

1 Introduction
Eukaryotic gene expression is a complex process (Fig. 1) that is regulated at transcriptional and post-transcriptional steps. It involves successive, but overlapping and interrelating, steps that are subjected to regulation and quality controls. The transcription rate (TR) is the rate at which mature molecules in the cytoplasm appear, while the rate at which RNA polymerase II transcribes a gene can be named “nascent” TR. Both rates describe the number of mRNA molecules being produced. Nonetheless, nascent TR is always higher than TR because, logically, many molecules are degraded during elongation or before leaving the nucleus. The actual proportion of this apparently unsuccessful transcription is currently unknown.
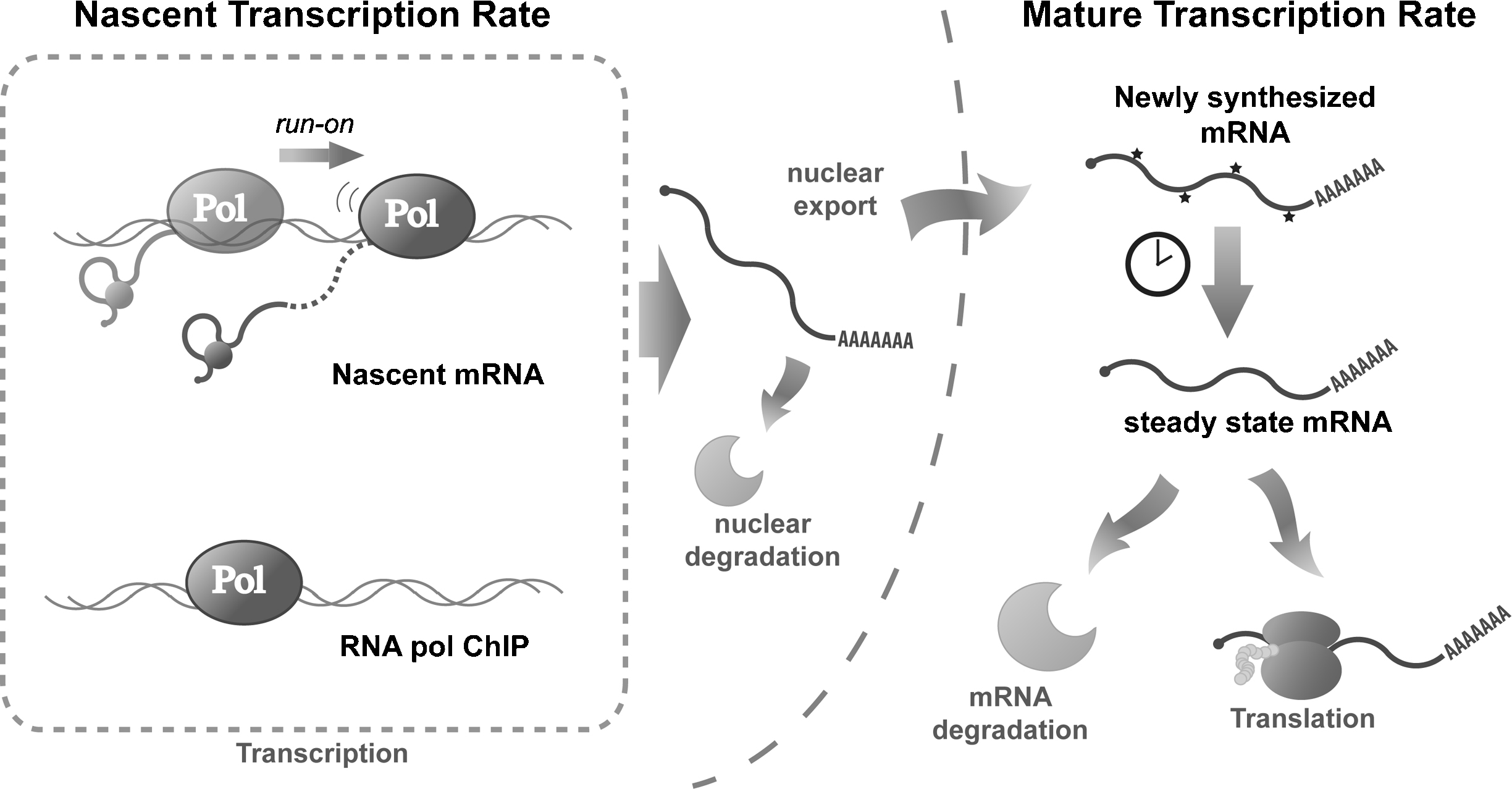
Methods to study the transcription rate at genomic scale. mRNA is transcribed, processed and transported from genes to the cytoplasm. The TR can be measured along all the different steps. For the direct measurement of the TR (nascent TR, left-hand side of the figure), it is possible to measure the physical association of polymerase (RNA pol ChIP), the presence of nascent transcripts or the elongation activity by run-on. It is also possible to measure the newly synthesised mRNA or to estimate mature TR either by pulse-labelling experiments or indirectly by assuming a dynamic equilibrium between its transcription (TR) and degradation (DR) rate (right-hand side of the figure).
