

1 Introduction
Filoviruses contain Ebolaviruses and Marburgviruses that have caused many hemorrhagic fever outbreaks in sub-Saharan Africa since a few decades, resulting in high case-fatality rates (25–90%) in humans and other primates, such as chimpanzees and gorillas. To date, six filoviruses were described in Africa, including four Ebolaviruses (Zaire, Sudan, Taï Forest, and Bundibugyo) and two Marburgviruses (Marburg and Ravn) [1,2] (Fig. 1). Two filoviruses were also detected outside of Africa: the Reston Ebolavirus in healthy humans and ill animals (macaques and domestic pigs) in the Philippines and China [3,4], and the Lloviu Cuevavirus, which caused massive die-offs in cave colonies of Schreiber's bat (Miniopterus schreibersii) in France, Spain, and Portugal in 2002 [5].
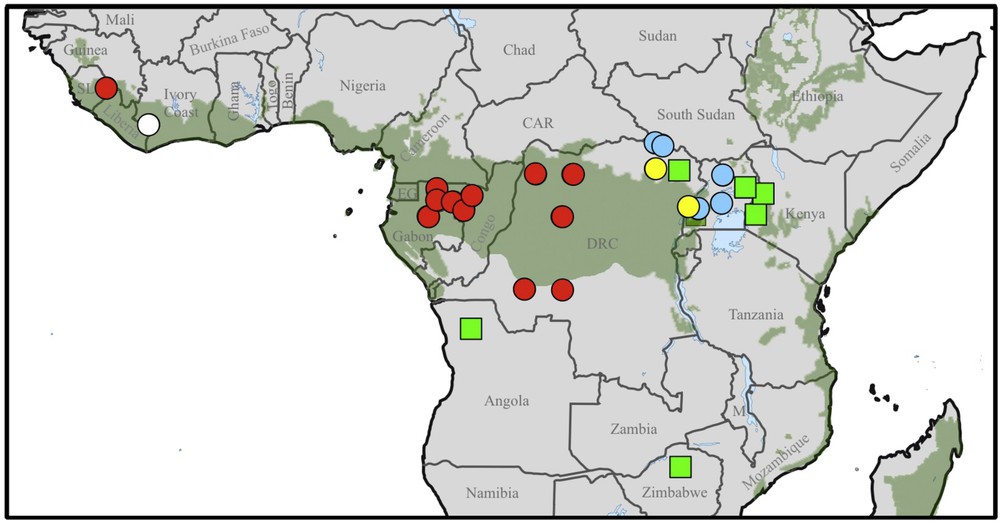
Location of Ebola hemorrhagic fever (circles) and Marburg hemorrhagic fever (green squares) outbreaks. The four species of Ebolaviruses are distinguished by colours: red for Zaire (ZEBOV), blue for Sudan, white for Taï Forest, and yellow for Bundibugyo. The tropical and subtropical moist broadleaf forests are highlighted in green (http://www.worldwildlife.org/science/wildfinder).
Since the 1970s, researchers have sampled thousands of arthropods and vertebrates to detect the presence of anti-filovirus antibodies or a direct evidence of filoviruses (RT-PCR or isolation) [2,6,7]. In 2005, Leroy et al. [8] provided the first molecular evidence that fruit bats may be the reservoir hosts for Zaire Ebolavirus (ZEBOV): the virus was detected by RT-PCR in several wild-caught and apparently healthy fruit bats belonging to three species of the family Pteropodidae: Epomops franqueti (Franquet's epauletted fruit bat), Hypsignathus monstrosus (hammer-headed fruit bat), and Myonycteris torquata (little collared fruit bat). Subsequently, bats have been intensively studied to better understand their role in the maintenance, transmission, and evolution of filoviruses. Two years after, Marburg virus was detected, using both specific antibodies and RT-PCR, in Egyptian fruit bats (Rousettus aegyptiacus) collected in northeastern Democratic Republic of the Congo (DRC) and Gabon [9]. In 2009, Towner et al. [10] isolated the virus from five R. aegyptiacus found in Kitaka Cave (Uganda), and detected highly divergent viral genomes (21%) corresponding to both Marburg virus and Ravn virus in the same colony, lending additional support to the idea that R. aegyptiacus represents a major reservoir host for Marburgviruses. The geographic distribution of R. aegyptiacus overlaps with that of Marburg and Ravn outbreaks.
The geographic range of the three species of Pteropodidae identified as potential host reservoirs of ZEBOV coincides with that of Ebola outbreaks (Fig. 1), but to date, no live Ebolavirus has been isolated from any bat. Therefore, it is difficult to know if they are the primary source of infection for this virus or if they are only involved with secondary transmission of infection to other species. Fruit bats seem, however, to play an important role as reservoir host of both Marburgviruses and Ebolaviruses. Particularly relevant is the fact that anti-Ebolavirus antibodies have been detected in five other fruit bat species in Africa: Eidolon helvum (African straw-coloured fruit bat), Epomophorus gambianus (Gambian epauletted fruit bat), Micropteropus pusillus (Peter's dwarf epauletted fruit Bat), Nanonycteris veldkampii (Veldkamp's bat), and R. aegyptiacus [11,12], as well as in the Asiatic R. leschenaultii (Leschenault's rousette) in Bangladesh [13] (taxa highlighted in Fig. 2).
