

1 Introduction
Among orthopterans, locusts are notorious for their population outbreaks and swarming capabilities and no continent is spared from locust outbreaks. Indeed, infamous among the species known to experience population outbreaks, we find Schistocerca gregaria and Oedaleus senegalensis in Africa and parts of Asia, Locusta migratoria and Oedaleus asiaticus in Africa and Asia, Melanoplus sanguinipes in North America, other Schistocerca species (such as S. Cancellata and S. americana) in South America and Chortoicetes terminifera in Australia. At the outbreak phase, each of these species can cover large geographic areas (Fig. 1), often with disastrous consequences. Indeed, millions of dollars are spent each year in order to control the population size of such species that, otherwise, might experience outbreaks and become even more costly. For instance, a document on the benefit-cost analysis of locust control operations for 2010–2011 by the Australian Plague Locust Commission (http://www.daff.gov.au/animal-plant-health/locusts) states: “Locust control operations this season are estimated to have avoided potential losses of $963 million. Total expenditure by all parties was estimated to be $50 million. The net benefits of control are therefore $913 million, with an estimated ratio of benefits to costs of around 19.2:1.”

Geographical distribution of Schistocerca gregaria populations sighted between 01-01-2000 and 01-01-2013. Green dots: solitary hoppers; blue dots: solitary adults; orange dots: gregarious hoppers; red dots: gregarious adults.
United Nations Food and Agriculture Agency (http://www.fao.org/ag/locusts/en/info/info/index.html).
Thus far, the solution to the outbreaks problem is largely based on prevention, with significant governmental personnel and resources dedicated to this task (e.g., the Moroccan anti-locust centre http://www.criquet-maroc.ma/, the pest research unit of the French CIRAD http://ur-bioagresseurs.cirad.fr/, and the Kenyan ICIPE http://www.icipe.org/). Coordination between countries is also needed and a section of the United Nations Food and Agriculture Organization is dedicated to this issue (see http://www.fao.org/ag/locusts/en/info/info/index.html). The strategies adopted for preventing locust outbreaks are proving helpful but, when they fail, the options are few. Although entomopathogenic fungi (such as Metarhizium anisopliae var. acridum) are currently available (e.g., http://www.lubilosa.org/) and, sometimes, used as a bio-control agents for treating the locust outbreaks, the pesticides option is still on the table and prevalent (e.g., Fipronil). No need to stress here that governments, society and local communities are increasingly aware of the dangers of the excessive use of pesticides (e.g., http://www.panna.org/). Neither it is necessary to extend on the issue of the non-specificity of the pesticides that, when used, affect the targeted locust as well as other organisms in the treated area, including humans, other mammals, bees… (e.g., [1–5]).
In my opinion, a species-specific treatment would be the ideal solution. For that, a deep knowledge of the molecular biology of the target species and its peculiarities is needed. Such knowledge would also offer precious data to many fundamental biological questions as well as to comparative studies (including the model organisms). Much is known about grasshoppers and locusts and the interest of researchers on this subject is continuously growing. In fact, a search for the word locust on the Pubmed database of the US National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/pubmed/) shows strong positive correlation between the number of published papers and the year of publication (r = 0.871). In fact, the numbers went from 25 papers published in 1970 to 221 in 2012, with 22 articles published just in January 2013 (Fig. 2a). However, these numbers are misleading, and their real meaning is only revealed when compared to the numbers from research on other organisms. For Drosophila, the number of published works on the NCBI database shows stronger correlation with the year of publication (r = 0.957). It grew from 331 papers published in 1970 to 3711 in 2012, with 406 papers published just in January 2013 (Fig. 2b). Strikingly, the ratio of locust articles to Drosophila articles per year shows that the number of publications on locusts represents just around 5% of the number of published works on Drosophila (Fig. 2c). Even more, the between-years increase in the number of research papers is much more irregular when it comes to locusts than it is for Drosophila (Fig. 2d). One might argue that comparing with a model organism may not be the right way to assess the state of the research on locusts. That is partially right, but the word locust includes several species. In addition, each of these species represents a serious (sometimes almost existential) threat to entire regions of the globe. One would thus expect research on these organisms to represent a bit more than a meager 5% of the research on Drosophila. Furthermore, the number of published works on locusts in 2012, for instance, is lower than that on ants (526), bees (580), and spiders (517).
