

1 Introduction
Despite an abundant biodiversity, all living beings are based on a similar cellular system, which is run by a population of self-organized molecules: proteins. Proteins catalyze, regulate and control most procedures that occur in a cell for the benefits of the whole organism. If we wish to understand how living systems work, it is important to understand how proteins function. The way life evolved facilitates this task in that we can compare not only organisms but also physiological processes and their components. Consequently, conclusions can be drawn from the findings and applied from one system to another. In the past decades, numerous methods – e.g., sequence comparisons, protein family prediction, detection of functional domains, conserved domains and altered amino-acid positions within these regions, motif searches, structure prediction or phylogenetic studies – and databases have been developed for the efficient prediction of a protein's function. However, such methods require the ‘correct’ amino-acid sequence as input; the retrieval of such input is still a challenging task [1]. In eukaryotes especially, the formation of the nascent amino-acid sequence implies the possible creation of a huge number of isoforms. Without further experimental evidence it is impossible to predict the existing and biologically relevant proteins from the total of all possible variants. The same is true for most of the other alterations in a protein's structure. What is more, there is still a long way to go to understand how polypeptides interact with each other in order to accomplish a specific task. In this respect, the study of inheritable diseases can be very informative and demonstrate how the smallest of deviations from a protein's structure can lead to its dysfunction and be the cause of severe disorders.
This article gives an overview on cellular processes underlying sequence variety and structural diversity. We have chosen to focus on eukaryotes to narrow down our discussion. The UniProtKB/Swiss-Prot protein knowledgebase [2,3] aims to record all protein variations and their functional impact. Throughout the text, examples of corresponding Swiss-Prot annotation are given and the reader is encouraged to look at further examples when the primary accession number is indicated (e.g. P12345). Complete Swiss-Prot examples are provided at http://www.expasy.org/sprot/tutorial/examples_CRB. Swiss-Prot entries can also be retrieved from the ExPASy server [4] by building the URL, e.g., http://www.expasy.org/uniprot/P12345.txt for the raw format, http://www.expasy.org/uniprot/P12345 for the NiceProt format, or by entering the accession number in the quick search at http://www.expasy.org/ (NiceProt format). Further examples of Swiss-Prot entries can be found via the relevant keywords. Details on the format of Swiss-Prot entries are provided in the user manual at http://www.expasy.org/sprot/userman.html.
2 Formation of the nascent amino-acid sequence
The vast majority of eukaryotic proteins are nuclear-encoded, as are most of the proteins which are the products of DNA-containing organelles. Mitochondria and plastids generate only a small fraction of their own proteome [5–10]. Regulatory mechanisms during protein synthesis can influence the concentration, destination, sequence variety, structural diversity and thus the functional options of the resulting protein. Out of a broad variety of possible mRNAs and protein sequences that can be generated from a single gene (Fig. 1), only one or a few are created at a time, dependent on the type of tissue or the stage of development.
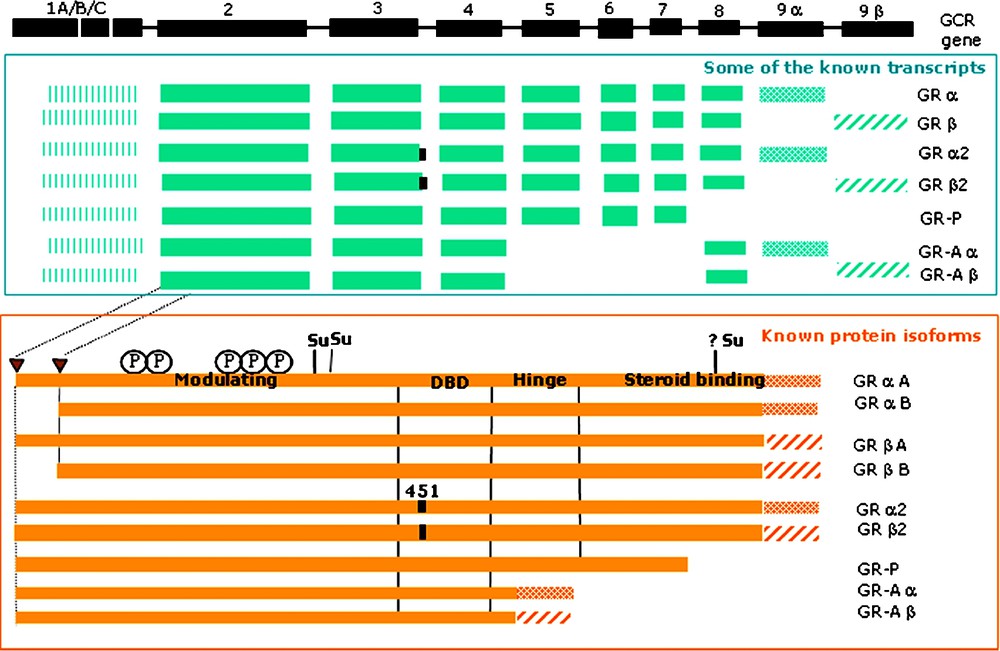
Known sequence isoforms of the human glucocorticoid receptor (GCR) (P04150). The formation of the protein isoforms is based on alternative splicing events, including partial intron retention at the
Known sequence isoforms of the human glucocorticoid receptor (GCR) (P04150). The formation of the protein isoforms is based on alternative splicing events, including partial intron retention at the
The initial step of transcription already gives rise to the production of different transcripts provided that a given gene expression is controlled by more than one promoter. Alternative promoter usage (Q9S7T7) can influence transcription initiation, mRNA stability as well as translation efficiency, thus causing the formation of distinct protein isoforms [11].