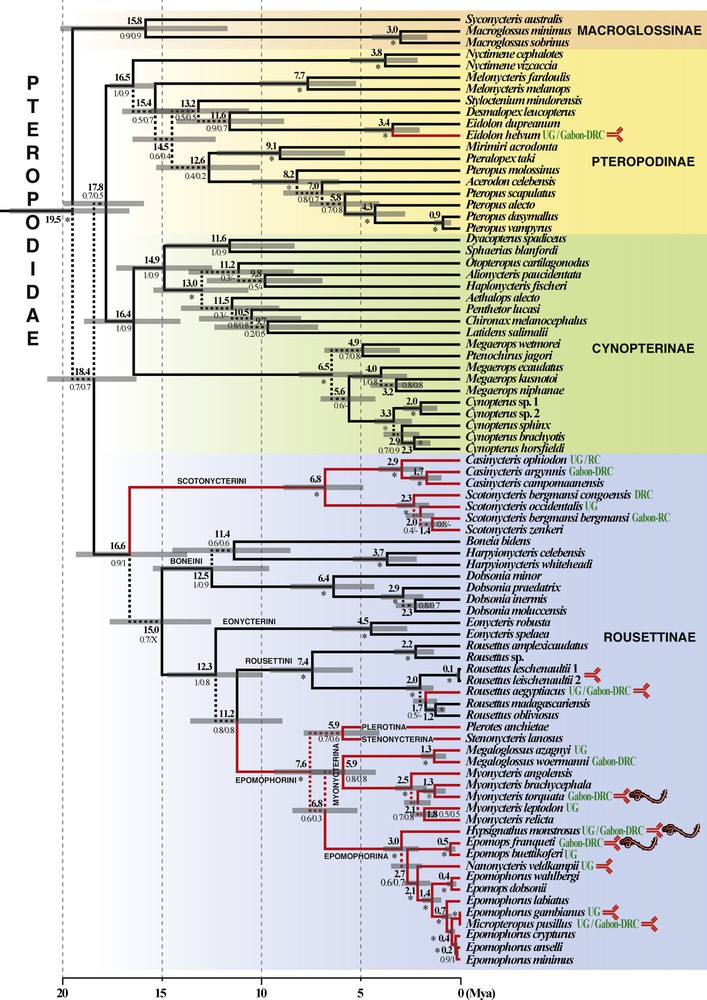
Chronogram of the family Pteropodidae based on complete mitochondrial cytochrome b gene sequences. Phylogenetic relationships and divergence times were first estimated using BEAST. The phylogeny was also inferred using MrBayes (see text for details). The ten outgroup taxa are not shown. For each node, the value in bold is the mean divergence time in millions years (grey bars indicate highest posterior density [HPD] intervals at 95%), whereas other values correspond to posterior probabilities calculated using either BEAST (at the left of the slash) or MrBayes (at the right of the slash). The symbol “–” indicates that the node was not found in the MrBayes analysis, but no alternative hypothesis was supported by PP > 0.8. The asterisk (*) shows that the node was supported by PP > 0.95 in both Bayesian analyses. A dotted branch indicates that the node was not highly supported (PP < 0.8) in the two Bayesian analyses. African species are highlighted with red branches, and those found (even occasionally) in the rainforests, UG (Upper Guinea) and/or Gabon-DRC, are indicated with green text. Red immunoglobulin symbols indicate taxa in which anti-ZEBOV antibodies were detected in previous studies (see main text for references). Pictures of Ebola virus show the three species from which ZEBOV RNA polymerase was sequenced [8].
Because of the massive outbreak in West Africa (Guinea, Liberia and Sierra Leone) in 2014–2016, it is crucial to know which fruit bat species might serve as potential reservoirs and circulating hosts of ZEBOV from Central Africa to West Africa or vice versa. Indeed, after four decades of ZEBOV outbreaks in Central Africa, it was only in 2014 that ZEBOV was detected in a country other than DRC, Gabon and Republic of the Congo (Congo). Of the 39 fruit bat species currently described on the African mainland, only 13 are present in West Africa, where they can be found in rainforests and forest-savannah mosaics [14,15]. Furthermore, phylogeographic analyses have shown that three species are in fact endemic to West Africa, i.e. Megaloglossus azagnyi (western Woermann's fruit bat), Myonycteris leptodon (western little collared fruit bat), and Scotonycteris occidentalis (Hayman's tear-drop fruit bat) [16,17]. The species Epomops buettikoferi (Buettikofer's epauletted fruit bat) is also considered endemic to West Africa [14,15], but no molecular data are currently available for this taxon. Of the nine species distributed in both West and Central Africa, only four species have been analysed for phylogeography structure, including the very rare Casinycteris ophiodon (Pohle's fruit bat) [17], as well as E. helvum [18], Myonycteris angolensis (Angolan fruit bat) [16], and R. aegyptiacus [19].
Here, we analysed the molecular phylogeography of African fruit bats of the family Pteropodidae to better understand their potential role as reservoirs and circulating hosts between Central and West Africa. The complete mitochondrial Cytb gene was sequenced for different populations of the following eight species to test for phylogeographic structure and recent gene flow from Central to West Africa: E. helvum, E. gambianus, E. buettikoferi, E. franqueti, H. monstrosus, M. pusillus, N. veldkampii, and R. aegyptiacus. Among these taxa, two are supposed to be the reservoir hosts of ZEBOV, i.e. E. franqueti and H. monstrosus [8], whereas R. aegyptiacus is considered to be the major reservoir host for Marburgviruses [10].
