

1 Introduction
Since the first sequencing of a whole eukaryote genome was completed (Saccharomyces cerevisiae in 1996 [1]), sequencing methods have greatly improved and costs have reduced significantly. This has opened the road to sequencing large genomes relatively quickly [2]. As a consequence, the number of organisms for which full genome information is available has vastly increased in the last decade. In plants, genomes of fourteen angiosperm species have already been completed (Arabidopsis, canola, grape vine, poplar, papaya, cucumber, rice, sorghum, maize, brachypodium, cassava, potato, soybean and African oil palm; on February 1st, 2010) and many additional genomes will be available soon (http://www.genomesonline.org). With predicted technological advances, the number of organisms completely sequenced is likely to grow exponentially [3]. This will open new avenues to comparative genomic analyses offering exceptional opportunities to better understand the mechanisms that shape genome organization [4], as well as shedding new light on the genetic mechanisms that led to the emergence of novel adaptations during evolutionary diversification [5]. In the following paragraphs, we detail the considerable value of full genome information for evolutionary studies in eucaryotes, and highlight the risks linked to the poor taxonomic coverage of full genomes, which will persist for a few years to come. Recent advances in the evolutionary genomics of the C4 photosynthetic pathway in grasses [6,7] are discussed to illustrate the advantages and highlight some limits of molecular studies based solely on whole genomic data.
2 Complete genomes to study evolutionary novelties
Identifying the genetic changes linked to the emergence of adaptive novelties is an important challenge contributing to our deep understanding of the evolutionary processes at the molecular level [8,9]. Comparison between related organisms that exhibit different phenotypes can help identify the genetic changes responsible for a novel adaptive trait as well as some genetic features promoting its evolution [10,11]. For example, the impact of gene duplication and polyploidy on phenotypic diversification is an attractive topic that is better addressed by comparison of genome portions between related species [12]. Moreover, when genes involved in a trait have been previously identified, comparative approaches can give strong insights into the constraints on the recruitment of particular genes for the new function [7,13]. The quality and spectrum of the data on genes and genomes is a key factor determining the accuracy of comparative evolutionary approaches and the high amount of information provided by full genomes projects will transform comparative genetics into comparative genomics, a step that is necessary for an integrative understanding of evolutionary biology.
Comparative analyses of multigene gene families are strongly facilitated when full genomes are available [8]. First, complete gene sequences are directly accessible, including introns and non-coding flanking regions that often contain promoters, while the sequencing of complete genes on a large panel of species is often time-consuming using PCR-based cloning and can be challenging [14,15]. In addition, full genomes provide information that would be almost unattainable with other techniques. For instance, the exact genomic location of the studied genes can reveal that two paralogues lie on duplicated chromosomes or are tandemly repeated and thus helps reconstruct the genomic mechanisms linked to genetic diversity [16,17]. Finally, a precise knowledge of the number of genes that compose any multigene family almost necessarily requires complete genomes, since demonstrating that one gene lineage is absent from a non-model organism is difficult [18], particularly with PCR-based methods [14]. When merging the genomic information with functional and evolutionary approaches, an exhaustive picture can emerge, bringing our understanding of evolution to a level that was never reached before.
3 The case of C4 photosynthesis in grasses
In plants, several of the most economical crops belong to Poaceae (or grass family) promoting intensive genetic and genomic studies in this family [19]. Poaceae is a worldly dominant family distributed in various environments from wet or dry tropical conditions to extremely cold habitats. The complete genome of four grass species, rice, sorghum, maize and brachypodium [16,20–22], is now available and others should be released in the next months (e.g., foxtail millet [19]), with a predicted burst of complete grass genomes in the coming years [19]. This high quantity of genomic data will be exceptional for a plant clade offering wonderful opportunities to understand evolution of traits at the molecular level. In particular, the multiplication of genomes will allow comparative analyses, shedding new lights on the molecular changes that gave rise to adaptive novelties, such as for developmental transitions to modulate flowering time or modify floral organ morphology [23,24], to change grain morphology [25], to develop new disease resistance [26] or photosynthetic adaptation, such as the C4 trait in tropical conditions [27].
