

1 Introduction
Aplastic anemia (AA) is a rare and a heterogeneous disorder characterized by the inability of the bone marrow (BM) to produce an adequate number of blood cells. The majority (70%) of these cases are categorized as idiopathic because their primary aetiology is unknown. In a subset of cases, a drug or infection can induce the bone marrow failure and in approximately 15–20% of patients, the disease is constitutional/inherited [1]. Fanconi anemia (FA) is the most frequent inherited cause of AA [2]. Affected individuals may have one or more somatic abnormalities such as short stature, microcephaly, microphthalmia, thumb and radius deformities, skin hyperpigmentation such as “café-au-lait spots”, cardiac, renal, genitourinary, and/or other malformations. A subset of FA patients (approximately a third) have no overt physical/somatic abnormalities [3].
There is considerable genetic heterogeneity in FA with 15 different FA genes (FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ/BRIP1, FANCL, FANCM, FANCN/PALB2, FANCO/RAD51C, and FANCP/SLX4) [3]. The most prevalent being FANCA, FANCC, FANCG, and FANCD2 [4]. Except for the very rare FANCB, which is located on X chromosome [5], all other FANC genes are autosomic and the disease is recessive.
FA cells characteristically show an abnormally high frequency of spontaneous chromosomal breakage and hypersensitivity to DNA cross-linking agents such as diepoxybutane (DEB) [6] and mitomycin C (MMC) [7]. Chromosomal breakage test with these agents is the technique of reference to distinguish FA from other AA [8].
In the majority of cases, a precise diagnosis can be made with careful history, physical examination, and a positive chromosomal breakage blood test.
An accurate diagnosis will influence the choice of therapy. Since FA patients are hypersensitive to all DNA cross-linking agents, they require a modified pre-transplantation conditioning regimen, with a lower than usual dose of cyclosphamide or lower doses of chemotherapeutic agents [9].
The main objective of the present research was to determine rational criteria for correctly and unambiguously diagnose patients with FA using MMC-test.
2 Patients and methodes
2.1 Patients and samples
Between 2004 and 2011, a total of 205 AA patients suspected for FA were referred to the cytogenetic laboratory at the Institut Pasteur de Tunis for chromosomal fragility evaluation.
Patients were selected for MMC-induced chromosomal breakage studies on the basis of AA, congenital abnormalities, and other hematological indicators known to be connected with FA, as well as family screening. Sensitivity and chromosomal instability of peripheral blood lymphocyte cultures, induced by MMC, were the main factors for dividing patients into two groups: MMC-sensitive (MMC+, FA) and MMC-insensitive (MMC−, non-FA) patients.
2.2 Chromosome fragility test
A cytogenetic study was carried out on peripheral blood cultures stimulated with phytohemagglutinin. Two cultures were set up for each patient: (i) cultures without alkylating agents and (ii) MMC-induced cultures. MMC-treated and untreated peripheral lymphocyte cultures of patients were prepared in the same conditions and at the same time as their counterpart culture of control subjects. MMC-test was carried out according to methods described in the literature with minor modifications [7,10]. In summary, the culture unit consisted of 1.6 ml of heparinized blood added to 5 ml of RPMI 1640 medium (GIBCO) supplemented with 20% fetal bovine serum (GIBCO), 1% penicillin-streptomycin solution (GIBCO), 1% L-glutamin (GIBCO) and 1% phytohemagglutinin (EUROBIO).
At initiation, MMC solution (SIGMA) was added at a final concentration of 50 ng/ml and incubated for 72 hours at 37 °C in a 5% CO2 atmosphere at high humidity.
This MMC concentration was optimized in order to not affect the mitotic index and to induce multiple chromosomal breaks in cells from FA patients while having little clastogenic effect on normal cells. Breakage analysis was evaluated on unbanded metaphases (stained with Giemsa). For chromosome fragility evaluation, at least 50 metaphases selected randomly were analyzed from each culture.
Each cell was scored for the numbers and types of structural abnormalities according to methods previously described [6]. Gaps were not counted as chromosome breaks, whereas single chromatid breaks, isochromatid breaks and acentric fragments were scored as one break. The other rearrangements and radials were scored as two breaks.