2 Materials and methods
2.1 Taxonomic sampling
Most of the fruit bat samples analysed in this study were collected by the authors using mist-nets (Ecotone, Gdynia, Poland) during field trips to Cameroon (AH), Central African Republic (AH, CN, EN and NN), Gabon (AH, EL and XP), Ivory Coast (BK and NN), Katanga Province of the DRC (AH, CPS, DT and NN), Liberia (BK), Orientale Province of the DRC (AH, GCG and PMA), Republic of the Congo (EL and XP), and Senegal (XP). Fruit bat species were identified morphologically using the key of Bergmans [20]. In addition, several tissue samples were obtained from specimens housed in the following museums: ‘Muséum national d’histoire naturelle’ (MNHN; Paris, France), ‘Muséum d’histoire naturelle’ of the City of Geneva (MHNG; Switzerland), and ‘Naturmuseum Senckenberg’ (SMF; Frankfurt, Germany). Names and geographical coordinates of all sampled localities are provided in Appendix A (Supporting information).
The number of individuals sequenced per species was 50 for E. helvum, 59 for E. buettikoferi, 14 for Epomops sp. (buettikoferi or franqueti) from West Africa, 146 for E. franqueti from Central Africa, 1 for Epomops dobsonii, 1 for Epomophorus anselli, 1 for Epomophorus crypturus, 12 for E. gambianus, 1 for Epomophorus labiatus, 1 for Epomophorus minimus, 76 for H. monstrosus, 79 for M. pusillus, 26 for N. veldkampii, 1 for Plerotes anchietae, and 47 for R. aegyptiacus (Appendices A and B). Eight Asian species were also sequenced for phylogenetic and molecular dating analyses (Fig. 2).
2.2 Molecular methods
Total DNA was extracted from muscle or patagium samples using DNeasy Tissue Kit (Qiagen, Hilden, Germany). The complete Cytb gene was amplified and sequenced using the primers detailed in Nesi et al. [21] and Hassanin [22]. The polymerase chain reactions (PCR) were carried out in a volume of 20 μl containing 3 μl of PCR buffer 10X with MgCl2, 2 μl of dNTPs (6.6 mM), 1 μl of each of two primers (10 μM) and 0.1 μl of Taq polymerase (2.5 U, Qiagen, Hilden, Germany). The PCRs were run using the C1000 Touch thermal cycler (BIO-RAD) as follows: 4 min at 94 °C; the denaturation/annealing/elongation process was set with five cycles of 30 s at 94 °C, 60 s at 60 °C, and 60 s at 72 °C, followed by 30 cycles of 30 s at 94 °C, 45 s at 50 °C, and 60 s at 72 °C. Final elongation followed for 7 min at 72 °C. PCR products were sequenced in both directions by the ‘Centre national de séquençage’ (Genoscope, Evry, France) or Eurofins MWG Operon (Ebersberg, Germany). Sequences were edited and assembled using Sequencher 5.1 (Gene Codes Corporation, Ann Arbor, Michigan, USA). The 523 sequences generated for this study were deposited in the GenBank database under accession numbers KX822797–KX823319.
2.3 Phylogenetic and dating analyses
The Cytb dataset used for phylogenetic analyses contains 1140 nucleotides and 92 taxa. Our new Cytb sequences were compared to those available in GenBank for other species of the family Pteropodidae (77 additional sequences; Appendix B). Ten outgroup species were used to root the pteropodid tree, representing three other mammal orders, i.e. Pholidota (Manis javanica), Cetartiodactyla (Bos javanicus), Perissodactyla (Ceratotherium simum), and seven other bat families, i.e. Emballonuridae (Taphozous melanopogon), Hipposideridae (Hipposideros armiger), Megadermatidae (Megaderma lyra), Natalidae (Natalus major), Nycteridae (Nycteris javanica), Phyllostomidae (Artibeus jamaicensis) and Rhinolophidae (Rhinolophus luctus). DNA sequences were aligned on Se–Al v2.0a11 (http://tree.bio.ed.ac.uk/software/seal/).
Divergence times were estimated using the Bayesian approach implemented in BEAST v.2.1.3 [23]. As no sufficiently accurate calibration point (fossil record or biogeographic event) is available for the family Pteropodidae, divergence times were estimated using a molecular calibration point corresponding to the age of the most recent common ancestor of Nyctimene and Pteropus (subfamily Pteropodinae in Fig. 2) estimated at 16.5 ± 1.5 Ma in Meredith et al. [24]. We applied a GTR + I + G model of evolution for each of the three codon positions (as selected under jModelTest 2.1.7 using the Akaike information criterion [25]) and a relaxed-clock model with uncorrelated lognormal distribution for substitution rates. Node ages were estimated using a calibrated Yule speciation prior and 108 generations, with tree sampling every 2000 generations, and a burn-in of 10%. Adequacy of chain mixing and MCMC chain convergence were assessed using the ESS values in Tracer v.1.6. The chronogram was reconstructed with TreeAnnotator v.1.7.5 and visualized with FigTree v.1.4.1 (http://www.tree.bio.ed.ac.uk/software/).
For comparison, phylogenetic relationships were also inferred using MrBayes 3.2.1 [26]. The posterior probabilities (PP) were calculated using four independent Markov chains run for 10,000,000 Metropolis-coupled MCMC generations, with tree sampling every 1000 generations, and a burn-in of 25%.
2.4 Population genetic analyses
Phylogeographic analyses were performed on eight species using mitochondrial Cytb sequences: E. buettikoferi, E. franqueti, E. gambianus, E. helvum, H. monstrosus, M. pusillus, N. veldkampii, and R. aegyptiacus. Mitochondrial DNA is particularly suitable for phylogeographic studies because it evolves with high rates of substitution and is transmitted maternally without recombination. However, discordant patterns between mtDNA and nuDNA markers can arise when mitochondrial introgression occurred due to secondary contact between closely related species, or when dispersal rates were higher in males than in females [17,21].