The next step – the processing of the primary transcript – is essential for pre-mRNA protection, mRNA export from the nucleus to the cytosol and for efficient translation and translation regulation [12]. In higher eukaryotes, most transcripts contain introns [13] that are removed co- and post-transcriptionally by way of RNA splicing [14,15]. The number of introns is highly variable from one gene to another, within the same organism and between species. In S. cerevisiae, for example, only about 255 of the 6200 expected genes contain introns (an average of one to two [16]) whereas most human genes are thought to contain at least one intron but often many more. The human titin gene contains 363 exons, but it has to be noted that it encodes for an unusually large protein of 38 138 residues [17]. Higher eukaryotes generally perform alternative splicing [18], which is a powerful mechanism for the creation of sequence variety. In humans, it has been estimated that 40 to 60% of the genes are alternatively spliced [19,20]. The production of an extensive variety of mRNA isoforms is achieved through alternative exon splicing, extension of the
The genetic information of a transcript can be further changed via mRNA editing thus possibly giving rise to functionally differing proteins [26]. In higher eukaryotes, the bases uracil and inosine are produced by the hydrolytic deamination of cytosine and adenosine, respectively [27], which could lead to the substitution of an amino acid in the polypeptide. The generation or modification of a stop codon within a reading frame of the mRNA creates an isoform that differs in its C-terminal portion (P04114). RNA editing can also modify the reading frame when bases within a coding region are deleted or inserted, as it has been reported for mitochondria in primitive eukaryotes [28] (Q07434).
After maturation, the mRNA is exported from the nucleus to the cytosol via the nuclear pores by a mechanism which has been conserved from yeast to humans [29]. Translation starts immediately at the cellular translation machinery [30,31]. For the great majority of mRNAs (90%), the initiation site is the first cap-proximal initiator codon located in the appropriate sequence context [30,32]. The initiator tRNA contains an anticodon which is specific for AUG (Met), rarely CUG (Leu), UUG (Leu) or GUG (Val) [33]. Whatever codon is used, methionine seems to be the first amino acid in the nascent polypeptides, except for the CUG-initiated MHC class I bound peptides, which can start with leucine [34]. The translation machinery can make use of alternative initiation codons. In particular, the usage of non-AUG codons at alternative initiation sites has been extensively observed in regulatory proteins such as proto-oncogenes, transcription factors, kinases and growth factors [35]. If the alternative translation initiation codon lies in the same reading frame, polypeptides which differ in their N-terminus are generated. If the long isoform includes an N-terminal transfer signal, which is missing in the shorter isoforms, the generated polypeptides will have distinct subcellular destinations (P08037). If the alternative initiation codon is not located in the same reading frame, the resulting polypeptide can be completely different, as shown for the Sendai virus P/V/C gene (P04862).
In the process of translation, non-standard decoding mechanisms can further manipulate the genetic information. Such mechanisms are known as ‘recoding’. Recoding events seem to be rare but probably exist in many organisms [36,37]. In a process named ‘programmed ribosomal frameshifting’, the translating ribosomes slide one base backward (−1 frameshifting) or forward (+1 frameshifting) at a specific site on the mRNA [36,38,39] (O95190). ‘Stop codon read-through’ is a mechanism by which the meaning of the stop codon is redefined. Well-studied examples are the incorporation of the amino acids selenocysteine (UGA) [40] (P24183) and pyrrolysine (UAG) [41] (O30642). According to our current knowledge, the latter has only been observed in prokaryotes [42].
In Swiss-Prot, all protein variants of a gene are generally described in a single database entry. Cellular mechanisms that lead to an amino-acid sequence differing from the one expected by standard translation of the nucleotide sequence are indicated. Examples of such annotation are given in Table 1. Unlike many other annotations, information on sequence variety is currently not transferred to the corresponding entries of closely related eukaryotes as the level of conservation of such processes in the course of evolution is not yet well defined.
Swiss-Prot annotation of cellular mechanisms leading to an amino-acid sequence different from the one expected by standard translation of the nucleotide sequence. Alternative promoter usage, alternative splicing and alternative initiation events are described in ‘CC ALTERNATIVE PRODUCTS’, with corresponding information on the sequence changes described in the FT lines (‘FT VARSPLIC’, ‘FT CHAIN’ and ‘FT INIT_MET’, resp.). RNA editing data are annotated in the ‘CC RNA EDITING’ line and, depending on the event, also in the ‘FT CHAIN’ or ‘FT VARIANT’ lines. Ribosomal frameshifting is annotated in the ‘CC MISCELLANEOUS’ line. A special FT key, ‘SE_CYS’, exists for stop codon readthrough with selenocystein incorporation. Currently, pyrrolysine integration is still indicated under the feature key ‘MOD_RES’. Each of these events has a corresponding keyword (KW line)
3 Protein sorting and associated sequence modifications
Once synthesized, proteins are usually further processed even if they remain in the same cellular compartment. The initiator methionine of many cytosolic proteins is co-translationally cleaved. This is frequently followed by the acetylation of the new N-terminus [43] (P07327). Such proteins fold immediately and are then transported to their final destination. Translocation to another subcellular compartment requires both protein transfer across at least one membrane and the existence of at least one translocation signal specific to the relevant trafficking mechanism. Major membrane transfer complexes are indicated in Table 2. During transport, nascent proteins are usually associated with chaperones that keep the former in a partly unfolded, soluble conformation whilst protecting them from abnormal folding and aggregation [60]. The chaperones and possibly other factors escort the polypeptide to the relevant membrane receptors of the translocation complex. Protein transfer signals can be proteolytically cleaved during their passage through the membrane (see Table 2), and polypeptides that have to cross several membranes to reach their destination might be cleaved more than once.