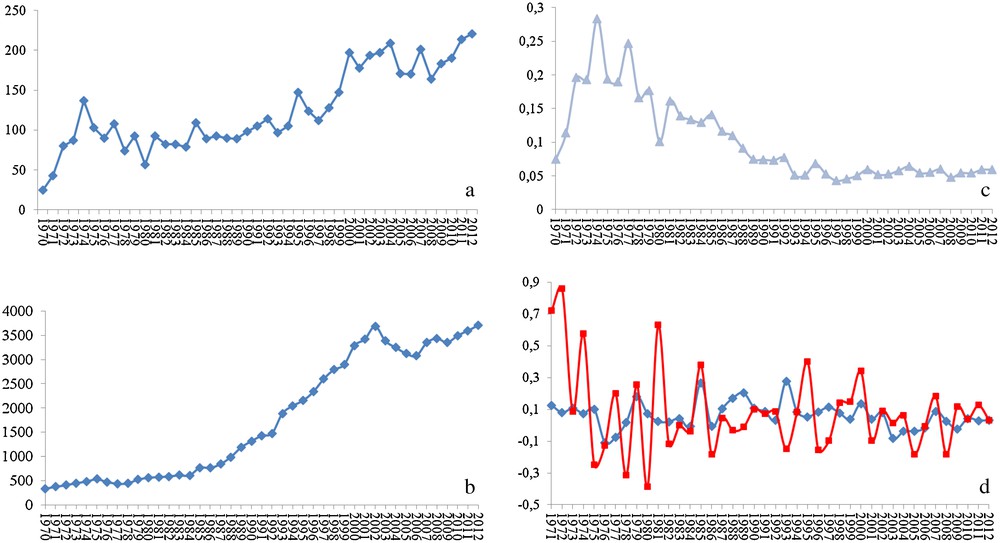
Statistics of the published peer-reviewed science on locusts and Drosophila. (a) Number of published articles on locusts per year, (b) number of published articles on Drosophila per year, (c) ratio of the number published articles on locusts to that on Drosophila per year, and (d) inter-annual increment in the number of articles published on locusts (red line) and Drosophila (blue line).
The number of published articles is directly dependent on the number of research groups and on the level of financing (the first also dependent on the second in a way that the more financing a research area, the more attractive it gets it to researchers). Discussing the reasons for this near-precarious state of the research on locusts and the ways to overcome it could by itself be a theme for a manuscript and would probably be better discussed in meetings, conferences and other scientific gatherings. What matters for us now is that we are still far from reaching the level of knowledge that should allow a deep understanding of the molecular mechanisms associated with locust population outbreaks, and efficient comparative studies to determine the species-specific processes and molecules that I hope might serve as target to those hopped for species-specific pesticides.
Fortunately, technological developments in the areas of molecular biology and genetics now make the possibility of a deep knowledge of the molecular aspects underlying the peculiarities of a species easier than ever. In genetics, the key words to such objective are comparative studies aimed at gene identification, gene location, analysis of gene expression, analysis of gene regulation and analysis of gene interactions and function. A battery of techniques is now available for such studies, and these include high-throughput sequencing (also called next generation or deep sequencing), in situ hybridization techniques, microarrays and quantitative PCRs, epigenetics study techniques and gene knockdowns. Combined together, and in the right order, these techniques should allow the characterization of the locust genes and of their functions. I therefore briefly introduce here the most relevant among these techniques as well as their potential usefulness for the locust research and a potential workflow that an average genetics laboratory could follow to get data on genes and their expression and function. My hope is that the combined research efforts would provide results that, when taken together, would provide valuable scientific knowledge and a good-enough basis for applied research on environmentally friendly and/or species-specific locust pesticides.