For each dataset, population genetic indices, including number of haplotypes (H), haplotype diversity (h) and nucleotide diversity (π), were calculated from Cytb sequences using DNASP v5.10 [27]. Mean, minimum, and maximum K2P distances were calculated with PAUP 4 [28]. Networks of Cytb haplotypes were constructed with the median joining method available in PopART 1.5 (http://www.popart.otago.ac.nz/) using equal weights for all mutations.
The genetic differentiation between pairs of populations (e.g., Central versus West Africa) was measured using both Fst and GammaST calculated with DNASP v5.10 [27].
3 Results
3.1 A Molecular timescale for pteropodid evolution
The Bayesian chronogram of Fig. 2 shows that species of the family Pteropodidae can be divided into four major clades (PP = 0.9 – 1) that we consider here as corresponding to the subfamilies Pteropodinae, Cynopterinae, Macroglossinae, and Rousettinae. According to our molecular dating estimates, these four groups diverged rapidly from each other during the Early Miocene, between 19.5 and 17.8 Mya.
All fruit bat species of Africa belong to the subfamily Rousettinae, except Eidolon helvum, a member of the subfamily Pteropodinae that is closely related to E. dupreanum (PP = 1), its congeneric species in Madagascar. The African species of the subfamily Rousettinae are further subdivided into three robust clades (PP = 1), here treated as three different tribes: (1) the tribe Scotonycterini, which contains six African species arranged into two genera, Scotonycteris and Casinycteris; (2) the tribe Rousettini, which includes a single genus, Rousettus, with only one African species, R. aegyptiacus, and several species from Asia (R. amplexicaudatus and R. leschenaultii), Madagascar (R. madagascariensis), and the Comoro Islands (R. obliviosus); (3) the tribe Epomophorini sensu lato, a large African clade composed of the four subtribes Epomophorina (with species of Epomophorus, Epomops, Hypsignathus, Micropteropus, and Nanonycteris), Myonycterina (with species of Myonycteris and Megaloglossus), Plerotina (Plerotes anchietae), and Stenonycterina (Stenonycteris lanosus). Our molecular estimates suggest that the three tribes Scotonycterini, Rousettini, and Epomophorini diversified synchronously during the Late Miocene, at around 8–7 Mya.
3.2 Phylogeographic networks
We analysed the nucleotide variation of 675 Cytb sequences (Appendix A) to explore the phylogeography of eight species of Pteropodidae. The number of sequences, segregating sites, haplotypes, as well as haplotype diversity, nucleotide diversity, and K2P distances are described for each taxon in Appendix C. Finally, a network of Cytb haplotypes was constructed for only six different datasets (Fig. 3). Indeed, preliminary analyses revealed that two pairs of species cannot be distinguished on the basis of their mitochondrial sequences: E. gambianus/M. pusillus [21] and E. franqueti/E. buettikoferi.
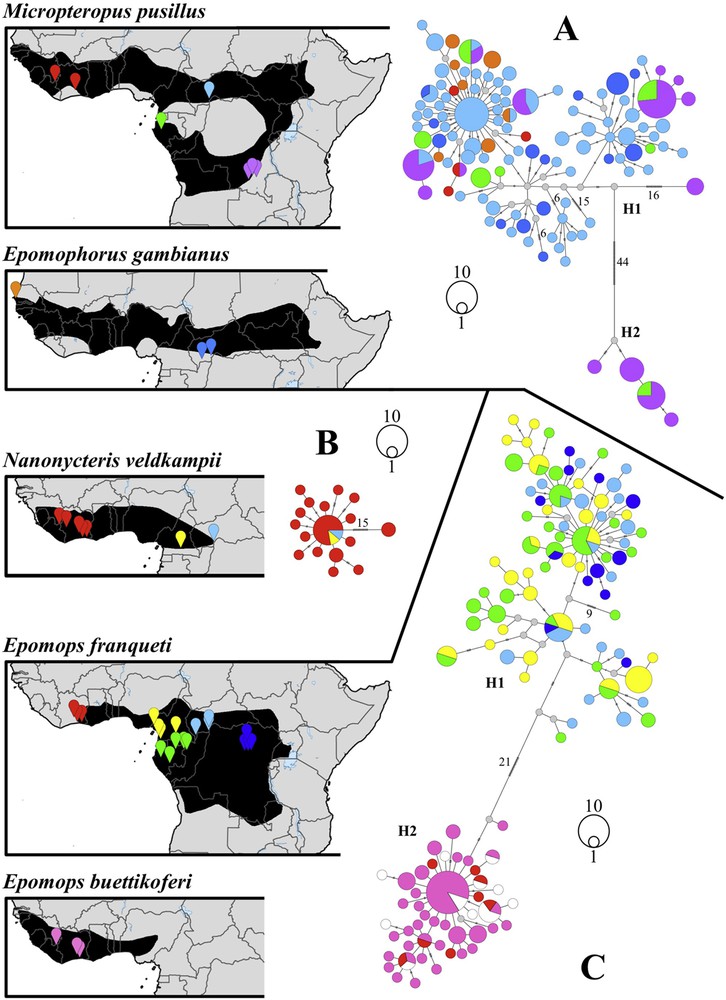
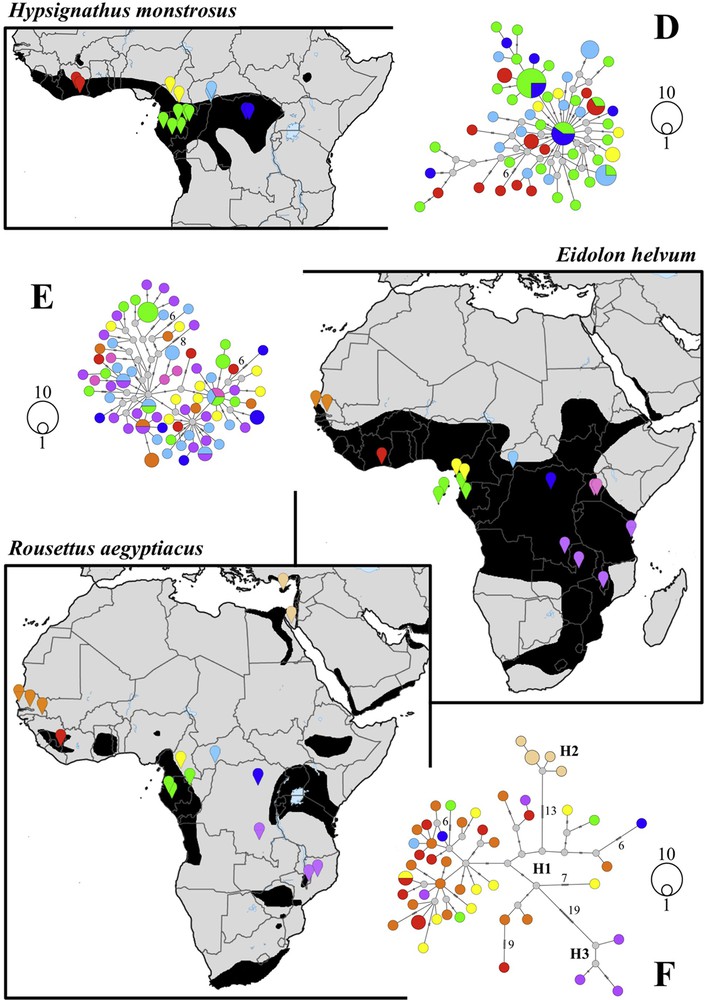
Phylogeographic patterns of fruit bat species distributed in both West and Central Africa. Median joining networks based on cytochrome b haplotypes were reconstructed for the six following datasets: (A) Micropteropus pusillus and Epomophorus gambianus (212 individuals); (B) Nanonycteris veldkampii (27 individuals); (C) Epomops buettikoferi and Epomops franqueti (221 individuals); (D) Hypsignathus monstrosus (76 individuals); (E) Eidolon helvum (88 individuals); (F) Rousettus aegyptiacus (51 individuals). The geographic distribution maps were extracted from the IUCN [14]. Localities of the haplotypes were classified in nine biogeographic regions (see details in Appendix A): Senegal (orange), Upper Guinean rainforest (red or pink), Cameroon (yellow), western Equatorial African rainforest (green), Central African Republic (light blue), eastern Equatorial African rainforest (navy blue), Uganda (pink), southeastern Africa (purple), and Mediterranean region (beige). In (C), white haplotypes were obtained from West African individuals that cannot be assigned to either E. franqueti or E. buettikoferi on the basis of the third palatal ridge.