Sixty percents of C4 species belong to the grass family (Poaceae), with several major crops, such as maize, sorghum or sugarcane [28]. The C4 pathway consists of a set of morphological and biochemical modifications that together allow concentrating CO2 around Rubisco and thus reducing photorespiration. The emergence of the C4 traits is an evolutionary puzzle since the establishment of such a CO2-pump has involved a high number of changes but occurred up to 18 times independently in grasses [29]. A key point to understand the evolution of this trait is that all enzymes involved in the C4 pathway already exist in the C3 ancestors, but are responsible for other functions [27]. In addition, the clustering of C4 origins in some plant clades strongly suggests that these groups of organisms possess attributes that increase the probability of C4 evolution [30]. C4 facilitators should be searched for in genomic properties, such as the propensity of some C3 lineages to create gene duplicates (particularly via polyploidisation) [27]. Besides theoretical works, genetic promoters of C4 evolution remained out of reach until recently. While comparative analyses of multigene families encoding C4 enzymes identified some changes in the protein sequences that are likely linked to C4 evolution [31–33], the lack of genomic information hampered our understanding of the genome dynamics that led to genetic diversity of these gene families. The recent release of sorghum genome [16], the first C4 plant to be completely sequenced, removed many obstacles on the road to C4 comparative genomics. A recent work by Wang et al. [6] used a comparative analysis of rice and sorghum genomes to test for the importance of gene duplications for C4 evolution and the action of adaptive evolution during the acquisition of C4-specific enzymes. These authors demonstrated that gene duplication (e.g., via whole genome duplication, tandem duplication or single gene duplication) was indeed an important step allowing evolution of most C4 genes, although it was not involved in the evolution of all enzymes of the C4 pathway (e.g., nadp-mdh). A long time lag between the availability of duplicates and the appearance of first C4 grasses, together with different genesis of C4 genes, also suggested that the transition process was very long before the establishment of fully C4 plants [6]. These results are key improvements of our understanding of C4 evolution and are a first step toward understanding the genetic factors linked to the recurrent evolutions of C4 photosynthesis in grasses, although their scope can be limited by the small number of species compared.
4 Toward an exhaustive taxon sampling
Nowadays, the low number of species completely sequenced limits the resolution of comparative genomics of C4 photosynthesis. Rice, brachypodium and Andropogoneae (e.g., sorghum, maize) are only very distantly related and their most recent common ancestor dates back to more than 50 million years ago [29]. Sorghum and maize belong to the PACMAD clade and share a common C4 ancestor, whereas rice and brachypodium belong to the sister BEP clade, which contains only C3 species [29]. The recent genomic comparison of rice and Andropogoneae, two distantly related C3 and C4 taxa, can be problematic and is unlikely to accurately resolve the genetic mechanisms directly linked to C4 evolution, since 50 million years of independent accumulation of genetic mutations can strongly blur any signal. For instance, the identification of orthologs between rice and sorghum-maize can be challenging, because independent losses of alternative homeologs could have occurred after gene duplication, as in the case of genes encoding the phosphoenolpyruvate carboxylases [6,32]. Erroneous assessments of orthology can mislead interpretations regarding the number of gene duplications and their nature (Fig. 1). Moreover, the comparison of highly divergent genes can bias the estimations of past selective pressures [34]. This highlights the limits of comparative analyses based on a few whole genomes in reconstructing an accurate evolutionary history of genes responsible for the emergence of a novel adaptive trait. In the next years, the number of species to be compared will strongly increase, as tens of grass genomes should quickly become available [19]. Unfortunately, sampling of species to be sequenced was driven by economical interests and did not take into account grass diversity and evolutionary issues. In particular, all sequenced C3 taxa belong to the exclusively C3 BEP clade whereas the PACMAD clade is represented by C4 species only [19], which will prevent a direct comparison of C4 species with their C3 sister taxa. Sequencing the whole genome of C3 PACMAD species would definitively suppress problems associated with taxon sampling, but is not yet realistic due to the low economical and agronomical interests of such plants. An alternative is to set up dense comparative analyses of specific gene families, and full genome information of the model species are useful to design appropriate methodologies to sequence genes on non-model species and help understand the genomic context in which the studied genes lie.