Well-studied membrane-transfer mechanisms. (1) N-terminal transfer signal peptides are typically cleaved but exceptions have been found (P05120). In some cases, the uncleaved signal peptide confers important functional properties to the protein [56] (P27170). Abbreviations: TOM = translocase of outer mitochondrial membrane, SAM = sorting and assembly machinery, TIM = translocase of inner mitochondrial membrane, TIM23 = presequence translocase, TIM22 = carrier translocase, PAM = presequence translocase-associated motor, TOC = translocon at the outer membrane of chloroplasts, TIC = translocon at the inner membrane of chloroplasts, TAT = twin-arginine translocation, NPC = nuclear pore complex, PEX = peroxin
| Translocation (from/to) | Transfer complex | Cleaved signal | References |
| Nucleus | |||
| Cytosol/nucleus (bi-directional) | NPC | No | [44] |
| Mitochondrion | |||
| Cytosol | |||
| → outer membrane | TOM/SAM | No | [45] |
| → inner membrane | TOM–TIM22/TIM54 | No | [46] |
| → matrix | TOM–TIM23/TIM17 | Yes | |
| → intermembrane space | TOM/Mia40 | No | [47] |
| Matrix | |||
| → inner membrane | Oxa1 | No | [48] |
| Oxa2 | No | [49] | |
| Chloroplast | |||
| Cytosol | |||
| → outer membrane | Spontaneous? | No | [50,51] |
| → inner membrane | TOC | Yes (1) | |
| → stroma | TOC/TIC | Yes | |
| Stroma | |||
| → thylakoid membr. | Spontaneous | Yes | [52] |
| ALB3 | Yes | ||
| SEC | |||
| → thylakoid lumen | TAT | Yes | [53] |
| SEC | Yes | ||
| Endoplasmic reticulum | |||
| Cytosol | |||
| → extracellular | Sec61 | Yes (1) | [54] |
| → type I membr. p. | Sec61 | Yes (1) | [55] |
| → type II membr. p. | Sec61 | No | [56] |
| Peroxisome | |||
| Cytosol | |||
| → membrane | PEX3/PEX16/PEX19 | No | [57] |
| → matrix | Unknown, | No | [58] |
| (>20 PEX) | [59] |
There are proteins – such as importins – that act in distinct subcellular compartments and have to cross the membrane in both directions. Such proteins make use of the bi-directional nuclear pore complex [44,61], whereas other membrane-transfer complexes support either the import or the export of polypeptides. Secretory routing via the endoplasmic reticulum (ER) [62,63] is accomplished by proteins which are designated not only for secretion but also for their incorporation into the organelles which are an integral part of the secretion path: the ER, the Golgi apparatus and the lysosomes. Such proteins generally have an N-terminal signal peptide which is removed during membrane transfer.
Various mechanisms have been described for the incorporation of proteins into different types of membranes (Table 2). Common examples are the type-I and type-II membrane proteins. The signal peptide of type-I membrane proteins is typically cleaved before the N-terminal of the polypeptide is transferred into the ER lumen. The C-terminal part of the protein remains in the cytosol. As for type-II membrane proteins, the exact contrary occurs: their C-terminal part is transferred into the ER lumen whilst the N-terminal part remains in the cytosol and the internal signal anchor functions as a transmembrane region. Polypeptides without a hydrophobic segment can still be anchored to intracellular or plasma membranes by the covalent attachment of a lipophil group such as glycosylphosphatidylinositol (GPI) (P04058), isoprenoid (P40855), myristate (P62330) or palmitate (O43687). As an example, prenylated proteins – that are estimated to represent 0.5% of all intracellular proteins – have in fact been found on the cytoplasmic surface of plasma membranes, peroxisomal membranes and nuclear membranes [64]. Transport mechanisms are generally well conserved throughout the kingdoms of life, and mitochondria as well as plastids contain translocation systems in their membranes, which point back to their bacterial origin [49,51,53,65]. Dysfunction in transport systems has been shown to be associated with a variety of diseases [66]. An example would be a dysfunction in the nuclear-cytoplasmic transport system, which has been found to give rise to distinct types of cancer [67].
Various aspects of protein sorting and associated protein processing are reported in Swiss-Prot. Extracts from Swiss-Prot entries show relevant annotation in Table 3.