The network reconstructed from the E. gambianus/M. pusillus dataset (Fig. 3A) revealed high genetic diversity, with 21 Cytb haplotypes for Epomophorus and 94 Cytb haplotypes for Micropteropus. There is no taxonomic coherence, as the haplotypes do not cluster according to species names, and there is no geographical structure among sampled populations. In addition, we found two Cytb haplotypes shared between the two species. In Micropteropus, we detected higher levels of nucleotide diversity (0.0171 versus 0.0094), with the existence of two highly divergent Cytb haplogroups differing by 44 mutations (named H1 and H2 in Fig. 3A): H1 was found in both northern populations (Ivory Coast and Central African Republic) and southern populations (Gabon and Katanga Province of the DRC); H2 was detected only in southern populations (Gabon and Katanga). All the four H2 Cytb haplotypes share 99–100% of identity with Cytb haplotypes of Epomophorus crypturus and E. wahlbergi collected in DRC (Katanga) and South Africa (unpublished data).
The dataset for E. franqueti and E. buettikoferi (Fig. 3C) also revealed high genetic diversity, with 91 Cytb haplotypes for E. franqueti and 35 Cytb haplotypes for E. buettikoferi. It was not possible to assign 14 individuals collected in West Africa to either E. franqueti or E. buettikoferi. The network of Epomops Cytb sequences revealed the existence of two divergent haplogroups separated by 21 mutations and geographically highly segregated: the first one includes all individuals of E. franqueti collected in Central Africa (Cameroon, Central African Republic, Gabon, Congo, and DRC), whereas the second one contains all individuals identified as E. franqueti or E. buettikoferi from West Africa (Liberia and Ivory Coast), with three haplotypes shared by the two species.
In all other species, there is no strong genetic structure between Central and West Africa, suggesting that populations were able to migrate extensively between the two regions. For N. veldkampii, the mtDNA analysis showed a star-like network (Fig. 3B), typical of recently expanded populations. The two Cytb haplotypes from Central Africa (Cameroon and Central African Republic) were found to be identical to the most common Cytb haplotype of West Africa. In R. aegyptiacus, the individuals collected in the eastern Mediterranean region (Cyprus and Egypt) differ by 13 mutations from populations of sub-Saharan Africa. In addition, some Cytb haplotypes of southeastern Africa were found to be highly divergent from the others (19 mutations) (Fig. 3F). By contrast, the population from West Africa does not differ significantly from that of Central Africa, as indicated by the low values of Fst and GammaST (< 0.1) (Appendix C). In both H. monstrosus and E. helvum (Fig. 3D and E), we found no evidence of geographic structure, and Fst and GammaST values confirmed the absence of differentiation between populations from Central and West Africa. This observation was corroborated by the discovery of shared Cytb haplotypes for each of the two species between these two regions of Africa. In Eidolon, one Cytb haplotype is shared by individuals from Senegal and Katanga (DRC). In Hypsignathus, one Cytb haplotype is shared between two individuals from Ivory Coast (collected in 2009) and an individual from Gabon (G5CHA068), from which Leroy et al. [8] sequenced a ZEBOV RNA polymerase.
