathway, a round-trip taking approximately 60 minutes [38]. The cleavage of a 22-residue C-terminal sequence in the ER and subsequent attachment of a GPI-anchor in the Golgi (Fig. 1) sequesters PrPC to a specific domain once it reaches the plasma membrane [39–41]. As with other proteins with GPI-anchors, PrPC is localized in areas rich in cholesterol and sphingomyelin known as lipid rafts or caveolae domains [39–41]. Once presented on the cell surface, PrPC is eventually internalized for either cycling back to the membrane or degradation, or converted to PrPSc.
Several independent studies demonstrate the importance of the membrane-bound state of PrPC and its possible role in the conversion process. Firstly, PrPSc aggregates are found bound to the plasma membrane, in late endosomes and in extracellular spaces [40–42]. Whilst PrPC can be cleaved from the plasma membrane using PIPLC, PrPSc is resistant to PIPLC and remains attached to the cell-surface [43]. Either the conformation of PrPSc itself is (sterically) preventing cleavage of its GPI anchor or it is interacting with the membrane in a manner that prevents access to the PIPLC cleavage site. When bound to the membrane PrPSc, like PrPC, is found in lipid rafts [39] and this localization of PrPC and PrPSc appears to be critical for efficient propagation of PrPSc. In scrapie-infected neuroblastoma cells, the GPI anchor of PrPC was replaced with transmembrane helices to prevent localization of PrPC in lipid rafts while still presenting PrP in a membrane-bound form on the surface of the cells [13]. This modification of the GPI anchor prevents propagation of PrPSc, as detected by a lack of proteinase K resistance [13]. Similarly, destruction of lipid rafts via cholesterol depletion inhibits PrPSc formation [13]. In vitro experiments corroborated the necessity of co-localization of PrPC and PrPSc for efficient production of PrPSc [44,45].
Besides the GPI-anchored form of PrP, several other forms of PrP have been identified. GPI-anchorless PrP can be released from the cell or form transmembrane PrP in which either the N- or C-terminus remains in the lumen [46–48]. The latter transmembrane forms inhibit PrPSc formation [49], possibly through a delocalization mechanism similar to that of GPI-replaced PrP [13]. Also to be considered are experiments using soluble, unglycosylated, GPI-anchorless rec-PrP in the absence of a membrane. Conversion, at least in vitro, can occur in the absence of the membrane. In cells, conversion occurs after PrPC reaches the plasma membrane [11–13], making membrane-association, whether through the GPI-anchor or through protein–lipid interactions, a necessary component of the disease process.
4.1 Soluble PrP
Morillas et al. observed the interaction of soluble synthetic HuPrP (23–231) with the acidic phospholipid membrane palmitoyloleoylphosphatidylserine (POPS) and a lack of interaction with the zwitterionic palmitoyloleoylphosphatidylcholine (POPC) membrane [50]. The PrP–POPS interaction is pH dependent, becoming stronger in acidic conditions. A slight increase in ordered secondary structure of HuPrP is observed by CD spectroscopy. This increase in secondary structure is hypothesized to occur within the N-terminus based on experiments with HuPrP (23–231), HuPrP (90–231) and a peptide spanning residues 23–145 of HuPrP. Both HuPrP (23–231) and the unstructured peptide gain structure upon interaction with the POPS membrane while the N-terminally truncated species, HuPrP (90–231), does not [50]. Binding of PrP to negatively charged membranes and not to neutral membranes has also been reported for soluble rec-SHaPrP (90–231) [51]. In addition to negatively charged phospholipid membranes, rec-SHaPrP was also found to bind to raft-like membranes at pH 7 and not at pH 5 [51]. Binding of rec-SHaPrPC to negatively charged membranes results in an increase in β-structure, membrane-lysis and formation of amorphous PrP aggregates. Binding of rec-SHaPrPC to the biologically relevant raft-like membranes results in an increase in α-helix content without destabilization of the membrane [51]. Binding between PrP and the membrane surface is believed to occur via hydrophobic and electrostatic interactions between the protein and the membrane [52].