2 Genomics
Genome sequences of a number of non-locust pest species are now available and some research centers are now engaged in deciphering even more genomes (e.g., The Insect Genome Research Unit of the Japanese Agronomics Research Center, http://www.nias.affrc.go.jp/eng/org/Agrogenome/Insect/index.html). Notorious examples of such genomes include those of the mosquito Anopheles gambiae [6], the red flour beetle Tribolium castaneum [7] and, recently, the pine beetle, Dendroctonus ponderosae [8]. However, obtaining locust DNA sequences, as we all know, means sequencing and, when it comes to that, we are faced with two main objectives: firstly, identifying the expressed sequences of the genome, and secondly, identifying the genome itself. The second question remains thus far untouched because of the difficulties that orthopteran genomes present. In fact, the genome of a species such as S. gregaria is two and half times the size of the human genome [9]; making any genome sequencing project too costly for the average genetics laboratory. In addition, the large size of the orthopteran genomes is due to its shared amount of repeated DNAs. From that comes the second difficulty; which is related to the challenge that represents assembling sequence reads that contain parts of the repeated DNA. Two sequencing reads of two separated genomic regions (even in different chromosomes) that share the same repeated DNA would be falsely assembled into one non-real sequence, due to the overlap between the repeated DNA parts of these reads. This issue could be surmounted only if:
- • the sequence reads were long enough (larger than the largest repeated DNA unit in the genome);
- • the sequencing depth were high enough as to sequence every possible genomic window of the same size as the sequencing read (that may mean a too costly sequencing effort at hundreds fold the size of the genome);
- • if the sequence assembly algorithms were able to discriminate between events of false joining of repeated DNA-containing reads and the correct assembly of genuine sequences containing the repeated elements.
For the size of the read, currently the 454 GS FLX plus system is probably the best technological option, as it offers up to 1000 bases per read (http://454.com/products/gs-flx-system/index.asp). Still, with just one million reads per plate, tens of plates are needed to get the desired sequencing depth, which, at a cost of around 15,000 US $ per plate, remains a very costly option for the average research laboratory. In addition, even the best assembly algorithms (e.g., ABYSS [10]) still need to be improved to deal with the assembly ambiguities that result from the repeated DNAs. Thus, as long as the algorithms for DNA sequence assembly do not deal with the issue of repeated sequences and/or the sequencing technology becomes even more affordable and produces even larger sequences, all what we can aim for at this point is to construct some large contigs of the genome or to specifically sequence genomic regions around a specific gene. The latter objective might be of great importance, as the genomic region of a gene is what contains its cis-regulatory elements and, thus, studying it could be key to characterizing that gene's activators and suppressors, as well as its interactions with other genes. For such objective, a chromosome walking strategy is needed and, although the technique was originally quite lengthy and technically demanding, several commercially available kits now make it both more affordable and attainable.
Tightly linked to the availability of genomic sequences are the epigenetic analyses. These are currently approached through detection of the methylation islands and of their variation between physiological conditions (for a review, see [11]). A couple of preliminary studies have recently been carried out on L. migratoria [12] and recently on S. gregaria [13]. In the absence of complete genomes, the first work used a ‘traditional’ enzymatic approach and the second used the bisulfate method on incomplete genomic DNAs. The bisulfite-sequencing technology is currently the tool of choice for methylation and epigenetics studies [14,15]. This method relays on converting the non-methylated cytosine residues of the DNA to uracil, while leaving the methylated cytosines intact. Subsequent sequencing of the genomic DNAs and the comparison of the untreated sequences to the treated ones (or of the bisulfite-treated DNA sequences from two physiological conditions) allows identification of the changes in methylation patterns associated with the studied physiological phenomenon. The availability of reference genomic sequences rather than expressed transcripts is of great importance to these methylation studies; as most of the epigenetic differences between the compared conditions (e.g., gregarious vs. solitary) would be due to either silencing or modification of the level of expression of certain genes. Both effects are largely due to modifications at the cis-regulatory elements (promoters and enhancers), which would not be available unless genomic DNAs are sequenced. Although the task is not easy, our lab is engaging in an initial locust genome sequencing project, whose first results should be available within the two coming years.
3 Transcriptomics
Contrarily to the genomic DNA, sequencing the transcripts of the expressed orthopteran genes is currently both possible and affordable. When it comes to the sequencing technology, sequencing using Illumina technology is the most cost effective and the Illumina Hiseq2000 system is probably the best among the currently available options (http://www.illumina.com/systems.ilmn). Although the read size is, for now, just up to 100 pb, this technique produces millions of reads (a full Hiseq2000 sequencing run produces 6 billion reads of 100 bases in 11 days). The sequencing reads are then easily filtered and assembled into expresses sequence tags (ESTs) and full transcripts. For this, sequence assembly software such as ABYSS [10], TRANSABYSS [16] VELVET [17] OASES [18] and TRINITY [19], are proving to be easy to handle and efficient in assembling expressed tag sequences from deep transcriptome sequencing. Of course, the reason for this difference between genome and transcriptome sequencing is the lack of the repeated DNA problem in the latter case, the average length of the sequences to assemble (genes vs. full chromosomes) and the absence of need for mapping and assigning sequences to chromosomes.