4 Discussion
4.1 Out of Asia: multiple origins of fruit bats in Africa
The fruit bats of the family Pteropodidae are only distributed in the Old World, and most of the species are found in the equatorial regions, where fruits are abundant throughout the seasons (Appendix D). Three different lines of evidence support a Southeast Asian origin of the family: (1) 94 species of Pteropodidae are found in this region [14], which represents more than 50% of the total diversity; (2) Sumatra, Borneo and Sulawesi contain the highest species density (> 15) (Appendix D); and (3) the oldest fossil of fruit bats has been described in the Late Eocene/Early Oligocene of Thailand [29].
With our expanded taxonomic coverage with respect to previous studies [30,31], the molecular analyses presented herein confirm the existence of four independent lineages of African fruit bats, all of which occupying a derived position with respect to species from Asia and Oceania (see red branches in Fig. 2): E. helvum is the sole African representative of the subfamily Pteropodinae, whereas the three other African lineages, i.e. R. aegyptiacus, Epomophorini and Scotonycterini, belong to the subfamily Rousettinae. The chronogram in Fig. 2 suggests that the African continent was colonized by at least four Asian ancestors: between 2.0 and 1.7 Mya for Rousettus, between 11.6 and 3.4 Mya for Eidolon, between 11.2 and 7.6 Mya for Epomophorini, and between 16.6 and 6.8 Mya for Scotonycterini. Anti-Ebolavirus antibodies have been previously detected in eight African species of Pteropodidae, representing three of these four lineages: E. franqueti, E. helvum, E. gambianus, H. monstrosus, M. pusillus, M. torquata, N. veldkampii, and R. aegyptiacus [8,11,12]. These results suggest therefore that several unrelated pteropodid taxa have developed a specific immunological response to Ebolavirus, and that the transmission of Ebolavirus between fruit bat species may have occurred through contacts in fruit trees.
4.2 Which are the fruit bat species able to disperse between Central and West Africa?
All species of Pteropodidae are highly dependent on plants for food. This explains why the species richness of fruit bats is higher in equatorial regions (Appendix D), where fruits and flowers are more diverse and available most of the year. Since pteropodids do not hibernate, they need abundant food all year round. To exploit seasonal food resources, some species have developed the ability for migrations or nomadic movements. Among African pteropodids, only E. helvum and N. veldkampii are considered as being migratory species, which means that they travel seasonally from one habitat to another using predetermined routes [32]. Populations of N. veldkampii are resident of the rainforest during the dry season, and both sexes migrate northwards towards savannah habitat types during the wet season. This small species seems to make an annual round-trip migration of 300–1100 km [32]. Males of E. helvum are known to make very long-range migrations across Central Africa (∼ 2500 km over five months), but the routes used by the populations during their annual migration remain to be discovered [33].
The following pteropodid species of Africa are considered to be nomadic rather than migratory, which means that their seasonal movements are irregular and unpredictable: most species of Epomophorus (including E. gambianus), M. pusillus, M. leptodon, M. woermanni, and R. aegyptiacus [32,34]. In addition, local hunters of Luebo and Mweka villages (DRC) have reported that E. franqueti and H. monstrosus have annual movements away from these regions [35]. Although field studies have indicated seasonal movements for H. monstrosus [36,37], to our knowledge, nothing was published on the migratory behaviour of E. franqueti. Only two species of African Pteropodidae have been the subjects of telemetry studies, i.e. E. helvum and R. aegyptiacus [12,33,38], and even for these taxa, seasonal movements remain poorly documented, as the marked individuals could not be tracked for at least one annual cycle.
Phylogeographic analyses therefore are particularly important to provide insight into which species disperse between Central and West Africa. Previous molecular studies on Myonycterina [16] and Scotonycterini [17] have identified a strong separation between populations from the two main blocks of African rainforest, the Upper Guinea forest in West Africa and the large Congo Basin forest in Central Africa. Accordingly, several species were described as endemic to each of these forest blocks: M. azagnyi, M. leptodon, and S. occidentalis in West Africa, and their sister-species in Central Africa, i.e. M. woermanni, M. torquata, and S. zenkeri/S. bergmansi, respectively. All these sister taxa have diverged in allopatry during two major glacial periods of the Pleistocene, at 2.8–2.5 Mya and 1.8–1.6 Mya. By contrast, a greater capacity for long-distance dispersals between these two rainforest blocks was proposed for the largest species of Scotonycterini, C. ophiodon, and for the sole cave-dwelling species of Myonycterina, M. angolensis [16,17].