Swiss-Prot annotation relevant to protein sorting and associated protein modifications. The location of the functional protein is annotated in the CC line topic ‘SUBCELLULAR LOCATION’. Cleaved transfer peptides are indicated in the feature table by the feature keys ‘SIGNAL’ for the signal peptide essential in the secretory pathway and ‘TRANSIT’ for the transit peptide which promotes transfer to mitochondria or chloroplasts. The extent of the mature protein is indicated in the feature key ‘CHAIN’ and the protein name given in the DE line is followed by the term ‘precursor’. The FT key ‘TRANSMEM’ is used to annotate both, transmembrane domains and signalling anchors; ‘MOTIF’ is used for short stretches of transfer signals which are not removed. Keywords (KW) refer to the subcellular location, the type of signal, and sometimes the routing paths
4 Protein folding and structure
Polypeptides fold spontaneously and some of them can attain their native conformation unaided in an extremely short lapse of time [68]. Many other proteins, however, require the assistance of chaperones and possibly additional folding factors and enzymes such as protein disulfide-isomerases or prolyl isomerases, which protect the polypeptides from aggregating and help them reach their native state [69,70]. Both the folding pathway and the three-dimensional structure are dictated by the amino-acid sequence of a polypeptide. Protein folding is driven by the free energy of conformation that is gained by going to a stable, native state [68]. Helices and beta-strands are rich in hydrogen bonds and therefore energetically favourable structures, which is why they form spontaneously in most unfolded proteins. Contacts between these elements give rise to local, native-like conformations and their nucleation facilitates achieving the final structure. This process implies a stochastic approach since the process of protein folding goes through a number of folding intermediates [68–72]. The native state is stabilized by favourable interactions both at the surface and inside the folded polypeptide chain, and involves direct contacts between amino-acid residues as well as contacts with water molecules or ions. Disulfide bonds strongly increase the stability of a protein; this is particularly important for proteins consisting of short chains, which have been derived from larger precursors [68]. Even though the protein structure is a stable construct, it can contain regions of conformational flexibility and many enzymes undergo allosteric transitions (P00489).
Protein structures are strongly conserved in evolution and are the basis for protein classification [73,74]. Most globular proteins contain both alpha helices and beta-strands; other classes of proteins have either only an alpha-helical structure or purely a beta-stranded one, such as aquaporin-CHIP (P29972) or the beta-barrel porin ompF (P02931), respectively. In the native structure, helices and beta-strands are often connected by beta-turns or mobile loops, thus forming structural motifs. Repeats consist of small structural elements, each of which is too short to be stable but multiple consecutive copies stabilize each other and form typical super-structures, such as the propeller-like structure consisting of Kelch repeats or the arch-like shape made up of leucine-rich (LRR) repeats [75]. Domains are stable, independent folding units, with a characteristic secondary structure topology. On an average, the smallest domains are about 35 amino acids long, but large domains can consist of several hundred amino acids. The average size of a domain is about 160 residues [76]. Usually, short domains such as the zinc-finger and EGF-like domains are stabilized by metal ligands or disulfide bonds. In a given protein, domains often have specific functions, as exemplified by the 40-kDa peptidyl-prolyl cis-trans isomerase (P26882) (Fig. 2). This enzyme contains two structural domains: a catalytic PPI domain and a region consisting of three TPR repeats that mediates interactions with heat-shock proteins.
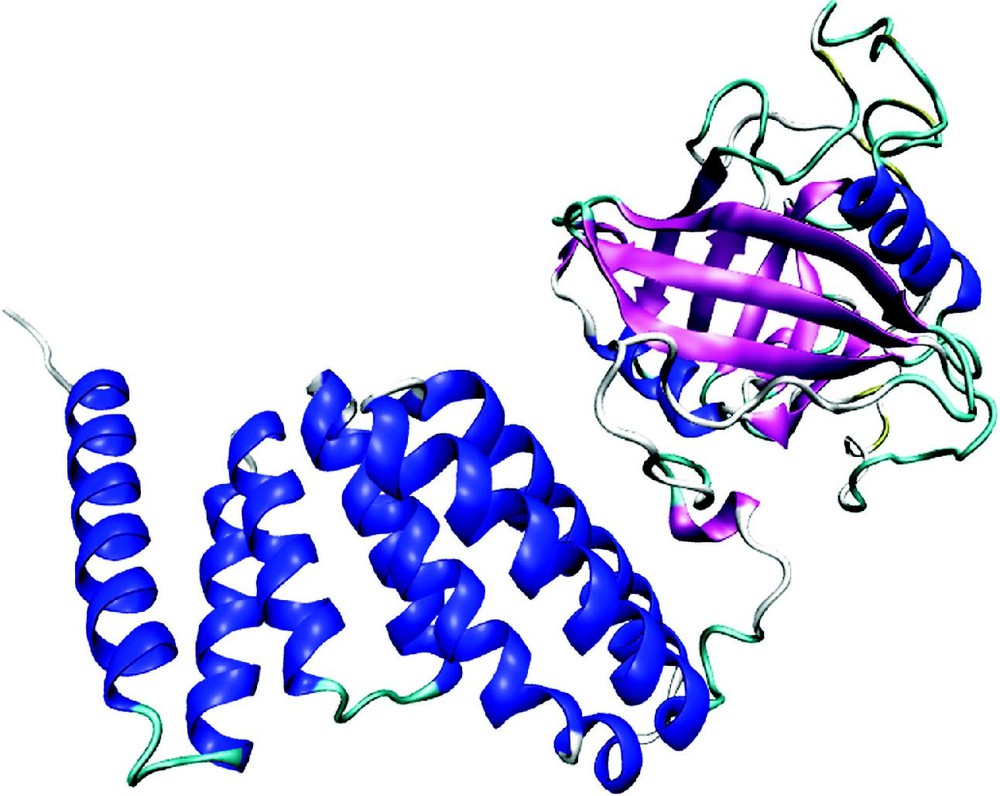
Ribbon plot of bovine 40-kDa peptidyl-prolyl cis-trans isomerase, generated from PDB 1IHG. On the right side the catalytic PPIase domain with a high content of beta-sheet, on the left side a second domain containing three tetratrico peptide repeats (TPR) that mediate protein–protein interactions. The TPR repeat is a degenerate repeat motif of ca. 34 amino acids, each containing 2 antiparallel helices. The minimal functional unit seems to be a structure containing three such repeats.