mRNA molecules do not live forever. This relative instability is the result of both the degradation of erroneous molecules and the biological need for mRNA turnover, which allows cellular re-adaptation to changing environments. The degradation of mature and correct mRNA molecules is conducted by specialised exonucleases and companion proteins (see Ref. [1] for a review). The mRNA degradation rate (DR) is regulated and controlled, and plays an important role at the cytoplasmatic mature mRNA level, as with the TR. Each mRNA species has its own characteristic stability (RS). Therefore, the amount (or concentration) of an individual mRNA (RA) is the balance between these processes: TR and DR.
When the environment does not change, it is logical to assume that most genes have a constant RA and are, therefore, in steady-state conditions. Thus in this situation, the TR and DR for each mRNA are equal. In other situations where the RA varies, this change could be due to variations in the TR, the DR, or in both. Traditionally, transcriptional responses have been studied only as changes at the RA level both on a single gene scale and on the genomic scale without differentiating the respective contributions of the TR and the DR. These approaches have been reviewed many times (e.g., [2]), whereas the genomic study of RS and the TR is more recent.
The use of genomic techniques to evaluate RA, TR and RS allows the study of general rules for the kinetic regulation of RA for groups of genes [3,4]. Moreover, the serial profiling of kinetic parameters during gene expression helps to identify functionally-related genes that could use common regulatory pathways at various levels in eukaryotic gene expression. The best-suited organism for genome-wide expression analyses is the yeast Saccharomyces cerevisiae. For this reason, this review focuses on the genome-wide protocols and data for RS and TR estimations with this yeast, and also on the general conclusions on yeast transcription and mRNA turnover drawn from them.
2 Methods for the genomic evaluation of transcription rates
Transcription rates can be defined as the number of mature mRNA molecules produced in a cell by a unit of time. However, the definition of a “mature mRNA” is flexible because mRNA is subjected to many maturation steps (Fig. 1); and several different populations of mRNA exist in the cell. Thus, the measured transcription rate can change according to the mRNA subpopulation being studied.
Several techniques have been developed to measure transcription (Fig. 1), some rely on the direct measurement of the transcriptional process, while others use indirect measures to mathematically derive the TR (see below). Although all the direct methods study the same process, the different experimental approaches used can produce slightly different results. This is due to not only the possible methodological biases of each technique, but also biological differences depending on the particular step of the transcription process being measured.
Most TR estimation techniques available do not directly measure the number of mRNA molecules produced in a given time, rather they determine the number of RNA polymerase II molecules present in a gene to compute nascent TR (Fig. 1, left-hand side). All these methods rely on the assumption of a constant RNA pol II elongation rate; however, this is a drawback because it is well-established that the elongation rate is subject to regulation [5].
The simplest approach is the use of the measurement of the polymerase bound to each gene as an indicator of its transcription level. This can be done using the well-known Chromatin immunoprecipitation techniques (ChIP) in their different variants, e.g., ChIP-chip [6,7] and ChIP-Seq [8]. When using an antibody against RNA pol II, ChIP-chip provides a measurement of RNA pol II density. The use of different antibodies can add selectivity to the type of immunoprecipitated RNA pol II molecules, such as using antibodies against phosphorylated forms of the CTD tail of Rpb1p to select elongating molecules [9,10]. However, the ChIP technique also presents several disadvantages (Table 1). This technique measures the DNA fragments that are bound by the polymerase. Thus, it is only able to detect the physical presence of the polymerase in that region, and not the activity itself. Moreover, as it studies bound DNA, it cannot provide information about the polymerase orientation.