Here, we used mitochondrial Cytb sequences to compare the phylogeography of E. helvum, R. aegyptiacus, and six species of the subtribe Epomophorina. For the two migratory species, E. helvum and N. veldkampii, we obtained a typical star-like network pattern, with no genetic structure across their geographic range (Fig. 3B and E). These results confirm that these two species have high dispersal capacity. Similar phylogeographic patterns, with no geographic structure between Central and West Africa, were also found for most species considered to be nomadic, i.e. E. gambianus, H. monstrosus, M. pusillus, and R. aegyptiacus. In R. aegyptiacus, we detected, however, divergent Cytb haplotypes in all individuals from the eastern Mediterranean, and in some individuals from Katanga (DRC) and Malawi, suggesting that these distant populations tend to be more isolated from the others. For several individuals from southern populations of M. pusillus (Gabon and Katanga; Fig. 3A), we detected very divergent H2 Cytb haplotypes (4.68–6.04%), which share 99–100% of identity with individuals of E. crypturus and E. wahlbergi collected in DRC (Katanga) and South Africa (unpublished data). Combined with the fact that the H1 Cytb haplotypes of M. pusillus cannot be differentiated from those of E. gambianus (Fig. 3A; see also Nesi et al. [21]), we suggest that several events of inter-specific hybridization have occurred between the different species of the Epomophorus–Micropteropus complex. We plan to further explore this issue with RADSeq data. Taxonomically, we recommend placing M. pusillus in the genus Epomophorus, as originally proposed by Peters in 1867 [39]. Our phylogenetic tree in Fig. 2 also indicates that the species E. dobsoni should be excluded from Epomops and rather treated as a species of Epomophorus. Morphologically, it is important to note that both M. pusillus and E. dobsoni have six thick palatal ridges (considering that the second and third ridges are partially fused in E. dobsoni), as in all other species of Epomophorus, whereas the species of Epomops (E. buettikoferi and E. franqueti) have only three thick palatal ridges [20]. Ecologically, both M. pusillus and E. dobsoni are more common in woodland savannahs, as all species of Epomophorus, whereas other species of Epomops (E. buettikoferi and E. franqueti) occur mainly in rainforest habitats.
Within Epomops sensu stricto, our Cytb analyses do not support the monophyly of the two species, E. franqueti and E. buettikoferii: all individuals from West Africa, identified as either E. franqueti or E. buettikoferii, share very similar or identical haplotypes, which are divergent from those sequenced for all individuals of E. franqueti collected in Central Africa (K2P distances: 2.24–3.63%). Taxonomically, this result shows that the single discrete character used in the key of Bergmans [20] for distinguishing E. buettikoferi from E. franqueti, i.e. the medial division of the third ridge in the palate, is not dependable. In agreement with that, it must be noted that the palatal pattern supposed to characterize E. buettikoferi has been previously described in some individuals of E. franqueti collected in Cameroon and Gabon [39]. To better understand the taxonomy of Epomops, we sequenced all the 12 nuclear introns analysed in Hassanin et al. [17] for two individuals representing each putative species and each geographic group, i.e. E. buettikoferi from West Africa and E. franqueti from Central Africa (data not shown). The nuclear divergence was only 0.08%, which is in the range of intraspecific distances found in other groups of Laurasiatheria, such as Myonycterina (< 0.21%) [16], Scotonycterini (< 0.13%) [17], or cattle and bison of the tribe Bovini (< 0.18%) [40]. The analyses of mitochondrial and nuclear data suggest therefore that the genus Epomops contains only one species, which can be divided into two subspecies: E. franqueti franqueti in Central Africa and E. f. buettikoferi in West Africa [32].
To summarize, our phylogeographic results suggest that all migratory or nomadic species known to be common in savannah woodland habitats, can disperse between Central and West Africa: E. gambianus, E. helvum, M. pusillus, N. veldkampii, and R. aegyptiacus. By contrast, all species restricted or largely restricted to the rainforests (C. argynnis, E. franqueti, M. azagnyi, M. leptodon, M. torquata, M. woermanni, S. bergmansi, and S. occidentalis), with the exception of H. monstrosus, do not disperse long distances, specifically across the Dahomey Gap, i.e. the savannah corridor in Ghana, Togo and Benin that separates the rainforest blocks of West and Central Africa [16,17]. Because of its larger body size, muscular strength, and higher wing loading [41], H. monstrosus can fly much faster and therefore farther than other fruit bats present in African rainforests, which may explain its greater dispersal capacity.
4.3 Consequences for the origin of ZEBOV in West Africa
On 21 March 2014, a circulating virus in humans was identified by the Institut Pasteur in Lyon (France) as ZEBOV, a strain previously detected in only three Central African countries (DRC, Gabon and Congo) between 1976 and 2014 [42]. Epidemiologic investigations have suggested that the first case of the West African outbreak was a 2-year-old child who died on 6 December 2013 in Meliandou, a small village in Guinea [43]. Subsequently, Guinea, Liberia, and Sierra Leone experienced the largest outbreak of Ebola ever recorded, with widespread and intense transmission between August 2014 and December 2014, after which case incidence declined. In total, 28,603 cases were identified, with deaths (as of 10 June 2016) [44].