Depending on the sequencing depth, also called coverage (i.e., the number of times any given nucleotide of the transcriptome is expected to be sequenced), Illumina sequencing also allows comparative analyses of the gene expression levels between physiological conditions – a method referred to as RNA-seq. The logic here is that there is a positive correlation between the relative number of sequencing reads that correspond to a gene and the relative level of expression of this gene. This way, the more reads of a sequencing experiment contribute to the assembly of an EST (or transcript), the more molecules of the corresponding gene must have been present in the sequenced library (indicative of the relative number of molecules per cell). Short-sequence alignment tools such as the Burrows–Wheeler aligner (BWA) [20] allow the alignment of the raw sequencing reads to the assembled contigs (ESTs and transcripts) and algorithms such as Cufflinks [21] and DESeq [22] do the comparative counts of the alignments events between the raw sequencing reads and the assembled ESTs and transcripts.
Indeed, we have separately sequenced the transcriptome of the desert locust S. gregaria from different tissues of both solitary and gregarious specimens (Fig. 3), and, with an estimated 120 × sequencing depth per tissue, we have assembled the expressed tag sequences and unigenes, and we are now in the phase of the comparative analysis of the gene expression levels. With this, we expect to identify genes that show differential expression between the harmless phase of this locust and their outbreak phase. This will be the first high-throughput comparative sequencing of S. gregaria transcriptome. Prior to this, an impressive comparative sequencing of the solitary and gregarious S. gregaria nervous system transcriptome was carried out using the by-now ‘old fashioned’ ABI sequencing technology [23]. However, the pioneering large-sequencing project was that of the L. migratoria transcriptome by Lee Kang's research team [24] – also based on ABI sequencing technology. Later, the same research group went for further sequencing of the L. migratoria transcriptome; this time using high-throughput technology [25].

Gregarious (left) and solitary (right) nymphs of the desert locust Schistocerca gregaria reared at our laboratory.
Of course, prior to the analysis of gene expression levels, one has to annotate the assembled sequences (annotation could also be done after analysis of the expression levels, but that would be counterintuitive). For this, a recommended initial strategy is to carry out local blast searches against the annotated sequences from organisms such as Drosophila melanogaster, Tribolium castaneum, Bomyx mori, Apis mellifera, Nasonia vitripennis, Aedes aegypti… These searches could be complemented by the use of the Blast2Go software [26], which provides further automated annotation steps.
In conclusion, the sharp drop in sequencing costs and the power and efficiency of the new sequencing technologies as well as the increasing sophistication of the bioinformatics algorithms and software used for assembling, annotating and analyzing sequences and their variation (isoforms) and levels of representation in the sample are making accurate and complete sequencing of the locust transcriptomes possible. Data from these sequencing efforts will no doubt pinpoint key genes for the locust outbreak behavior and/or for specific targeting of the locust of interest.
4 Characterization of gene expression
With the deep sequencing data of locust transcriptomes available, a suite of techniques for gene mapping and analysis of expression are now possible. These include in situ hybridization, immune-staining, quantitative (or real-time) Polymerase Chain Reactions (qPCR) and microarrays. Thinking of in situ hybridization means to deal with two variables: fluorescent versus non-fluorescent and on chromosomes versus on tissues. Detecting repeated DNAs on chromosomes is already routine technique for many orthopteran research groups (e.g., [27,28]). Detecting a single-copy gene on chromosomes is possible (e.g., [29,30]) and, in a way, has been applied to locusts [31]. Still, this remains a highly challenging task that requires a sufficient sequence length and a good fluorochrome, so the adaptation and use of some of its variations, such as branched DNA in situ hybridization [32] or PCR in situ hybridization [33], might be a better option. The other in situ hybridization, on tissues, sections and whole mounts, uses fluorescence or non-florescence labeled complementary single-stranded DNA or RNA to detect the transcript of one or several given genes in the tissue or the organism in general (see [34–38]). Analysis of gene expression using in situ hybridization is often used locusts (e.g., [39–42]). Except from the fact that one technique physically detects the location of a gene on the corresponding chromosome whilst the other detects the cells where this gene is actually expressed, and that the former technique needs fluorescence and is more challenging, the in situ hybridization principle is the same and its protocol is significantly similar (for instance, compare protocols in the references herein).