Summary of genome-wide techniques to measure TR and mRNA stability.
| Transcription rates | |||||||
| Advantages | Limitations | References | |||||
| Radioactive Run-on-based methods (GRO) | Direct estimation of elongating RNA pol II densities Nascent TR |
Average TR estimation throughout the coding region | Assumes a constant elongation rate for RNA pol II | [3,46,50] | |||
| Non-radioactive Run-on-based methods. | Allows for tiling array and HTS single nucleotide resolution | Direct estimation of elongating RNA pol II densities Nascent TR |
Less sensitive than radioactive methods. May require nascent RNA purification | Assumes a constant elongation rate for RNA pol II | [17] Not yet implemented for yeast | ||
| Chromatin immunoprecipitation-based methods | Different antibodies allow the differentiation of RNA pol II states | Direct estimation of RNA pol II densities Nascent TR |
A fraction of RNA pol II molecules does not elongate | Assumes a constant elongation rate for RNA pol II | Has no strand specificity. | [51] | |
| Indirect estimation from RA and RS | No need for experimental protocol | Mature mRNA TR | Error can be increased by mathematical calculations | Relies on calculations from other experimental values | Assumes steady-state conditions for mRNAs | [22] | |
| In vivo labelling with UTP precursors | Fluorescent labelling | Mature mRNA TR | Requires a time lapse | Requires newly synthesised RNA purification | [20,21,32] | ||
| Isolation of nascent chromatin-bound transcripts | Allows for tiling array and HTS single nucleotide resolution | Direct estimation of elongating RNA pol II densities Nascent TR |
Long and demanding protocol A fraction of RNA pol II molecules does not elongate |
Assumes a constant elongation rate for RNA pol II. Requires newly synthesised RNA purification | [11,12] | ||
| mRNA stabilities | |||||||
| Advantages | Limitations | References | |||||
| rpb1-ts | Simple method | Involves heat shock to cells | Difficult to use under dynamic conditions | Requires a time lapse | Requires a mutant strain | [29,30] | |
| RNA pol II inhibitors | Simple method | Involves toxic shock to cells | Difficult to use under dynamic conditions | Requires a time lapse | [30,52] | ||
| Simple indirect estimation from RA and TR | No need for experimental protocol True in vivo natural growth conditions No stress |
Instantaneous measurement | Assumes steady-state conditions for mRNAs | Error can be increased by mathematical calculations | Relies on calculations from other experimental values | [3] | |
| Indirect estimation from RA and TR | No need for experimental protocol True in vivo natural growth conditions No stress |
Does not assume steady-state conditions for mRNAs | Error increased by complex mathematical calculations | Relies on calculations from other experimental values | Needs several time points. | [33] | |
| Pulse and chase with labelled UTP precursors | True in vivo natural growth conditions No stress |
Does not assume steady-state conditions for mRNAs | Long and demanding protocol | Requires a time lapse | Needs several time points | [19,20] Not yet implemented for yeast | |
| Pulse with labelled UTP precursors | True in vivo natural growth conditions No stress |
Assumes steady-state conditions for mRNAs | Long and demanding protocol | Requires a short time lapse | [31,32] |
Another approach, similar to ChIP techniques, relies on the direct detection of nascent chromatin-bound transcripts. This can be done because the high stability of ternary complexes formed in vivo between genomic DNA, RNA pol II, and nascent RNA, allows its study either by means of co-immunoprecipitation of RNA with RNA pol II antibodies without the need for crosslinking [11] or by purification of chromatin and, then, of the nascent RNA bound to it [12]. It has the advantage of measuring only those polymerases that have a nascent transcript hanging from them, thus providing a more direct measure of transcriptional activity. However, the fact that one RNA polymerase is associated with an mRNA does not necessarily imply that it is transcriptionally active as the polymerase can also be backtracked or paused [13].