4.2 Membrane-bound PrP
PrPC is found primarily bound to membrane, not in the soluble form, and therefore membrane-PrPC interactions in this state may be of greater biological relevance. To examine the interactions of membrane-bound PrPC with lipid mixtures and to differentiate between the interactions of the membrane-bound and soluble forms of PrPC with lipid bilayers, Eberl et al. considered both soluble and PrPC covalently bound to liposomes [53]. PrPC was covalently bound to liposomes via a C-terminal extension with a free thiol group, which was then coupled to a thiol-reactive lipid in the liposome. Two different mixtures of lipids were used: a mixture of POPC and cholesterol (1:1 molar ratio), and a raft-like mixture of POPC, phosphatidylethanolamine, sphingomyelin, cerebrosides and cholesterol (1:1:1:1:2 molar ratio). Far-UV spectral analysis indicates similar secondary structure for membrane-bound PrP and soluble PrP at both pH 7 and pH 5. When membrane-bound and soluble PrP in the presence of a membrane are subjected to proteolysis, similar digestion products form, suggesting that membrane-bound and soluble rec-PrP have a similar conformation [53]. In addition to far-UV and proteinase K digestion, antibody mapping has been used to probe the structural differences between soluble rec-PrPC and membrane-bound PrPC [54]. While this study indicates discrepancies between antibody binding in the two forms of PrP, it is attributed to the conformational heterogeneity of the membrane-bound PrP and not to specific differences between soluble and membrane-bound forms [54]. The exception to this conclusion is the antibody that targets residues 138–141. This antibody shows a significant decrease in binding in the membrane-bound form as compared to the soluble form, although it is not determined whether this is due to a conformational change in PrPC, interactions between PrPC and other molecules [54], or the orientation and proximity of the epitope to the membrane.
4.3 Computational studies
To gain a better understanding of the effect of lipid membranes on PrPC structure, we have begun molecular dynamics simulations of diglycosylated HuPrP (90–231) anchored to a POPC bilayer via its GPI anchor (Fig. 4C). Preliminary results, in agreement with experimental observations, indicate that the structured region of HuPrPC does not undergo a conformational change at the membrane surface. The N-terminal unstructured region transiently interacts with the lipid head groups and traverses the surface of the membrane, remaining largely unstructured. There are no significant changes in either secondary or tertiary structure of the membrane-bound protein as it maintains a conformation similar to what is observed in the NMR structure of rec-HuPrP [9].
4.4 Summary
Sensitivity to experimental design has been blamed for the discrepancies between published reports of PrP's ability to bind to certain membranes. We can find consensus among experiments by narrowing the focus to the two most important membrane structures, raft-like membranes and POPC membranes, the former being where PrPC is located in vivo and the latter being the major lipid component of the membrane region near PrP in neuronal cells [55]. For POPC membranes, there is no observed binding of rec-PrP (whether soluble or membrane-bound) to the membrane. For raft-like membranes, there are indications that PrPC gains α-helical structure as it binds to the membrane in its soluble form and that membrane-bound PrP may also experience a slight conformational change.
5 Copper and other metal ions
To determine the function of PrPC, Prnp knockout mice (mice lacking the gene encoding PrP) were genetically engineered [56–58]. Clues as to the function of PrP could have been drawn from the differences between the normal (PrP-expressing) and knockout mice; however, the knockout mice displayed little difference between their wild-type counter parts during development. In some cases, older-aged mice provided clues as Prnp knockout mice had impaired synaptic inhibition, memory functions and motor coordination, and a loss of cerebellar Purkinje cells [58–60]. Besides providing evidence for possible functions of PrP, these studies were critical for establishing the necessity of host PrP production for infection and disease progression, that is, knockout mice were resistant to PrPSc infection. Not long after the first knockout studies, it was demonstrated that synthetic PrP peptides [61,62] and PrP in vivo bound copper [63]. These experiments led to the hypothesis that PrP is involved in copper homeostasis.