Chromosomal breakage score was expressed as percentage of aberrant cells, breaks per cell and breaks per aberrant cell. Results were compared with healthy controls.
2.3 Statistical analysis
Statistical analysis of cytogenetic data was performed using STATA software to compare chromosomal instability between patients and controls. Categorical variables were compared using Student's t-test. A P-value less than 0.05 was considered to indicate statistical significance.
3 Results
Chromosomal fragility test was performed for 205 AA patients. The mean age of patients at presentation in the present study was 11.87 ± 9.32 years (range: 2 months to 48 years).
A slight male predominance was observed with a sex-ratio M/F = 1.13.
Induced chromosomal breakage study was successful in 171 out of 205 patients, and in all healthy controls. For the remaining 34 patients, patient's blood cells showed no growth in culture.
According to the sensitivity to MMC at 50 ng/ml (Table 1), 38 patients (22.22%) aged between 11 months and 33 years were diagnosed as affected (MMC+) and 132 patients (77.17%) aged between 2 months and 42 years were diagnosed as unaffected (MMC−). Somatic mosaicism was suspected in an 11-year-old patient with a FA phenotype.
Chromosome breakage frequency in Fanconi anemia patients (FA), mosaic-FA patient (FA-mosaic), and non-Fanconi anemia patients (non-FA).
| Groups | Number | % of aberrant cells | Breaks/aberrant cell | Breaks/cell | |||
| Mean ± SD | Interval | Mean ± SD | Interval | Mean ± SD | Interval | ||
| FA | 38 | 93.55 ± 10.94 | 66–100 | 13.02 ± 8.44 | 3.56–34.92 | 12.61 ± 8.73 | 2.36–34.92 |
| Mosaic-FA | 1 | 52 | 3.65 | 1.9 | |||
| Non-FA | 132 | 21.98 ± 11.38 | 0–58 | 1.46 ± 0.44 | 0.42–2.54 | 0.48 ± 33.33 | 0.02–1.61 |
| Controls | 205 | 11.57 ± 7.64 | 0–34 | 1.19 ± 0.39 | 0.04–2.16 | 0.24 ± 0.269 | 0.02–1.42 |
Twenty-six siblings of FA patients were also evaluated for induced chromosomal fragility. On the basis of MMC-test, five of them (19.23%) were diagnosed as FA.
FA patients showed a significantly elevated level of chromosome breakage compared to controls with a percentage of unstable mitoses between 66% and 100%.
All MMC-induced cultures of the 38 FA patients exhibited a significantly (P < 10−4) high frequency of mean chromosomal breakage per cell (12.61 ± 8.73), mean chromosomal breakage per aberrant cell (13.02 ± 8.44) and mean percentage of aberrant cells (93.55 ± 10.94) compared respectively to controls (0.24 ± 0.269, 1.19 ± 0.39, 11.57 ± 7.64).
Spontaneous chromosome fragility was also analysed and was detected in 62% of FA patients.
There was no difference in the chromosomal breakage frequency among the 132 non-FA patients and the controls. As shown in Fig. 1, there was a clear discrimination between FA and non-FA subgroups with no overlapping.
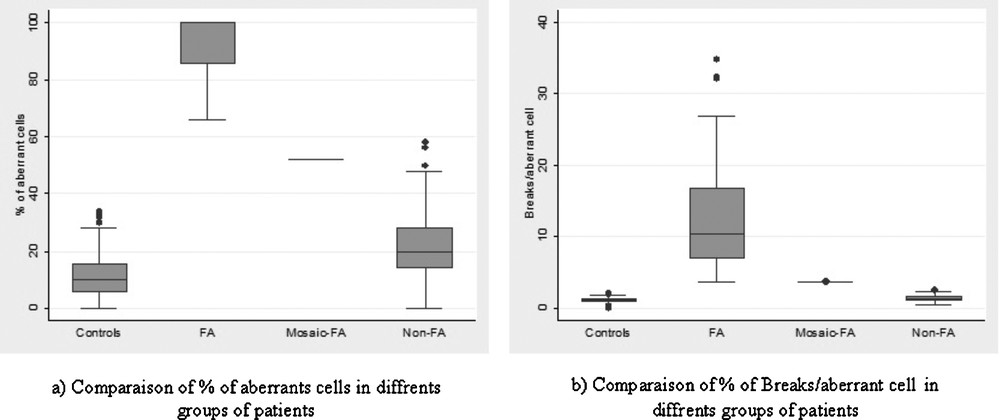
MMC-induced chromosome fragility in Fanconi anemia (FA), mosaic-FA and non-Fanconi anemia (non-FA) groups expressed by percentage of aberrant cells (a) and breaks/aberrant cell (b).