Phylogenetic analyses of Ebola virus genomes have suggested that the new ZEBOV variant from West Africa diverged from lineages of Central Africa around 2003–2004 [45,46]. An introduction into Guinea from a human traveller seems unlikely, because the epicentre region of Guéckédou is a remote area of the Upper Guinean rainforest, which is far from the Congo rainforest where all previous ZEBOV outbreaks occurred [47]. By contrast, migratory bats may have carried ZEBOV from Central Africa to Guinea. Among fruit bats, eight species are commonly found in the Congo rainforest [14,15]: C. argynnis, E. helvum, E. franqueti, H. monstrosus, M. woermanni, M. torquata, R. aegyptiacus, and S. bergmansi. In 2005, Leroy et al. [8] obtained ZEBOV RNA polymerase sequences from liver and spleen samples of H. monstrosus (19%; 4/21), E. franqueti (4.3%; 5/117), and M. torquata (2.8%; 4/141), suggesting that these three species represent natural reservoir hosts of Ebola viruses. In Gabon and Congo, ZEBOV-specific antibodies were detected in all fruit bat species tested for a sample size > 125 individuals: R. aegyptiacus (7.8%; 24/307), H. monstrosus (7.2%; 9/125), E. franqueti (4.5%; 36/805), M. torquata (3.3%; 19/573), and M. pusillus (2.0%; 4/197). In addition, two insectivore bat species were found to be ZEBOV positive: Hipposideros gigas and Mops condylurus [11]. In West Africa, ZEBOV antibodies were detected in four pteropodid species: E. gambianus (13.5%; 5/37), H. monstrosus (12.5%; 2/16), E. franqueti (10.7%; 3/28), and E. helvum (0.4%; 1/262) [12,48]. All these data suggest that ZEBOV is a circulating virus across Africa and that it was transmitted not only in bats endemic to rainforests, but also to those generally found in savannah woodlands, such as E. gambianus and M. condylurus. Transmission of ZEBOV between bat species may have occurred through contacts in fruit trees, involving either fighting for food resources, or indirect contamination via infected body fluids (saliva, blood, faeces, and urine) deposited on branches and fruits [6].
Our phylogeographic analyses revealed that only three fruit bat species were able to disperse directly ZEBOV from the rainforests of Central Africa (where ZEBOV was endemic until sometime before 2014) to those of West Africa (where ZEBOV suddenly appeared in 2014): E. helvum, H. monstrosus, and R. aegyptiacus (Fig. 3). The species E. helvum seems, however, to be associated with secondary transmissions from a zoonotic reservoir: it showed a very low ZEBOV prevalence (0.4%); and its implication in human outbreaks appears unlikely, because it typically lives in large urban colonies (e.g., Bangui, Yaoundé) and is a source of bushmeat in many African regions [49]. By contrast, H. monstrosus and R. aegyptiacus represent the two leading candidates for explaining the dispersal of ZEBOV from Central to West Africa. First of all, H. monstrosus was identified as one of the three reservoir hosts of ZEBOV in the rainforests of Central Africa [8,35], whereas R. aegyptiacus was identified as the main reservoir host of Marburgviruses [10]. The highest seroprevalences against ZEBOV were found for R. aegyptiacus and H. monstrosus (7.8 and 7.2%, respectively) [11]. In Uganda, the study of a large population of Marburg-virus-infected R. aegyptiacus fruit bats has evidenced that the two biannual birthing seasons represent times of increased infection among older juvenile bats (≈ six months of age) that roughly coincide with historical dates of Marburg virus spillover into humans [50]. In Gabon and Congo, most human ZEBOV outbreaks have occurred through contact with infected animal carcasses, especially great apes and duikers. It has been suggested that the transmission to great apes and duikers have been initiated by the consumption of fruits contaminated with blood and placentas during parturition of infected fruit bats [11]. All these elements suggest that the parturition of infected fruit bats may have increased ZEBOV prevalence in fruit bat populations, and therefore the contamination of other species, including humans. This hypothesis is also supported by the fact that the first case of the West African ZEBOV outbreak was infected in November or December, a period that coincides perfectly with one of the two birthing seasons of H. monstrosus and R. aegyptiacus in West Africa [32,51].
5 Conclusion
Our genetic analyses have shown that three fruit bat species have dispersal movements between Central and West Africa, and, hence, are capable of acting as dispersal agents for this virus. Among them, only H. monstrosus is restricted to rainforest habitats, where all ZEBOV outbreaks have occurred, while E. helvum and R. aegyptiacus are also commonly found in savannah woodlands. Since fruit bats often eat on the same trees, inter-species infections of ZEBOV are expected to be frequent, in particular during the two biannual birthing seasons of the main reservoir host species. Because of that, we cannot completely rule out the hypothesis involving that ZEBOV was dispersed indirectly in West Africa, i.e. through nomadic or migratory species of savannah woodlands, i.e. E. gambianus, M. pusillus and N. veldkampii, which may have encountered the main reservoir host species of the Congo Basin in the rainforest–savannah mosaics.
Acknowledgments
We thank the numerous individuals who helped us to collect tissue samples during field missions: Philippe Blot, André Délicat, and Jean-Pierre Hugot in Gabon; Jérôme Fuchs, Philippe Gaubert, Flobert Njiokou, and Anne Ropiquet in Cameroon; Alain Le Faou in Central African Republic; Christiane Denys, Mireille Dosso, François Jacquet, and Stéphane Kan Kouassi in Ivory Coast; and Raphaël Colombo, Laurent Daudet, Benjamin Dudu, Tamas Görföl, Keunen Hilde, Ros Kiri Ing, Jean-François Julien, Vuong Tan Tu and Peter Vallo in DRC. We are very grateful to collection managers and curators who provided samples from museum specimens: Katrin Krohmann and Virginie Volpato (Naturmuseum Senckenberg), Anne-Marie Ohler and Jean-Marc Pons (MNHN). AH would like to thank Kevin Racine and the World Bat Library (Geneva, Switzerland) for bibliographic assistance. We also acknowledge the two anonymous reviewers for their helpful comments on the manuscript. This work was supported by the MNHN, CNRS, ‘PPF Taxonomie moléculaire, DNA Barcode and gestion durable des collections’, ‘PPF Biodiversité actuelle et fossile’, ‘Société des amis du Muséum national d’histoire naturelle et du Jardin des plantes’, LabEx BCDiv 2012-2013, and ‘Consortium national de recherche en génomique’. It is part of agreement No. 2005/67 between the Genoscope and the MNHN on the projects ‘SpeedID’ and ‘Bibliothèque du Vivant’.