The copper binding ability of PrP was first linked to the octarepeat region, which is common to all mammalian prion species. The eight-residue sequence, expressed in a variable number of repeats depending on the species (humans have five octarepeats), contains residues P(H/Q)GGGWGQ and can coordinate copper. There are also residues downstream of the octarepeat region, His-96 and His-111 that have been implicated in forming a copper coordination site [64] as well as more C-terminal locations [65]. NMR structural studies of PrP constructs containing the octarepeat region reveal that, at least in the absences of metal cations, the N-terminal residues, approximately residues 23 to 124 (depending on the species), are highly flexible and unstructured [9,27,66,67]. Copper and other metal ions are found in relatively high concentrations in the brain, making the structure of PrPC in the presence of these cations of interest with respect to function and also disease pathology.
5.1 Recombinant PrP
The ‘unstructured’ region of PrP is, in fact, partially structured in the presence of copper [68]. Peptides of the octarepeat region, like the octarepeat region in the full-length protein, are unstructured in the absence of copper and gain structure in the presence of copper [69]. The coordination of copper by PrP (and water) is shown in Fig. 5A [70]. While the structure was solved for a peptide from the PrP sequence binding copper, based on EPR data for the peptide-copper complex [70] and EPR data from the full-length protein [71], copper coordination by the peptide and the full-length protein are similar. It has been well established that copper causes a gain in structure in the octarepeat region, but what about the rest of the protein?
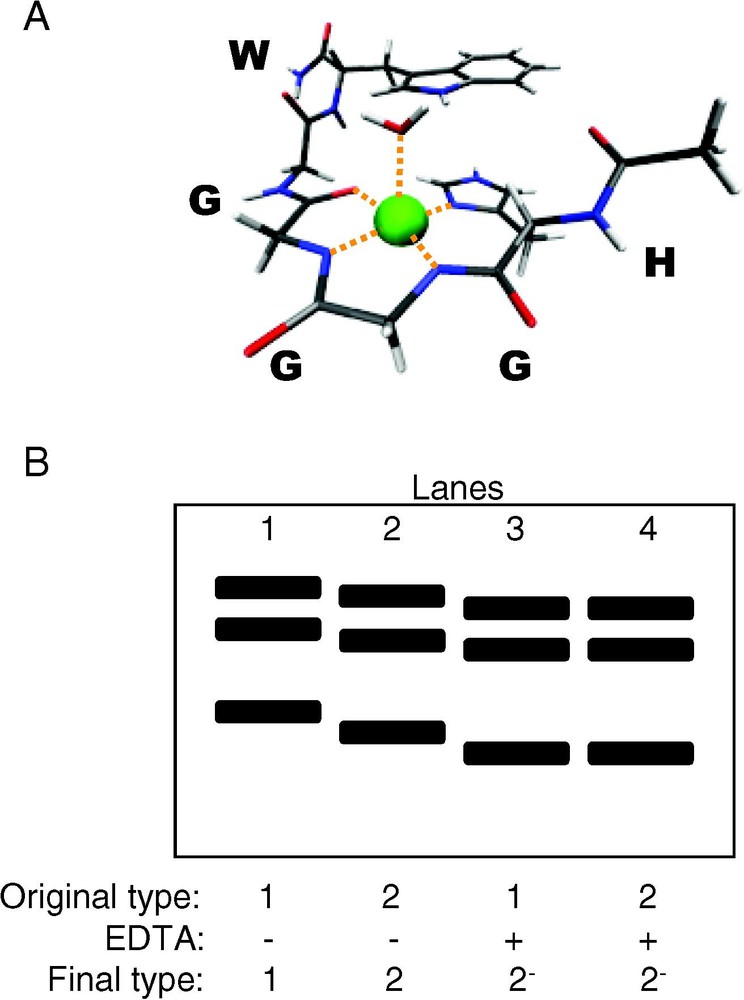
Copper binding to PrPC and PrPSc. (A) X-ray crystal structure at 0.7-Å resolution of a peptide from the PrP octarepeat region, HGGGW, and water, coordinating copper. (B) Western blot of proteinase K digestion fragments of brain-derived PrPSc strain-types 1 (lanes 1 and 3) and 2 (lane 2 and 4), with and without incubation with EDTA prior to proteinase K digestion (recreated from [78]). After incubation with EDTA, differences in digestion profiles between type-1 and type-2 PrPSc are abolished, demonstrating the ability of metal ions to modulate PrPSc structure.