Diagnosis of somatic mosaicism was strongly suspected in one patient. In this case, 52% of metaphases showed chromosomal breakage and elevated number of chromosomal breakage per aberrant cell (3.65). Discrimination between FA-mosaic patients and non-FA is the principal difficulty found when making an FA diagnosis. However, when considering only breaks/aberrant cell (Fig. 1b), chromosome fragility level of mosaic patients is equivalent to that seen in FA patients.
The RHG-banded metaphases of patients suspected with FA were found to have normal karyotypes.
A significantly (P = 10−6) high frequency (93%) of consanguinity was noticed among FA patients compared to non-FA patients (43%).
4 Discussion
We have investigated a large group (205) of AA patients using MMC-chromosomal breakage test. Patients were categorized according to cytogenetic criteria. A total of 43 patients were identified as FA (38 patients suspected for FA and 5 siblings). Their age range was 11 months and 32 years. In about 95% of cases, congenital abnormality(s) such as skin pigmentation, skeletal abnormalities and dysmorphic faces were obvious.
Consanguinity was observed with a high frequency (93%) in FA families, whereas, it concerned only 43% of non-FA families. In Tunisia, the rate of consanguineous marriages was estimated at 36% in the north [11] and more than 70% in the south [12].
FA is traditionally considered as a paediatric disease The median age at diagnosis of FA in literature cases is under 7 years. Only 9% of the published cases were diagnosed as adults [13]. The discovery of the disease after the age of 20 years is a very rare event. However, in our study, six cases (15.7%) were found.
FA has long been considered as a spontaneous chromosome fragility syndrome [14]. Results obtained in this study show that 62% of FA patients have a spontaneous chromosome fragility level within the normal range of non-FA patients. Therefore, spontaneous chromosome fragility cannot be used as a diagnostic tool for FA.
It is well known that patients with FA are highly sensitive to DNA cross-linking agents such as DEB and MMC [8]. In our study, patients were tested for MMC sensitivity in lymphocyte cultures. All patients clinically diagnosed as FA showed significantly elevated levels of chromosomal breakage compared with controls. However, we have also retained FA diagnosis for five asymptomatic cases. Thus, absence of physical abnormalities does not rule out the diagnosis of FA and apparently unaffected siblings should be tested for FA.
The major breakthrough of this study was the differentiation of mosaic-FA from non-FA patients using the chromosomal breakage test. This discrimination is based on the combination of percentage of aberrant cells and breaks/aberrant cell.
The somatic mosaicism was suspected in a patient with clinical signs characteristic of FA such as facial dysmorphism (microphtalmia, triangular face) short stature and urogenital malformations. The diagnosis of this patient is likely a reverted form of FA because of the intermediate % of aberrant cells (52%) associated to the presence of two populations of peripheral T cells: one with no breaks comparable to the non-FA patients and the other with increased chromosomal breakages typical of FA patients. Therefore, the value of breaks per aberrant cell for mosaic patients is important and nearly similar to that seen in FA patients. This value is a powerful parameter for the diagnosis of mosaic patients. However, to confirm this hypothesis, it is necessary to test skin fibroblasts. As opposed to lymphocytes, fibroblasts do not undergo the phenomenon of genetic reversion [15] For this patient, molecular investigation has been conducted and showed a compound heterozygous state, which is in favor of a mosaic state.
It is important to diagnose this particular form of FA to achieve an adapted conditioning regimen that is more moderate than that recommended for other AA patients. Indeed, the presence of non-FA cells among FA hematopoietic cells has been considered as a risk factor for engraftment in SCT (stem cell transplantation) from alternative donors because cross-linking agents-resistant T-cells may increase the risk of graft rejection [16].