The proteome of higher eukaryotes contains a high proportion of multi-domain proteins and a multitude of different domain combinations, which are hypothesized to have arisen by gene duplication and exon shuffling events. These mechanisms are thought to be a driving force for the rapid evolution of new proteins and more complex organisms. [77,78]. Alternative exon usage can change the number and type of domains present in the protein and has profound effects on a protein's function, such as its capability to interact with other proteins and ligands [79] (Fig. 1).
In a Swiss-Prot entry, the extent of structural elements is recorded in the feature table (Table 4). Cross-references to structure-related databases facilitate access to complementary information.
Swiss-Prot annotation relevant to the protein structure, extracted from the entry P26882. Experimental information is indicated in the RP line of the relevant reference. Similarity to domains or a protein family is given in the comment (CC) line topic ‘SIMILARITY’. Cross-references to structure-related databases are recorded in the DR lines. The keyword ‘3D-structure’ (KW) is added to an entry whenever the 3D-structure of the protein or part of it has been resolved experimentally. The boundaries of repeats, domains, helices, beta-strands and turns are annotated in the feature table (FT)
5 Function-related protein modifications
The majority of protein modifications occur post-translationally, i.e. once the protein has undergone folding, and is typically catalyzed by specific enzymes found in the ER, the Golgi apparatus, the cytoplasm or the nucleus. In the literature – as in Swiss-Prot – the term PTM (Post Translational Modification) is often used in a rather general sense and includes both co- and post-translational modifications. Protein alterations can be relevant to the transport of the nascent protein, to protein folding, and to the activity and function of the native protein. This section is addressed to function-related protein modifications.
Various native proteins, including structural proteins, hormones, neuropeptides and secreted enzymes, are cleaved to achieve their mature form. As an example, receptors of the notch signalling pathway (P46531) are cleaved after ligand-binding, to release the cytoplasmic domain which then acts as a transcription factor in the nucleus. Various seed storage proteins are cleaved once they have been transported into protein storage vacuoles; cleavage will bring on a new conformation necessary for their deposition (P09802).
In addition to these specific cleavages, more than 300 distinct types of amino-acid modifications have been identified to date in prokaryotic and eukaryotic proteins [80], of which each is specific to one or more amino acids. According to the RESID database (release 41) [80], cysteine is the amino acid which undergoes the most possible kinds of alterations (Fig. 3). One or more distinct types of modification can occur in a protein – and in various combinations – thus effectively extending the structural variety of a gene product. Many sequence modifications are irreversible, thus changing the property of the protein irreversibly too. Reversible protein modifications such as phosphorylation, S-nitrosylation or O-glycosylation can alter the dynamics of a protein and are considered to be major mechanisms for the fast and intricate regulation of metabolic enzymes [81] and hence signalling pathways [81–84]. Competition for different PTMs at a single site in response to distinct upstream signals, can provide a fine-tuning mechanism for signal integration. A good illustration for this kind of mechanism is the ‘histone modification code’, where the acetylation of Lys-9 in histone H3 would lead to transcriptional activation, or its tri-methylation would lead to transcriptional repression [85]. Transcription factors are also regulated through different and exclusive lysine-directed PTMs: Acetylation can stimulate or inhibit their activity, monoubiquitination generally enhances it, SUMOylation controls their subnuclear localization, and polyubiquitination signals their destruction by the proteasome [86].