With the gene sequence on hand, another possibility for gene expression analysis is to detect expression after the transcript has been translated into protein. The technique of choice here is immunofluorescence staining (e.g., [43–46]). The procedure starts by inferring the amino acids sequence from the sequenced transcript then synthesizing it and producing an antibody against the synthesized amino acids sequence (called primary antibody) in a host animal (e.g., mouse, goat). When infused into tissues and cells, this antibody should specifically bind to the protein produced by the gene of interest. A labeled second antibody (called secondary antibody), which depends on the host used for producing the primary antibody, is then used so that it can bind to and reveal the primary antibody through its labeling (fluorescent normally). Although immune-staining is a powerful technique that has been used for locusts in many occasions (e.g., [46,47]), it remains expensive and, like in situ hybridization on tissues and whole mounts, does not provide quantitative results. The use of this technique could thus be replaced by the more affordable in situ hybridization, unless a primary antibody is available and tested for specificity, or if what the researcher is looking for is to infer the site of action of the gene product (membrane localized, cytoplasmic, nuclear…). It is worth mentioning here that many of the antibodies used for Drosophila as well as other invertebrates are known to work for Orthoptera (e.g., [48]).
The availability of transcripts, from deep transcriptome sequencing projects, therefore provides a good set of potential sequences to either map on the chromosomes or detect and follow the evolution of their expression between tissues, stages and physiological conditions. However, if in situ hybridization could be done with just a single-gene (normally part of it) that might even be obtained through amplification using degenerated primers (thus no need for deep sequencing), one of the powerful techniques of gene expression analysis is microarrays [49–52] and, this necessarily needs a good set of sequences (such as those provided by deep sequencing). As stated above, high-throughput sequencing, especially Illumina Hiseq2000, allows analysis of gene expression levels (RNA-seq). Still, these data are usually further validated through the use of microarrays. The latter comes in two flavors. The by-now ‘old fashioned’ on-slide microarrays that relays on the synthesis of oligonucleotides (between 60 and 70-mer), each of them complementary to one end of one transcript (generally the 3′ end). Here, the oligonucleotides are printed as spots on the slide and the labeled cDNA of the transcriptome of interest are then hybridized against the printed oligomers using a similar protocol to the one used for the in situ hybridization of chromosomes and tissues. This technique allows comparative analysis of the relative gene expression levels between samples and conditions as one can label each DNA sample using a different fluorochrome (red versus green), mix the cDNAs in equal quantities and let them compete for hybridization against the spots of oligos printed on the microarray slide. This is a nice and powerful technique for gene expression analysis that has provided precious data on various research topics, but it suffers from two drawbacks: the cost of the oligos and the physical limitations of the slide that do not allow for as much sequences as a transcriptome may require. At the near cutting-edge of the technology, we now can opt either for Roche NimbleGen or Agilent's costume gene expression microarrays, the latter in its SurePrint HD or SurePrint G3 formats. With this technique, microarray chips can now take up to one million sequences and the costs are lowered down as, instead of synthesizing the oligos and printing them on the slide, one can now opt for building the oligos strait on the array chip. In addition, the great development in the optics and image analysis algorithms is making the microarrays even more sensitive. When it comes to locusts, the first, pioneering, microarray work was that of Le Kang group on L. migratoria [53] and, with our transcriptomic sequencing data ready, we are planning a microarray experiment for S. gregaria.
Another technology that allows detection and comparison of gene expression levels is the qPCR. I would not go into much detail about this technique as it is currently used almost routinely in most molecular biology and genetics laboratories. It is based on an estimation of the number of cDNA molecules corresponding to a particular transcript by real-time detection of the dynamics of a PCR amplification of its double-stranded molecules. A series of normalizations and comparison with the amplification dynamics of few control sequences (usually three housekeeping genes) thus allow estimation of the relative abundance of the molecules corresponding to our gene of interest in the cDNA library being analyzed. Detection of the number of double-stranded DNA molecules and the following of the PCR dynamics is possible thanks to the detection of fluorescence levels of a fluorochrome that emits more fluorescence when it is associated with double-stranded DNA molecules than when it is associated with single-stranded DNA molecules (SYBER Green). Although this technique is relatively old and cannot produce masses of data, as it detects sequence by sequence (we cannot usually mix different transcripts in the same reaction tube), it remains an almost necessary tool for validation of both deep sequencing and microarray data. In fact, no deep transcriptome sequencing or microarray project would be complete without validation of some of the data (selected genes) via qPCR.