The standard approach to directly quantify the density of elongating RNA polymerases is the transcription run-on assay (TRO) [13]. This method is based on allowing the extension of nascent transcripts by cellular RNA polymerases in the presence of labelled (usually radioactive) nucleotides. Afterwards, RNA is extracted and nascent-labelled transcripts are hybridised to a single filter containing multiple gene probes (Genomic Run-on, GRO), allowing quantitative comparisons. GRO takes advantage of the fact that not every polymerase sitting on DNA is able to perform run-on in the presence of labelled nucleotides to discriminate between actively elongating polymerases and polymerases stopped without mRNA or unable to elongate for some other reason (e.g. backtracking). Under the assumption that RNA polymerases elongate at a constant rate, the quantification of their density provides a TR measure at the time of RNA labelling [13].This technique has been applied to S. cerevisiae to perform accurate kinetic measures of TR in both standard growing conditions [4,14], and in plenty of different stress responses [3,15,16]. One of the advantages of yeast is that the GRO assay can be performed on whole cells, which allows very rapid and accurate measurements. The possibility of combining non-radioactive labelling (e.g., BrUTP, Biotin-UTP) of run-on samples by high-throughput sequencing [17] or microarray technology [18,19] has only been recently shown.
Another possibility to estimate the transcription rate, which is not based on the study of RNA polymerase presence/activity, is measuring the appearance of a new mRNAs during a known time lapse. This can be done by introducing a labelled nucleotide precursor, for example pulsing with thiouracil [20] or thiouridine [21]. Thiolated RNA is then purified by affinity chromatography and used for microarray analyses. This method is suitable for in vivo applications, and may overcome some of the disadvantages of the run-on and ChIP assays in higher eukaryotes to determine transcription rates. As the measured TR needs a long pulse with the precursor, it is not instantaneous, but corresponds to an average in the time-labelling period (Table 1). One important difference between this technique and those based on the detection of RNA polymerase presence/activity is that the required time lapse incubation prevents the detection of transcripts with a very low stability; for instance, cryptic transcripts or other transcripts subject to nuclear degradation.
Finally, the TR can also been calculated indirectly from the knowledge of both the steady-state level of each mRNA species (the transcriptome) and the half-life of those species. This approach needs to assume the existence of a steady-state condition for the transcriptome [4], but the advantage here is that no experimental method is required. However, it does have its drawbacks as it relies on having accurate data for both RA and RS. As RS measures are especially challenging (see below), the use of these data to indirectly compute the TR can imply more experimental errors. Despite these problems, the use of indirect calculations for the TR [22] is widely generalised in the literature [23,24].
3 Effect of cryptic transcription on different TR measures
In the last few years, the development of genome-wide techniques has revealed an unexpected complexity among eukaryotic RNA populations in which unstable and overlapping transcripts are more common than previously thought [25]. The function of this plethora of non-coding transcripts is still not well-known [26]. However, these findings evidence that cryptic transcription is a more widespread process than formerly thought. As most of these non-coding RNAs possess a very low stability, the use of methods for measuring nascent TR will reveal the contribution of the cryptic transcription that will not be seen in the measured transcriptome.
One drawback of nascent TR methods is that cryptic transcription is added to the value of the regular one. Therefore, to be able to express the calculated TR, it is necessary to assume that the percentage of cryptic transcription is small. Methods that measure the increase of mature mRNA (e.g., direct pulse-based techniques or indirect TR estimations) are not affected by cryptic transcription. The comparison with methods that measure RNA polymerase activity (e.g., run-on) can provide information about the existence of cryptic transcription, and also about the early post-transcriptional process for the mRNAs pointing at groups of genes that are subject to post-transcriptional regulation (e.g., mRNA maturation and export, etc.) [14].
4 Methods for the genomic evaluation of mRNA stabilities
Several different techniques have been used for years for single gene mRNA stability determination in S. cerevisiae (reviewed in [27,28]); some are not applicable to genomic studies, while others are scalable for genomic use. The most used methods are those based on transcription stopping by means of either transcription inhibitors (mainly thiolutin or phenantroline) or the temperature-sensitive mutant rpb1-1 of the largest RNA pol II subunit (RPO21). After the transcription shutoff, the remaining RA is determined by DNA chip analyses to calculate the decay of every single mRNA over a time interval using the known decay kinetics (Fig. 2). These analyses have established that the mRNA half-lives for yeast range from 3 to 300 min, and that 23 min is the average time [29]. The yeast genes belonging to the same functional category tended to have similar RS and can be regulated at the mRNA stability level [30].