The influence of copper on PrP structure may be observed beyond the limits of the octarepeat region. Several studies have implicated copper and other metal cations in a conformational conversion of PrPC. Copper has been found to induce an increase in β-structure and impart proteinase K resistance [72–75]. In rec-SHaPrP (residues 29–231), it was found that copper binding to PrP facilitates the temperature triggered conversion of PrPC from a predominantly α-helical state to a β-rich isomer, as observed by near-UV CD spectroscopy [72]. Using rec-MoPrP peptides of the unstructured region of PrPC, binding of copper correlates with an increase in β-like extended structure [74]. Another hallmark of the PrPC to PrPSc conversion, acquisition of proteinase K resistance, has been observed in PrP-Cu2+ complexes [75–78]. Using two Ovine (Ov) PrP sequences, PrPARR, associated with scrapie-resistant phenotype, and PrPVRQ, associated with scrapie-susceptible phenotype, folded rec-PrPC was incubated with a nanomolar concentration of copper [75]. While both sequences bind copper, only the disease-susceptible PrPVRQ converts to an isoform high in β-like extended structure. In both predominantly α-helical PrPARR-Cu2+ and β-like PrPVRQ-Cu2+ complexes, copper binding provides increased resistance to proteinase K digestion [75]. It is also interesting to note that neither of the proteinase K resistant proteins have digestion profiles similar to natural OvPrPSc [75]. Another in vitro study using rec-MoPrP and different methods highlights the difficulty in garnering consensus among PrP experiments as rec-MoPrP refolded in the presence of copper is completely digested by proteinase K [77]. Rec-MoPrP refolded in the presence of magnesium is partially proteinase K resistant and eventually converts to an isoform with predominantly β-structure [77].
5.2 Brain-derived PrP
Perhaps more convincing evidence that metal ions can alter PrP structure are experiments using metal-ion-containing PrP from homogenates of human brain tissue of CJD autopsy patients, which show that copper and zinc can have a significant impact on the conformational properties of PrPSc [78]. Patients with distinct disease phenotypes produce PrPSc that can be characterized into strain-types based on conformational variations (assessed by proteinase K digestion) and glycosylation patterns of PrPSc [79]. Type-1 and type-2 PrPSc can be separated by their respective digestion patterns after exposure to proteinase K. In the experiment by Wadsworth et al., both types of PrPSc were treated with the metal chelator ethylene diamine tetraacetic acid (EDTA) and subsequently subjected to proteinase K digestion [78]. The use of the metal chelator abolished differences in digestion fragment profiles of type-1 and type-2 PrPSc (Fig. 5B), showing that metal-ion occupancy can influence conformational properties of PrPSc [78]. Metal ions can facilitate conversion of PrPC to a β-rich form, induce proteinase K resistance and dictate conformational properties of PrPSc.
Metal ions can also alter PrP expression and metabolism. These metal-concentration-driven events may increase the likelihood of other factors altering the conformation of PrP. For instance, high levels of extracellular copper trigger PrPC expression into overdrive [80]. Overexpression of PrPC has deleterious effects, as demonstrated by the decreased incubation times of scrapie-infected transgenic mice overexpressing PrP [81,82]. Metal ions can also interfere with the normal metabolism of PrP. High extracellular concentrations of Cu2+ [83], as well as Zn2+ [84,85], rapidly (and reversibly) stimulate PrP endocytosis. Both changes in expression and metabolism have the potential to promote disease progression. Increased cell-surface/endocytic cycling of PrP would cause an increase in the amount of PrP in the low pH environment of early endosomes [85], an environment that has been linked to conversion. Changes in PrP expression could have the greatest influence by providing more substrate for the conversion. In vivo, the amount of PrPSc formed has been directly linked to the expression levels of PrPC [86].
5.3 Summary
Copper and other metal ions appear to have a significant impact on PrP structure. Copper can induce structure, locally, as it binds to the octarepeat region. It can also alter secondary and tertiary structure outside of the octarepeat region, causing PrPC to form a β-rich and proteinase K resistant species. Copper can modulate the difference between PrPSc strain-types, and affect metabolism and expression of PrPC.