5 Functional genetics
With the transcriptome and/or the genomic DNA sequenced, the genes of interest selected, mapped, and their expression analyzed both in space (tissues) and in time (conditions or stages), what else can a geneticist do? The answer could be the analysis of the function of the gene.
When at this point, the researcher would already have had a large set of genes. Some of these would be identified as potentially involved in the researched phenomenon (based on the quantitative differences of their expression levels). The expression of some of these genes would also have been linked to organs, tissues or cell types. What remains is to study the effects of these genes and their potential interactions. This could be done by altering the gene of interest or its expression level. For this, one can think of two potential methods: transgenesis and gene knockdown.
The locust genome is full of transposons of all kinds. Indeed this could be suspected from the very large size of the orthopteran genomes and was, in fact, confirmed in L. migratoria [54,55] as well as in Saga pedo, Eyprepocnemis plorans, Dociostaurus maroccanus, and S. gregaria (Data in preparation from our multiple transcriptome sequencing projects). We also saw that transposases are highly expressed in locust cells (in preparation). While this may be an interesting fact for research on genome instability, unfortunately, it means that transgenesis would be a very challenging task as one has to find a transposon whose transposase is not naturally expressed in the locust genome. Furthermore, the large size of the locust genome means that the transposition events would be very likely to land on chromosomal regions of repeated DNAs, where the transformed sequence may not be expressed or may present inconsistent expression patterns because of the position effect variegation.
Fortunately, there is a technique that is now proven to allow altering gene expression levels. RNA interference-mediated knockdown of gene expression is increasingly used for decreasing transcript levels by up to 90% and, thus, inferring gene function and interactions from the resulting phenotypes. Knockdown of a gene expression through the use of RNA interference technique (RNAi for short) relays on the introduction into the cells of double-stranded RNA molecules that correspond to part of the target gene. Using a well-known set of enzymes (see [56–59]), the cell fragments the introduced double-stranded RNA and uses the resulting fragments as templates for fragmenting the messenger RNAs that contain such template. This normally results in the degradation of the messenger RNA of the targeted gene and, thus, the prevention of its translation and protein synthesis. The final result is hence the abolition of the target gene function and interactions (for a review on RNAi see [60,61]). Delivery of the double-stranded RNA into the cells is easier than it sounds given the presence of a cross-membrane RNA transport protein (called SID1 [62,63]). Fortunately, locust cells express this protein, as proven by the success of RNAi in grasshoppers and locusts. The pioneering RNAi work on an orthopteran species was by Dong and Friedrich [64] on S. americana and, since then, the technique was proven successful in other species such as L. migratoria [53] – in our laboratory, RNAi has proven useful in the two species where we tested it, E. plorans and S. gregaria (e.g., [65]).
If knocking down a gene's expression would allow inferring its function, a microarray study of the RNAi-treated transcriptomes should provide information on the genes lying downstream of the studied one in the gene network. Unfortunately, we are all too aware that the average laboratory cannot financially afford a microarray study after each gene knockdown experiment. So, unless one wants to go for qPCR and test individual genes whose interaction with the gene of interest was previously reported or inferred from the phenotype observed after the RNAi experiment, at this point, one can take the study of the gene of interest as completed since the gene of interest would have been identified, its expression characterized both in quantity, space and time, and its function inferred.
The use of RNAi as a tool for dealing with pest insects is debated since few years now (e.g., [66,67]) and screening for potentially useful genes to use as pesticides was already done for few species (such as Beet Armyworm, Spodoptera exigua [68]) and a patent on controlling pests using RNAi that includes locusts (patent number EP1934357 B1) was granted in 2011 to Els Van Bleu et al.