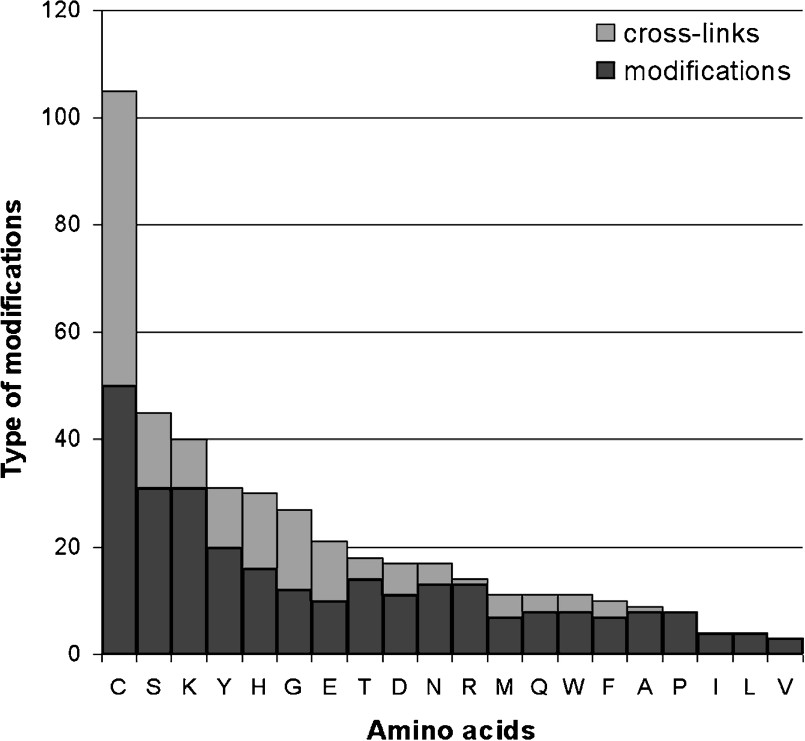
Number of different types of amino-acid modifications and cross-links between distinct amino acids based on the RESID database (release 41).
Protein post-translational modification is a powerful mechanism to enhance the diversity of protein structures and to modify protein properties. Thus, it is hardly surprising that missing or abnormal modifications of a given protein can be the cause for dysfunction (see also Section 7) [87–90]. In particular, the highly dynamic modifications in glycoproteins are known to be associated with or are the reason for many distinct diseases [91]. Similarly, various abnormal post-translational modifications have been observed in aging tissue [92].
In Swiss-Prot, modified amino acids are annotated in the feature table. The type of feature key used depends on the nature of the modification and the exact name of the modified amino acid is indicated in the description field (Table 5). A list of the controlled vocabularies is available at http://www.expasy.org/sprot/userman.html#PTM_vocabularies. Modifications can also be described in the comment (CC) lines under the topic ‘PTM’. More than 40 keywords (KW) are used to describe protein modifications (see http://www.expasy.org/cgi-bin/get-entries?cat=PTM).
Swiss-Prot annotation relevant to function-related protein modifications. Position-specific information is indicated in the feature table. Additional details can be provided in the comment (CC) line topic ‘PTM’
6 Protein–protein interactions
Cellular processes are generally carried out by protein assemblies rather than by individual proteins [93]. Protein complexes consist of at least two subunits, but can also be formed by dozens of polypeptides or more. Complex associations may last only a fraction of a millisecond but they can also form stable cellular structures. Many proteins are part of various complexes, each of which may act in a distinct functional context. As an example, the regulatory subunit of the cAMP-dependent protein kinase A (P10644) also interacts with the 40 kDa subunit of the replication factor C (P35250) [94]. A function is also frequently attributed to a complex rather than to the individual chains that make up the complex. In the case of homologous quaternary structures, the number of subunits can vary between taxonomic groups – as has been shown for DNA clamps which consist of a three-domain dimer in bacteria but a two-domain trimer in eukaryotes and Archea [95].
The formation of complexes is based on direct protein–protein interactions (PPIs) between the subunits, mediated by electrostatic interactions, hydrogen bonds, van der Waals attraction and hydrophobic effects. The minimal contact surface appears to be around 800 Å2 [96] and the average interaction surface is 1600 ± 400 Å2 [97]. The interaction strength between two proteins is quantitatively characterized by the equilibrium constant
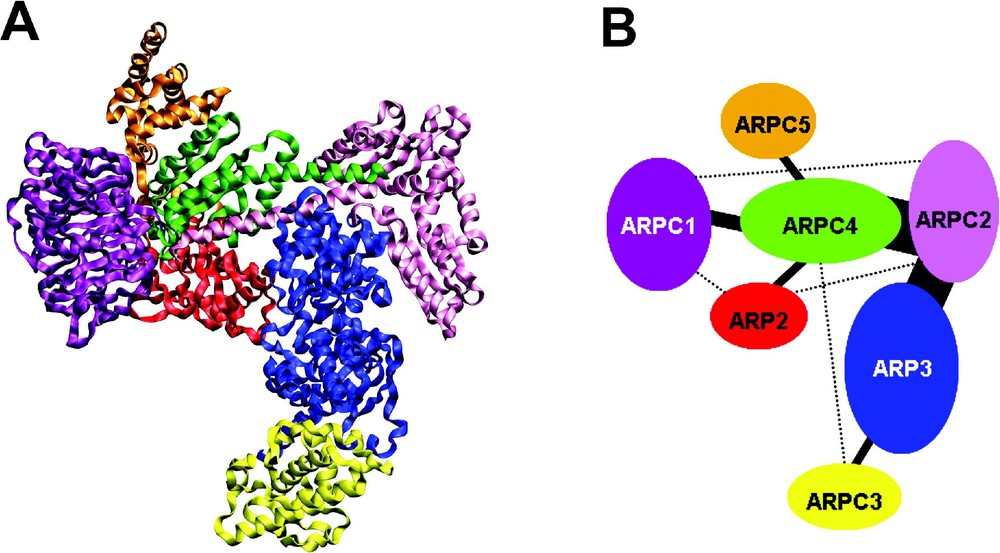
The ARP2/3 complex. (A) Ribbon plot, generated from PDB 1K8K [100], shows the 7 subunits of the complex in distinct colours; (B) Schematic interaction diagram of the 7 subunits. Analysis of the complex structure reveals 6 binary interactions (threshold:
The ARP2/3 complex. (A) Ribbon plot, generated from PDB 1K8K [100], shows the 7 subunits of the complex in distinct colours; (B) Schematic interaction diagram of the 7 subunits. Analysis of the complex structure reveals 6 binary interactions (threshold: ... Lire la suite
The ‘classical’ types of contact between proteins are domain–domain interactions in which two independently folded domains – usually complementary in shape and charge – form the interface between the proteins [104]. In general, a central region protected from the solvent contributes the most to the binding energy. Sequence mutations in this region have a large impact on the interaction. Much research in domain–domain interaction has been carried out on serine proteases and their inhibitors. Another common contact type is the domain–peptide interaction: a domain of one protein interacts with a small portion of a second protein, which is unstructured in the absence of its binding partner. Typical examples are the binding between the major histocompatibility complex and the antigen, and regulatory modules of intracellular signalling cascades, like the SH2 domain which interacts with phosphotyrosine-containing target peptides in a sequence-specific manner. Protein–protein interactions can also induce conformational changes, as is often observed in signal transduction mechanisms. An example would be the tyrosine kinase Src, which contains SH2 and SH3 domains that mask the catalytic site of the enzyme. The Src-activating ligands contain specific SH2- and SH3-binding motifs; while the kinase and its ligand interact, the inhibition of the catalytic site is relieved [105].