6 Small molecules
While pH, glycoforms, membranes and metal ions are factors that PrP ‘naturally’ encounters, also of interest are small molecules as potential therapeutics for prion diseases. Therapeutic targets are often designed in an attempt to disrupt PrPSc structure and hopefully regain the PrPC structure or any other innocuous isoform. For example, β-sheet breaker peptides were designed specifically to destroy the β-rich amyloidogenic PrPSc species to slow or reverse disease progression [87,88]. Disrupting PrPSc aggregates (breaking them down into smaller aggregates) is not sufficient to inhibit disease and this strategy may increase propagation of PrPSc and infectivity [89,90]. Targeting only PrPSc may not be a sufficient therapeutic strategy, as stabilization of the PrPC conformation also appears to be a requirement.
Small molecules that have shown to be effective in the destruction/protection strategy include the protective osmolytes trimethylamine N-oxide (TMAO), dimethyl sulfoxide (DMSO) and glycerol [91]. When added to scrapie-infected mouse neuroblastoma cells, these osmolytes reduce PrPSc formation, with TMAO being the most effective [91]. While these osmolytes cannot be used as actual treatments, since the necessary efficacious dose is toxic, exploiting the mechanism by which they are effective may yield clues for the design of new (less toxic) compounds. These small molecules, known as chemical chaperones for their ability to affect protein folding, are thought to stabilize the PrPC conformation. To test this hypothesis, MD simulation of PrP and TMAO were run [92]. To observe the destructive abilities of TMAO on PrPSc structure, simulations starting with a misfolded PrPSc-like species from a previous MD simulation (see Fig. 3B, wt-SHa [25]), were run under amyloidogenic conditions (low pH) and with TMAO (Fig. 6) [92]. During these simulations, the extended-sheet is disrupted, PrPSc regains some native contacts (that had been previously lost during conversion) and PrP is restored to a more globular, PrPC-like form. A simulation beginning with PrPC in the presence of 1 M TMAO at low pH, demonstrates the protective nature of the osmolyte (Fig. 6). In these simulations, the native structure of PrPC is protected in the amyloidogenic environment as no conversion is observed. PrP in TMAO simulations starting with either a PrPC or PrPSc conformation converged onto a PrPC-like structure with an omega-loop in the region of the unstructured N-terminus. By acting primarily on the solvent surrounding the protein and not on the protein itself, TMAO can protect PrPC from conversion and rescue misfolded species along the conversion pathway.
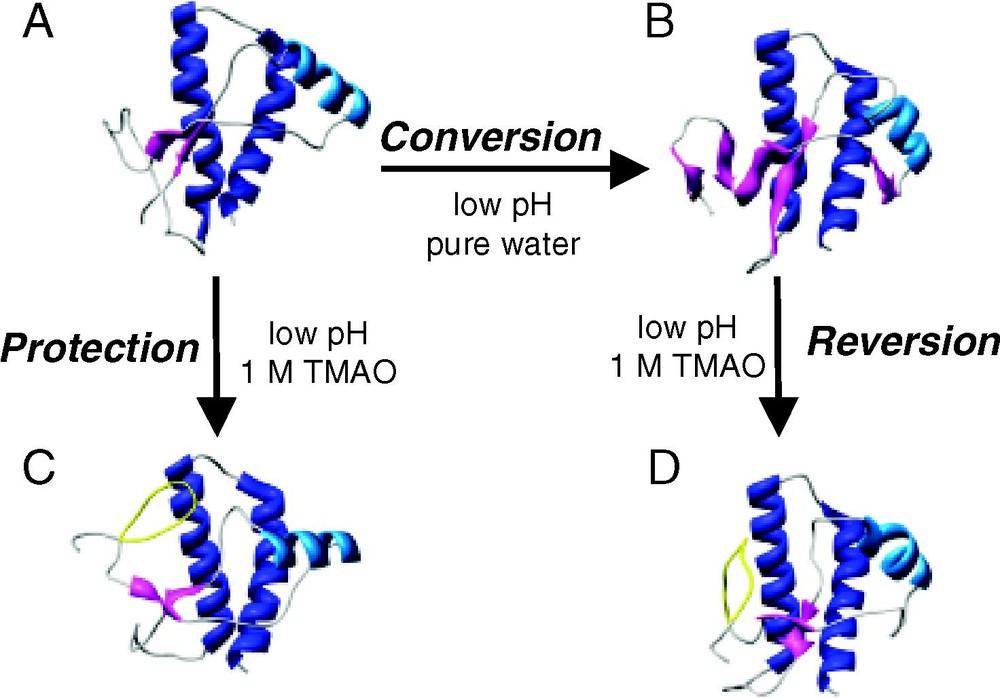
Snapshots from MD simulations of hamster PrP (residues 109–219) and the chemical chaperone TMAO. (A → B) The conversion simulation: low pH triggered conversion of PrPC to a PrPSc-like species. (A → C) The protection simulation: TMAO protected PrPC from conversion under amyloidogenic conditions. (B → D) The reversion simulation: TMAO rescued a misfolded species. (A) Minimized Syrian hamster NMR structure of PrPC. (B) PrP after 10 ns of MD simulation at low pH yielding a PrPSc-like structure rich in extended structure. (C) A snapshot of the 10 ns structure from MD simulation at low pH in 1-M TMAO, which used the NMR structure (A) as the initial coordinates. (D) A snapshot of the 10-ns structure from MD simulation at low pH in 1-M TMAO using (B) as the starting structure. The omega-loop formed in the presence of TMAO (residues 113–123) is colored yellow.