The chromosomal breakage test is the technique of reference for FA diagnosing [6–8,17]. Many variations of this test are used in different cytogenetic laboratories. In fact, there is a great variability in the sensitivity of FA cells depending on the type of alkylating agent, drug concentration and the exposure time [10]. Therefore, there is a great need for a clearly described reliable protocol for an accurate diagnosis of FA patients. The experience is also essential for a good reproducibility of this type of test.
Other FA diagnosis tests include cell-cycle analysis [18], and evaluation of FANCD2 mono-ubiquitination, which can positively diagnose FA core patients [19]. Once FA diagnosis is established at the cellular level, FANC gene mutations can be screened. The most frequent gene (FANCA) is analyzed first then FANCG, FANCC and so on [20]. In Tunisia, previous studies on FA patients have shown that FANCA gene is implicated in 94% of FA patients [21].
It is crucial for a therapeutic center to accurately identify patients with FA. Moreover, due to a hypersensitivity to chemotherapy agents, patients with FA will often die of toxicity if treated by a conventional conditioning for hematopoietic stem cell transplantation (HSCT) [9,13,20].
5 Conclusions
From this study, a standard protocol for FA diagnosis was developed. It is routinely used as a diagnostic test of FA in Tunisia that helped confirm diagnosis of FA patients among AA and hence give better classification in the Tunisian Fanconi Anemia Registry (TFAR) [22].
The present data illustrates that MMC-test provides a powerful test to differentiate between FA and idiopathic AA. The separation of these two groups is important for therapy planning. Results of this study also indicate that it is extremely important to rule out FA in all siblings, in particular if a sib would be an eventual bone marrow transplant donor.
Disclosure of interest
The authors have not supplied their declaration of conflict of interest.
Acknowledgments
We are thankful to the patients and their families for their collaboration. We are also grateful to the Tunisian Fanconi Anemia Study Group. This work was supported by the Tunisian Ministry of Higher Education and Scientific Research (Laboratory of “Biomedical Genomics and Oncogenetics” LR11IPT05) and the Tunisian Ministry of Public Health.
The Tunisian Fanconi Anemia Study Group:
F. Talmoudi, O. Kilani, W. Ayed, A. Amouri, Laboratory of Histology and Cytogenetics, Institut Pasteur de Tunis, Tunis, Tunisia.
O. Messaoud, M. Ben Rekaya, S. Abdelhak, Laboratory of Biomedical Genomics and Oncogenetics, Institut Pasteur de Tunis, Tunis, Tunisia.
L. Kammoun, S. Hdiji, M. Elloumi, Haematology Department, Hedi Chaker University Hospital, Sfax, Tunisia.
L. Torjemane, A. Lakhal, N. Ben Abdeljelil, S. Ladeb, T. Ben Othmen, Department of Haematology and Transplantation, National Bone Marrow Transplantation Centre, Tunis, Tunisia.
M. Ouederni, N. Dhouib, F. Mellouli, M. Bejaoui, Department of Peadiatric Immuno-Haematology, National Bone Marrow Transplantation, Tunis, Tunisia.
L. Aissaoui, Y. Ben Abdennebi, H. Ben Abid, Haematology Department, Aziza Othmana Hospital, Tunis, Tunisia.
S. Mougou, H. El Ghezal, A. Saad, Cytogenetic Department, Farhat Hached Hospital, Sousse, Tunisia.
Y. Ben Youssef, A. Khelif, Haematology Department, Farhat Hached Hospital, Sousse, Tunisia.
F. Amri, Paediatric Department, Ibn El Jazzar Hospital, Kairouan, Tunisia.
M. Hachicha, Paediatric Department, Hedi Chaker Hospital, Sfax, Tunisia.
M. Frikha, J. Feki, Medical Carcinology Department, Habib Bourguiba Hospital, Sfax, Tunisia.
F. Fakhfakh, Laboratory of Human Molecular Genetics, Faculty of Medicine, Sfax, Tunisia.
H. Bellaj, Intelligent Control, Design & Optimization of Complex Systems, National Engineering School of Sfax, Tunisia.
M. Meddeb, Laboratory of Medical Genetics, Tunis, Tunisia.
I. Safra, Department of Haematology, Institut Pasteur de Tunis, Tunisia.
S. Hmida, National Blood Transfusion Center, Tunis, Tunisia.