For a long time, PPIs were studied individually using genetic, biochemical and biophysical techniques. More recently, genome-scale studies provide vast interaction datasets from high-throughput experiments. Yeast two-hybrid screens detect binary PPIs, without giving further information on functional complex associations [106–110]. Comparative analysis revealed not only an important amount of false-negative interactions (Fig. 4B), but led to the estimation that 30–50% of the reported interactions from large-scale projects do not exist [111–115]. Affinity purification with mass spectrometry detects the components of a complex without registering direct PPIs [116,117], however. Remarkably, stable complexes have rarely been found, and the composition and functionality of many of the detected transient complexes are not yet well understood. Proteome-wide interaction networks have been established for yeast [118], C. elegans [119] and fruit fly [109] and strive to assign functions to uncharacterized proteins. The yeast proteome consists of approximately 6200 proteins and would account for a minimum of 30 000 interactions. The number of existing PPIs is probably much higher since interactions occurring during distinct developmental stages or responses to different external conditions have also to be taken into account [111]. X-ray crystallography still provides the ‘golden standard’ in terms of accuracy for the structural analysis of protein complexes (Fig. 4B) but is not scalable to unravel a complete cell ‘interactome’. Understanding the functional protein assembly in a cell will require an integrated approach by combining different experimental and theoretical methods [120].
The quaternary structure and any type of interaction with other proteins or protein complexes are described in Swiss-Prot entries. Cross-references to the IntAct database [121] facilitate access to complementary information including experimental details (Table 6).
Swiss-Prot annotation relevant to PPIs. Literature-derived protein–protein interactions are annotated in the CC line topic ‘SUBUNIT’. Binary interactions in the CC line topic ‘INTERACTION’ are automatically derived from the IntAct database. The FT keys ‘REGION’ and ‘VARIANT’ are used to specify an interaction
7 Protein function and disease association
Once a polypeptide is functional, it participates in the coordination or maintenance of cellular processes, such as metabolism, transport, communication, growth, cellular biosynthesis or apoptosis. Dysfunction or, on the other hand, the simple lack of a given protein can lead to a disease status, but in many cases a combination of causes result in a disorder: genetic predisposition, environmental factors, infections and aging. Due to the complexity of living systems, understanding disease mechanisms may require moving from the global view of the organism as a whole to a more focused view of specific components at the molecular level.
Genetic mutations are one of the causes of protein alterations. Missense mutations, which ultimately lead to amino-acid substitutions, are the most frequent type of mutations related to disease [122,123]. A major challenge in medical genetics is to distinguish disease-causing missense mutations from neutral polymorphisms with no clinical relevance. Several criteria can be used to assess the pathogenicity of a mutation: de novo appearance of the mutation, segregation of the mutation with the disease within pedigrees, absence of the mutation in control individuals, change of amino-acid polarity or size in the protein, change in a domain which is conserved between species and/or shared between proteins belonging to the same family. If the function of the protein is known, the effect of the mutation can be assessed by in vitro mutagenesis and functional assay. Generally, missense mutations affect amino-acid residues with a relevant functional and structural role. They may have a deleterious effect in that they can lead to protein mistargeting, unstable miss-folded proteins, alteration of normally post-translationally modified sites, disruption of catalytic sites, or disruption of protein complexes. A mutation in the sulfatase modifying factor 1 (SUMF1; Q8NBK3) which activates all sulfatases (e.g., P15848, P15289, P08842) by transforming a conserved cysteine to 3-oxoalanine (Table 7) [124] is an example of a disorder based on missing PTMs. The lack of such a modification leads to multisulfatase deficiency (MSD), a severe multisystemic disorder which combines all the effects observed for each single sulfatase defect [125,126].