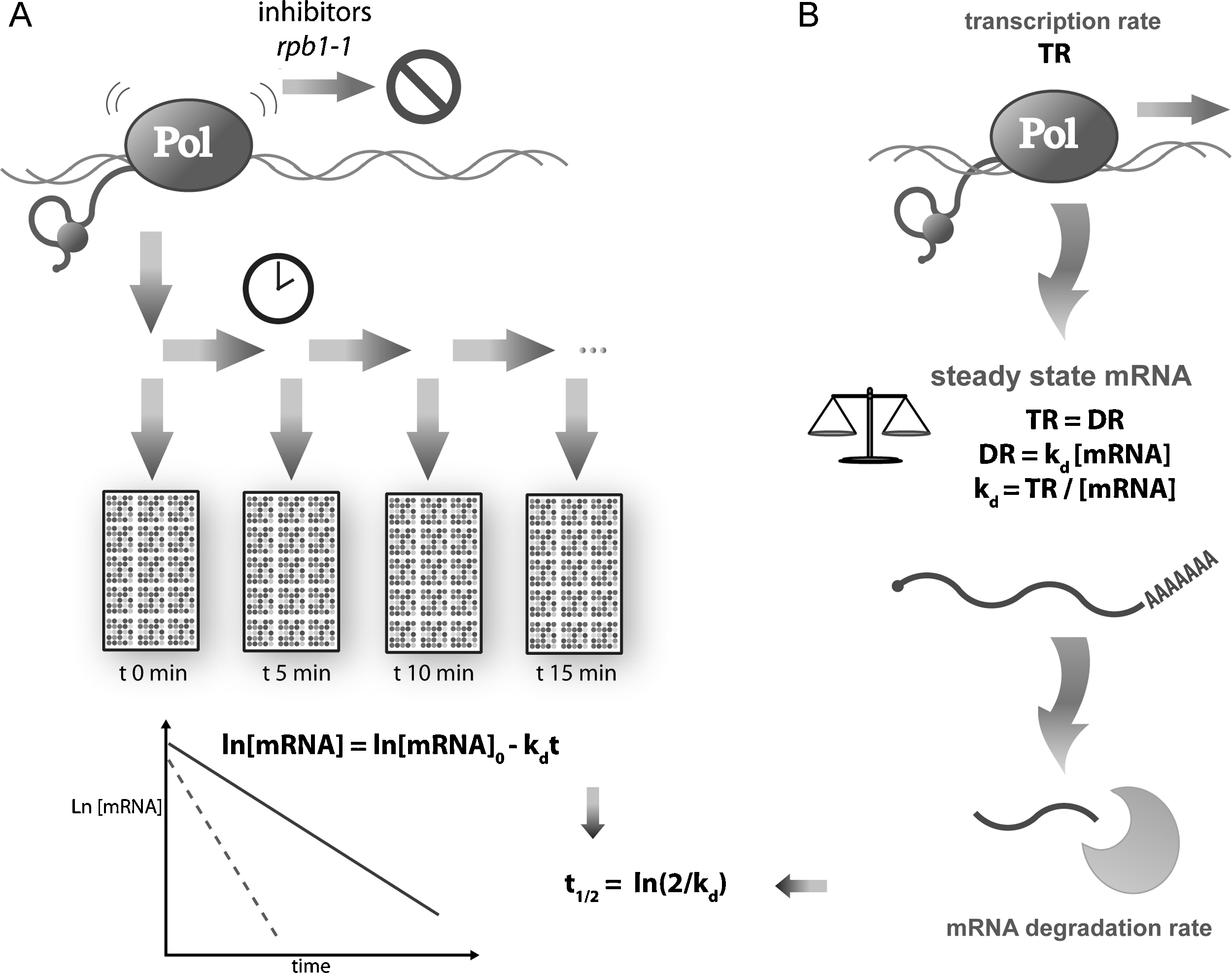
Methods to study mRNA stability at genomic scale. A) mRNA stability can be measured directly by inhibiting the transcription (using inhibitors or thermosensitive mutants) and then following the disappearance of mRNAs over time. B) Alternatively, it can be also computed indirectly by assuming a steady-state for mRNA production and degradation or by applying kinetic laws for non-steady-state conditions (see the main text).