Other small molecules that have shown promises as therapeutic agents include Congo-red derivatives, polyphenols, polyene antibiotics, polyamines, cyclic tetrapyrroles, lysosomotropic antimalarial compounds (readers are directed to the accompanying references for a review of potential therapeutics) [93,94]. Efforts are currently underway to determine how these small molecules act on PrPC or PrPSc to inhibit disease progression.
7 Effects of non-protein moieties on protein misfolding diseases
A handful of the possible environments that could influence PrP structure and in turn either impede or facilitate the disease process have been discussed above and include: pH, carbohydrates, lipid membranes, metal ions and small molecules. All of these factors can have a significant impact on the conformation of PrP with the exception of glycosylation, which does not appear to significantly influence PrPC structure. Glycosylation does, however, play an integral role in disease, as glycosylation affects distribution and trafficking of PrPC and deposition patterns of PrPSc aggregates. More studies of glycosylated PrPC, like NMR relaxation experiments and molecular dynamics simulations, may yet reveal dynamic effects associated with the glycosylation of PrPC.
Observation of other non-protein moieties affecting PrP conformation does not preclude the central hypothesis in prion disease; that it is a protein-only disease. As proof of the protein-only hypothesis, (1) PrPSc would have to be synthesized directly from PrPC, and then, (2) proved to be infectious, triggering disease in vivo. The first step in the proof of the protein-only hypothesis has already been accomplished; rec-PrPC can be triggered to form a misfolded species that shares the properties of purified PrPSc from diseased brains. The second step has proved to be more difficult. At present time only one successful experiment demonstrating infectivity of synthetic PrPSc has been published [19] and these results need to be reproduced and confirmed. The inefficiency of conversion in vitro and the difficulties in demonstrating infectivity do not necessarily invalidate the protein-only hypothesis, but may serve to highlight the complex interactions that allow for the efficient unfolding, misfolding and aggregation of PrP in vivo.
Through continued study and identification of environmental factors that cause misfolding of PrP in vivo, general therapeutic strategies could be designed to protect against disease. Conditions like low pH and high concentrations of metal ions may not be dangerous for just PrP, as other proteins and peptides linked to disease are also susceptible to misfolding and aggregation in similar environments. For example, metal ions can increase the rate of formation of Aβ fibrils [95], the peptide implicated in neurodegeneration associated with Alzheimer's disease. A low pH environment can trigger misfolding and aggregate formation of many proteins including Aβ [96,97], transthyretin [98], lysozyme [99], and superoxide dismutase [100]. Protein misfolding diseases appear to have much in common, similar environments can trigger comparable events, even the formation of misfolded species with similar structural characteristics [101–103]. As such, identification and modulation of environmental factors that either facilitate or inhibit the progression of prion diseases may have an impact on misfolding diseases in general.
Acknowledgements
We are grateful for financial support provided by the National Institutes of Health (GM 50789 to V.D. and Pharmacological Sciences Training Grant GM 07750 to M.L.D.).