Swiss-Prot annotation related to protein function and associated disorders. In the given example, the dysfunction of the SUMF1 impedes the activation of all sulfatases, thus causing MSD. In the Swiss-Prot entries, the protein function and related disease information is indicated in the comment (CC) lines. The sequence positions of variants are recorded in the feature table (FT). For disease mutations, the corresponding disorder is indicated in the description field of the feature key ‘VARIANT’; if no additional information is given, the substitution refers to polymorphism. The keyword ‘Polymorphism’ is assigned to an entry if protein sequence variants are not associated with a disease status: the keyword ‘Disease mutation’ indicates disease-linked mutations. A specific disease term is used as keyword only when a disorder is associated with mutations in more than a single protein. A list of medical-oriented keywords is available at http://www.expasy.org/cgi-bin/get-entries?cat=Disease. Cross-references are provided to databases relevant to medical genetics such as Online Mendelian Inheritance in Men (OMIM) [129], dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), GenAtlas (http://www.genatlas.org/) and locus-specific databases
The mutation-disease relationship is not trivial. One same mutation can cause different phenotypes in individuals depending on the genome and the environment. Disease mutations in the fibroblast growth factor receptor (P21802) are a cause of certain craniosynostoses. In particular, the Cys342Tyr mutation is associated with two different craniosynostoses: Crouzon syndrome and Pfeiffer syndrome [127]. On the other hand, mutations in more than one gene may be necessary for disease manifestation, as shown for the Bardet–Biedl syndrome [128]. In order to elucidate the complex relationship between genotype and phenotype, great interest is devoted to the identification and cataloguing of single nucleotide polymorphisms (SNPs) that are expected to facilitate large-scale association genetics studies.
Swiss-Prot stores information related to protein function and detailed information is given on an enzyme's activity (Table 7). Disorders associated with the dysfunction of a protein are also indicated. Swiss-Prot aims to record all known single amino-acid substitutions, with an emphasis on human disease-related variants and their functional effects.
8 Concluding remarks
Even though Swiss-Prot entries generally correspond to a gene rather than a protein, it stores a wealth of information regarding protein variety and functional diversity. As shown for a number of proteins above, protein variants can be essential for an organism and protein dysfunctions can cause severe disorders. Information, such as sequence variety, protein complexes or PTMs that are not related to a conserved domain, is difficult to predict in a reliable fashion, – or indeed difficult to predict at all – and thus has to be annotated manually on the basis of experimental results. As knowledge on protein variety is essential for the understanding of cellular systems, Swiss-Prot aims to record all such data. For human entries, annotation on the naturally occurring polymorphisms and disease mutations provides valuable information on a protein's function. In Fig. 5, statistics on some relevant annotation is given based on the Swiss-Prot data from UniProt release 5.0 of 10 May 2005.
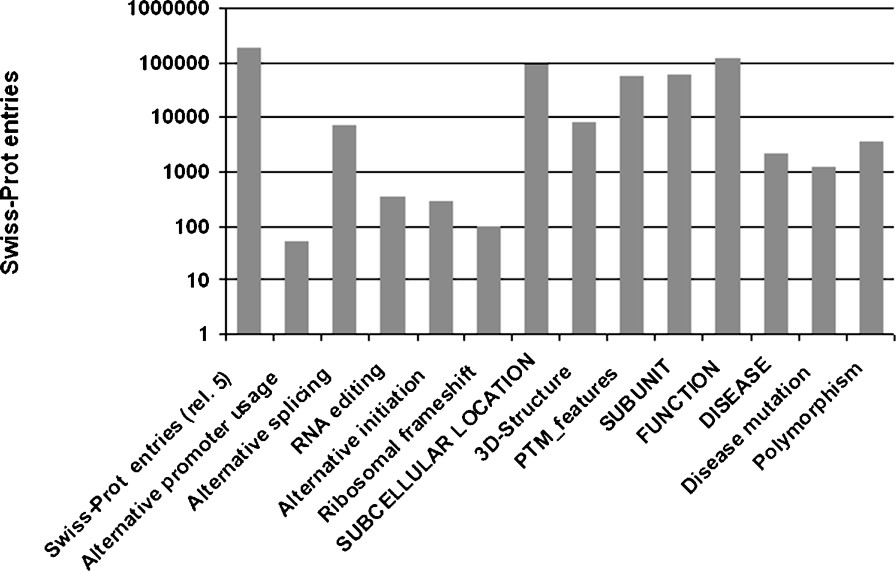
Statistics on selected annotation items. The first column shows the number of entries in Swiss-Prot of UniProtKB release 5.0, other values on the x-axis indicate certain annotation items: terms in uppercase are comment (CC) line topics, terms in mixed case indicate keywords, except for ‘PTM_features’, which summarizes all feature keys relevant to post-translational amino-acid modifications.
Acknowledgements
The authors would like to thank Vivienne Baillie Gerritsen for the correction of the manuscript. We are grateful to Anne Estreicher for stimulating discussions on protein synthesis. This work is partially funded by the Swiss Federal Government through the State Secretariat for Education and Research and the National Institutes of Health (NIH) grant 1 U01 HG02712-01. Apologies to authors whose work was not cited due to space limitation.
Vous devez vous connecter pour continuer.
S'authentifier