However, the use of transcription shutoff procedures involves several problems (Table 1). The first is that mRNA half-lives are calculated from the data collected over a considerable time interval (up to 120 min). Therefore, the calculated half-life is an average along the time interval. Second, these methods cause a cell stress response due to a temperature shift or a drug addition needed to block transcription, which changes the expression of some genes or possibly alters the mRNA degradation mechanisms during the experiment, as has been shown for human cells in culture (discussed in [31]). Such studies are not appropriate for monitoring stress-induced genes because an instantaneous increase in the mRNA level occurs before the general shutoff, which prevents the decay curve for those mRNAs from being calculated [29,30].
Other direct methods used to calculate RS are those based on the metabolic labelling of mRNA. The most simple method consists in using a pulse with RNA precursors, such as bromouridine [19] or 2,4-dithiouracil [20], which allows to purify nascent RNA with affinity columns. The chase of labelled RNA at different time points after the pulse allows the direct determination of RS from the decay curve. This method has been developed for Toxoplasma gondii [20] and mouse cells [19] in culture because these cells efficiently incorporate those UTP precursors. Recently, it has been shown that it is possible to incorporate 4-thiouridine into yeast cells [32].
To avoid transcription shutoff problems, alternative strategies are needed. One such strategy is based on the properties of the steady-state conditions and on chemical kinetics laws. Since the degradation rate follows first-order kinetics (DR = kd RA) in steady-state conditions (TR = DR):
In most papers, mRNA stability is given as a half-life (referred to here as RS) instead of kd, therefore:
| (1) |
This allows us to calculate values for either the TR or RS whenever the respective values and RA are available. Experimentally determined TR values are needed for RS determination (see the previous section). This method offers the advantage of not needing a time lapse in which circumstances may vary. However, it has two important drawbacks: it is necessary to assume steady-state conditions for each mRNA species (which is not usually easy to demonstrate [4]), and it may imply the mathematical amplification of experimental errors (Table 1).
Since steady-state conditions cannot be guaranteed for most genes in many circumstances, a different mathematical approach can be applied. In those cases where Eq. (1) is not operational, a differential equation could be used:
Another possibility is labelling cells in culture with a pulse of labelled UTP precursors and isolating a single time point after it. The comparison of nascent mRNA with total (mature) mRNA isolated from the same sample allows the determination of the incorporation rate of nascent mRNA into the mature mRNA pool and to, therefore, assume steady-state conditions to mathematically calculate the RS for it (reviewed in [20]). An application of this method, known as Dynamic Transcriptome Analysis (DTA) has been recently developed for yeast [32]. DTA has estimated that most transcripts have a very low synthesis rate per cell and cell cycle, and that their half-lives have a median of about 11 min. Interestingly, this disagrees with the median half-life, 23 min, calculated by direct methods [29]. This may mean that the direct estimations are affected by the stress caused by stopping transcription (see above). A drawback of this method is that the time for pulse-labelling the cells (from few minutes in yeast to several hours in higher eukaryote cells) limits its use for fast-changing situations.
5 The steady-state transcriptome: a snapshot of yeast transcription
By using the GRO method, we have found that the median TR in yeast genes is about 7 mRNAs/h; 90% of them have TRs of between 2.33 and 29.7 mRNAs/h, and the total transcription for RNA pol II in a yeast cell growing in standard conditions is about 60,200 mRNAs/h [14].
Gene functional groups tend to have similar TRs. Histone genes are the highest transcribed genes with a median TR of 206 mRNAs/h, if we take into account they are only transcribed during the S phase. The statistical distribution shows that less than 1% of yeast genes have more than one molecule of elongating RNA pol II/gene. Therefore, we can state that productive transcription on a yeast gene is a rare phenomenon in an actively growing cell. In any case, the existence of “unproductive transcription” (cryptic transcription, see above) inside and outside coding regions [25,35,36] may alter these figures. Therefore, we calculate that about 690–1200 RNA pol II molecules actively transcribe in a snapshot of an average cell during exponential growth in YPD medium [14]. On the other hand, as our data are population averages and since cell populations tend to be quite variable [37], it is most likely that some individual cells in some genes have higher densities than others.
The number of mRNA molecules in a yeast cell has been determined by different methods. Published values oscillate between 15,000 [38] and 60,000 [37]. When an intermediate value is used, 26,000 molecules/haploid cell, the turnover of the global yeast transcriptome in a exponentially growing cell is about 2–3 times per generation. This is obviously an average of very different turnovers for different mRNA species. Some are replaced many more times per cycle. Others, however, can be inherited from the cell mother, or even from grandmothers [39].