6 A final note
Genetics is a vast and fast evolving science and the tools it offers are currently used for solving questions ranging from taxonomy to physiology and molecular biology. The genetics techniques and the experimental strategies a researcher can adopt are countless and the workflow described in this article (Fig. 4) is just one of the many possible ways one can adopt. In addition, we tend to venerate the new developments but, in my opinion, it is worth mentioning that, by no means modern genetics techniques make classical genetics techniques obsolete.
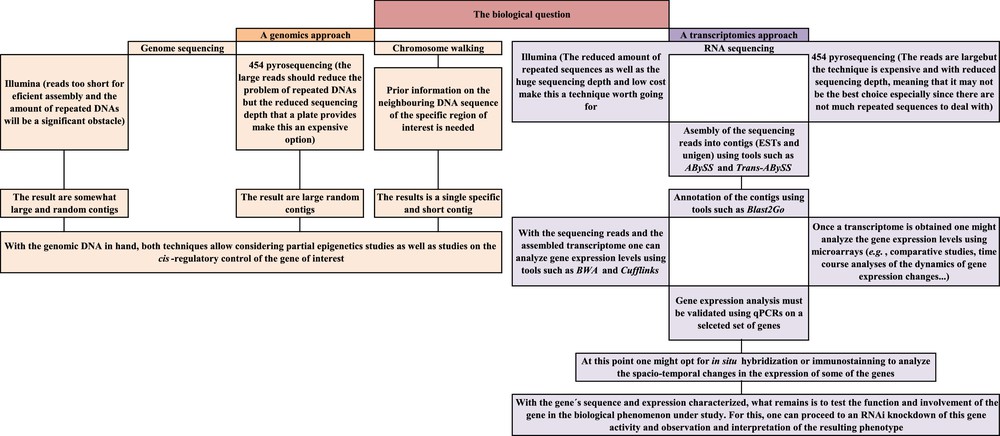
Schematic summary of the modern genetics workflow suggested for a deep analysis of the genetic basis of phenomena shown by locusts (such as the capacity to shift from solitary to gregarious behaviors).
I started this essay by introducing the potentially ‘applied’ aspect of researching locust genetics and the reader may be left with the feeling that Orthoptera research is of no use unless it is to deal with the problems that these organisms cause. Of course, increasing the research effort on the orthopteran molecular biology and genetics would, and I expect will, provide useful information and ideas to those of us who are interested in developing alternative pesticides; which I think should be both environmentally friendly and species-specific. However, orthopteran chromosomes and genetics have been used as key material not only for scientific research (especially on chromosomes and repeated and non-coding DNAs), but also for teaching the science of genetics. Due to the large size of the chromosomes and the ease of observing them under the microscope, Orthoptera are a material of choice for the teaching of cytogenetics and meiosis. Even more, the upcoming stream of genetics and molecular biology data on locusts and other orthopteran species should serve for comparison with and validation of the scientific facts obtained from research on model organisms.
Indeed, locusts and other orthopteran species do not make it as model organisms for the genetics studies as they are ‘too complex’ and not as easy to rear and study as model organisms are (i.e., too mundane). But they are closer to most of the ‘real’ organisms than model organisms are. In fact, model organisms are chosen as such precisely because of characteristics that make them unique and less representative of most of the living beings (i.e., most of the organisms that the model one is meant to represent do not have simple traits, their genomes are not small, they take time to reproduce and do not produce as many progeny as the model organisms do). It is my personal reflection that, with the advances in genetics and molecular biology tools, it is probably time to move from the model organism to more ‘realistic’ ones. Orthopteran species are both very ‘realistic’ and economically important (in the negative sense). They therefore have the potential to be a material of choice for the post-model organism era which I predict is about to emerge.
Disclosure of interest
The author declares that he has no conflicts of interest concerning this article.
Acknowledgements
I am thankful to my wife Pernille Lavgesen for her continuous understanding and support. I am also thankful to Prof. Josefa Cabrero Hurtado and Prof. Juan Pedro Martinez Camacho for their support and helpful comments on this manuscript. This work was supported by a Spanish Ramon y Cajal Followship and a grant from the Spanish Ministerio de Ciencia e Innovación (BFU2010-16438).
This article is dedicated to my mother, Fatima Allal Errahhaoui, and children, Nadia Lavgesen Bakkali and Elias Lavgesen Bakkali, and in memory of my father Abdeslam Bakkali (1945–2011).