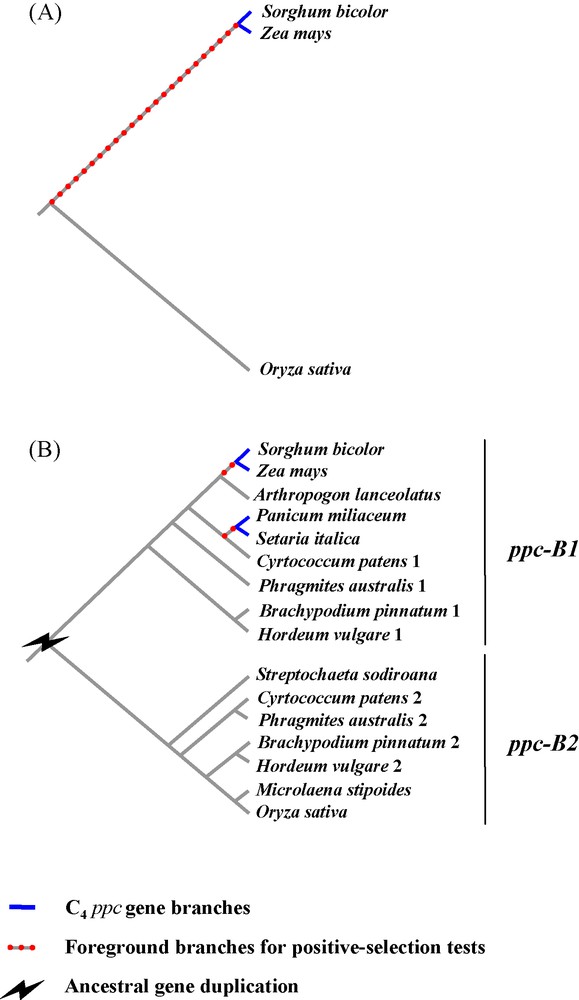
Limited taxon sampling leading to erroneous statement of orthology and reducing the power of positive-selection tests. The case of the ppc gene family in grasses (i.e., gene duplicates ppc-B1 and ppc-B2) is taken as an example [31,32]. A. First, when only complete genomes of rice, sorghum and maize are considered, one ppc-B copy is detected in each species. The gene of rice (Ehrhartoideae, C3) is distantly related from the gene of sorghum and maize (Panicoideae, tribe Andropogoneae; C4). As a consequence, the branch at the base of the C4 specific gene, used for positive-selection tests, is long; B. Second, by analyzing ppc-B gene segments (generated by PCR) on a species sample covering different grass subfamilies, two gene clades (ppc-B1 and ppc-B2) are identified in the phylogram. Gene duplication in the ancestor of these species is inferred from this topology (for a topology based on a more comprehensive species sampling see [31,32]). Gene ppc-B2 was not isolated in some subfamilies such as Ehrhartoideae (e.g., Oryza), while gene ppc-B1 was absent from numerous PACMAD species (in particular, Sorghum and Zea). This means that genes ppc-B1 and ppc-B2 were independently lost in different grass lineages. These observations were further confirmed by the presence of both genes in the complete genome of Brachypodium distachyon [22], as well as the distinct genomic locations (non-collinear regions) of ppc-B1 and ppc-B2 in rice and sorghum, respectively [16,32].
In a recent study, such an approach was used to assess the genetic diversity of genes encoding NADP-malic enzyme (nadpme) in three model grasses (rice, sorghum and Brachypodium distachyon) [7]. Long fragments of nadpme were then sequenced from about 50 other grass species chosen to represent the different subfamilies and a variety of photosynthetic types. The joint analysis of genes extracted from full genomes and those isolated via PCR showed that four nadpme lineages appeared through recurrent gene duplications before grass diversification. The encoded enzyme of one of these lineages (nadpme-IV) acquired a plastid-specific localization through the acquisition of a first exon containing a transit peptide long before the different C4 origins. Interestingly, this gene lineage became involved in C4 photosynthesis at least five times independently, and it is strongly suggested that its plastid expression, which is necessary for the C4 pathway, predisposed it for the C4 function. On the other hand, the supposed absence of this nadpme-IV gene lineage in genomes of Chloridoideae may have prevented the evolution of the C4 biochemical subtype based on NADP-malic enzyme in this large grass subfamily [7]. We are looking forward to the future release of additional C4 grass genomes for exploring such hypotheses about C4 evolutionary genetics.
5 Conclusion
While full genome sequencing projects bear great promises for evolutionary biology, we must keep in mind that the low taxonomic coverage they offer limits the scope of comparative genomics. In particular, the very long branches in the phylogenetic trees that include only genes from distantly related organisms can blur the signature of the past selection pressures. Similarly, the long evolutionary gap between completely sequenced organisms hampers causation between observed genetic differences and known phenotypic divergence. To maximize the impact of full genome projects, comparative analyses should be complemented by the sequencing of genes from non-model organisms of interest, to reduce the branch lengths in phylogenetic trees and obtain a taxon sampling suited for each research question. This can improve the accuracy of selection tests and, in the case of C4 photosynthesis, already gave strong and novel insights into the genetic mechanisms linked to the recurrent origins of this complex and highly adaptive trait [6,7].
Acknowledgments
G.B. and P.A.C. were respectively funded by the Intra-European Fellowship PIEF-GA-2008-220813 and the Swiss National Science Foundation grant PBLAP3-129423.