6 The changing transcriptome: strategies for mRNA turnover in response to stress
The most interesting application for the kinetic study of gene expression is the case of changing environments. The GRO method allows the instantaneous determination of the TR and RA and, by mathematical approaches (see above), the determination of RS values for mRNAs. Following this strategy, we have studied yeast responses to change in carbon sources [3], and in oxidative [15], osmotic [16] and heat [34] stresses. In all cases, we have found that changes in both the TR and RS contribute to changes in RA for most stress-responsive genes. In many cases, responses are homodirectional, i.e., increases in the TR together with increases in RS (decreases in kd) bring about an increase in RA for stress-induced genes and, correspondingly, the opposite applies to stress-repressed genes (mostly translation-related genes). In those cases, the TR seems to be the main factor that determines changes in RA [3,34]. However, there are interesting differences in RS profiles, which indicate that changes in mRNA stability are used to finely tune the response. In other cases, changes in RS partially compensate changes in the TR. For instance, in changes in glucose for galactose, a general downshift in the TR is compensated by general mRNA stabilisation which keeps the mRNA concentrations relatively high while cells re-adapt to the new metabolism [3]. This case illustrates the possibility of changes in RS for the purpose of saving mature mRNAs, even when they are not being translated [40] for future use. Other authors [41] have used their own and our published data on oxidative stress [15] to reveal that stress-induced genes display a paradoxical decrease in RS. This has been interpreted as a way to speed the transcriptional response because unstable mRNA is able to change its RA faster in accordance with chemical kinetics laws [33]. Another interesting application for the kinetic study of gene expression is the use of TR changes to cluster the genes during a stress response. This is a better method for the determination of gene regulons than RA profiles [42] because, as stated above, the transcription factors controlling gene transcription and RA profiles depend not only on TR changes, but also on RS changes. RS profiles, on the other hand, can be used to look for RNA binding proteins (RBP) which could be responsible for changes in the stability of their target mRNAs [43,44]. We have used these profiles during the heat stress response to find the putative RBPs involved in the regulation of mRNA stabilities in some gene groups [34].
For the first time in any organism, the existence of genomic data for the three variables involved in transcription (RA, TR and RS) in yeast allows a detailed study of the strategies followed by yeast genes to cope with the functions they perform. However, mRNA should be translated into protein, which can also be controlled at the stability level. A comprehensive study of gene regulation should use all six variables in gene expression (TR, RA, RS, translation rate, protein amount and protein stability) throughout a changing physiological situation to evaluate the respective contributions of each one to the expression of each gene. Currently, however, protein variables are only known for a single condition: exponential growth in a complete medium. Nevertheless, the study of steady-state conditions may shed light on the strategies that genes may adopt during physiological changes. We have used published protein variables data, together with those of the mRNA variables, to evaluate the general strategies of yeast genes under this steady-state condition. We have found that functionally-related genes follow similar strategies. We have also drawn the conclusion that regulation at the transcription level is quantitatively more important than at the translation level, and that RS plays a distinctive role for gene expression: to modulate the response speed [45]. Once again, this highlights the importance of the kinetics in gene expression strategies.
7 Concluding remarks: studies in other organisms
Although techniques for the genomic evaluation of RA have been widely developed, the state of the art for the TR and RS in organisms other than S. cerevisiae is much less advanced. mRNA half-lives are short for free-living organisms (a few minutes), and are much longer for complex eukaryotes (up to many hours). For TR determination, run-on techniques are only possible in eukaryotes. In higher eukaryotes, TRO should be performed on isolated nuclei, causing a lag between the actual physiological state of the cell and the capture of nascent RNA. Moreover, studies in which a comparison between RAs and TRs has been made offer only qualitative results [46–50]. Thus, yeast S. cerevisiae is currently the best-suited organism to perform a genomic view of mRNA turnover.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
The authors are grateful to the members of their laboratories for discussion and support and to Dr Anick Pierce for her French translation. V.P is supported by an EMBO fellowship and A. Jordán-Pla by a FPI fellowship from the Spanish MCINN. J.E.P-O. was supported by grants from the Spanish MCINN (BFU2007-67575-C03-01 and BFU2010-21975-C03-01), and from the Regional Valencian Government (Generalitat Valenciana - ACOMP/2009/368).