


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Our genome is particularly vulnerable during the S phase of the cell cycle. During this process, the two strands of the double helix are separated and the complementary strands are faithfully synthesized so that the mother cell can pass two identical copies of the genome to the daughter cells. This task is performed by thousands of molecular micromachines called replisomes, which polymerize DNA at structures called replication forks [1, 2]. This process is initiated at well-defined genetic elements called replication origins [3]. It depends on the assembly of pre-replication complexes on origins during the G1 phase of the cell cycle and their activation in S phase under the control of CDK and DDK kinases [4, 5, 6]. Origin activation allows the recruitment of the CMG helicase complex, consisting of the CDC45 protein, the MCM2-7 hexamer and the GINS tetrameric complex. The CMG helicase interacts with the Fork Protection Complex (composed of Claspin, Timeless, Tipin), as well as DNA polymerases, and various accessory factors of the replisome [1, 7, 8]. Replication origins are activated sequentially throughout the S phase, following a precise spatiotemporal program [9, 10]. The replication forks progress at a rate of 1–2 kb per minute along the chromosomes until they encounter forks progressing in the opposite direction or reach the end of the chromosomes [11]. The two replisomes are then disassembled in a process called replication termination [12]. Correct execution of the replication program allows each portion of the genome to be replicated once and only once during the S phase and ensures faithful transmission of genetic material to daughter cells. However, the replication forks encounter various obstacles during their progression along the chromosomes, causing what is commonly referred to as replication stress [13].
2. Mechanisms of response to replication stress
Blocking replication forks can induce chromosomal breaks and rearrangements. In fact, replication defects represent the major source of endogenous genomic instability at the origin of cancers. Stalled forks are unstable structures that must be rapidly stabilized and restarted to avoid the formation of toxic recombination intermediates and the induction of chromosomal rearrangements [11]. During evolution, complex surveillance mechanisms have emerged in eukaryotic cells, the main one being the S phase checkpoint mediated by the proteins ATR and CHK1 [14].
The ATR kinase is recruited to the forks via its ATRIP subunit, which interacts with RPA, a complex that protects single-stranded DNA. ATR is activated by TopBP1, a factor recruited to junctions between single- and double-stranded DNA [15]. Once activated, ATR phosphorylates the effector kinase CHK1, which allows the amplification and propagation of the stress signal. CHK1 activation is mediated by the Claspin protein, which is part of the replication fork protection complex [16, 17, 18]. The ATR-CHK1 pathway acts at multiple levels to coordinate fork repair processes, prevent premature entry into mitosis, and enable completion of DNA replication [14].
Fork restart depends on the concerted action of numerous enzymes including homologous recombination factors, helicases, nucleases, translocases and chromatin remodelers [19]. Under replication stress conditions, these factors ensure extensive remodeling of the fork structure. Specifically, this involves a remodeling of nascent DNA strands to form a four-way junction called a reversed fork [20, 21]. Fork reversal allows for the controlled degradation of nascent DNA strands by the nucleases MRE11, DNA2, and EXO1 [22] and the recruitment of the recombinase RAD51 to initiate fork repair by homologous recombination [23]. Excessive nascent strand resection is prevented by BRCA1 and BRCA2, two repair proteins frequently mutated in breast and ovarian cancers [24, 25]. The inability of cells to respond effectively to replication stress results in chronic genomic instability and contributes to tumorigenesis.
3. Main sources of replication stress
Cellular responses to replication stress have been particularly studied in the presence of drugs such as hydroxyurea (HU) or aphidicolin, which block replication by inducing nucleotide pool depletion or inhibiting DNA polymerase activity, respectively. Replication stress is also induced by most drugs commonly used in chemotherapy, such as alkylating agents, antimetabolites and topoisomerase inhibitors [26]. These drugs induce uncoupling between helicase and polymerase activities and increase single-stranded DNA, which is recognized as a universal stress signal by the ATR kinase [13, 27]. Interestingly, recent evidence indicates that HU can also slow down fork progression independently of dNTP pools by inducing oxidative stress and displacing Timeless from the replisome [28, 29].
Under normal growth conditions, cells are also exposed to endogenous sources of replication stress. This stress can originate from protein complexes strongly associated with DNA, secondary structures of DNA (G-quadruplexes, hairpins, etc.), or lesions induced by cellular metabolism byproducts [13, 19]. These obstacles are generally well managed by the cell and their impact on replication remains limited. A more complex problem concerns the availability of deoxyribonucleotides (dNTPs) during S phase. Indeed, the production of dNTPs must be closely coordinated with DNA synthesis to ensure optimal progression of replication forks [11]. Thus, it has been shown that a deficit of dNTPs slows down the forks and an excess increases their speed [30]. A too large or unbalanced pool of dNTPs can also increase the frequency of mutations. Maintaining a balanced pool of dNTPs throughout S phase requires a tight coupling between the cell cycle and cell metabolism [31]. Thus, cells must be able to detect ongoing replication in order to produce dNTPs at the right time. Recent evidence indicates that this is an essential function of the ATR-CHK1 pathway [32]. In addition, ribonucleotides (rNTPs) can be mistakenly incorporated during DNA synthesis. These ribonucleotides are normally removed by RNase H2 [33, 34, 35]. In the absence of RNase H2, excision of ribonucleotides by topoisomerase I (Top1) generates non-repairable DNA lesions leading to deletions and double-strand breaks [36].
Another major source of endogenous replication stress results from conflicts between replication and transcription. This is due to the fact that DNA and RNA polymerases must share the same DNA substrate as they move along the chromosomes, making conflicts inevitable [37]. These conflicts can manifest in different ways, such as head-on collisions between the transcription and replication machineries, accumulation of topological stress along the DNA or the formation of R-loops [38]. R-loops are three-stranded nucleic acid structures that form during transcription when nascent RNA reanneals with the template DNA strand, leaving the non-coding strand unpaired [39, 40]. Recent evidence indicates that R-loops represent barriers to replication, but the molecular mechanisms involved in this interference are still poorly understood [38].
In the absence of exogenous sources of replication stress (Figure 1A), the low level of endogenous replication stress is properly managed by the cell. Indeed, it enters S phase only when its metabolic state allows it, regulates dNTPs pools and coordinates transcription and replication programs to limit conflicts [11]. In contrast, aberrant activation of oncogenes in pre-tumor cells disrupts this coordination (Figure 1B). Endogenous replication stress increases, which induces genetic instability and contributes to cancer development [41].
Major sources of endogenous replication stress in normal and cancer cells. (A) Replication fork progression depends on a constant supply of deoxyribonucleotide triphosphate (dNTPs) to fuel the activity of DNA polymerases α, δ and 𝜀. The cell must also avoid collisions between replication forks and transcription complexes (RNAPII). Under normal growth conditions, this coordination is achieved by tight coupling between replication and transcription programs, cell cycle progression, and cell metabolism. (B) Deregulation of oncogenic pathways increases transcription and R-loop formation, alters the G1/S transition, and disrupts cell metabolism. This leads to deregulation of dNTP pools, increased replication–transcription conflicts and accumulation of chromosomal rearrangements.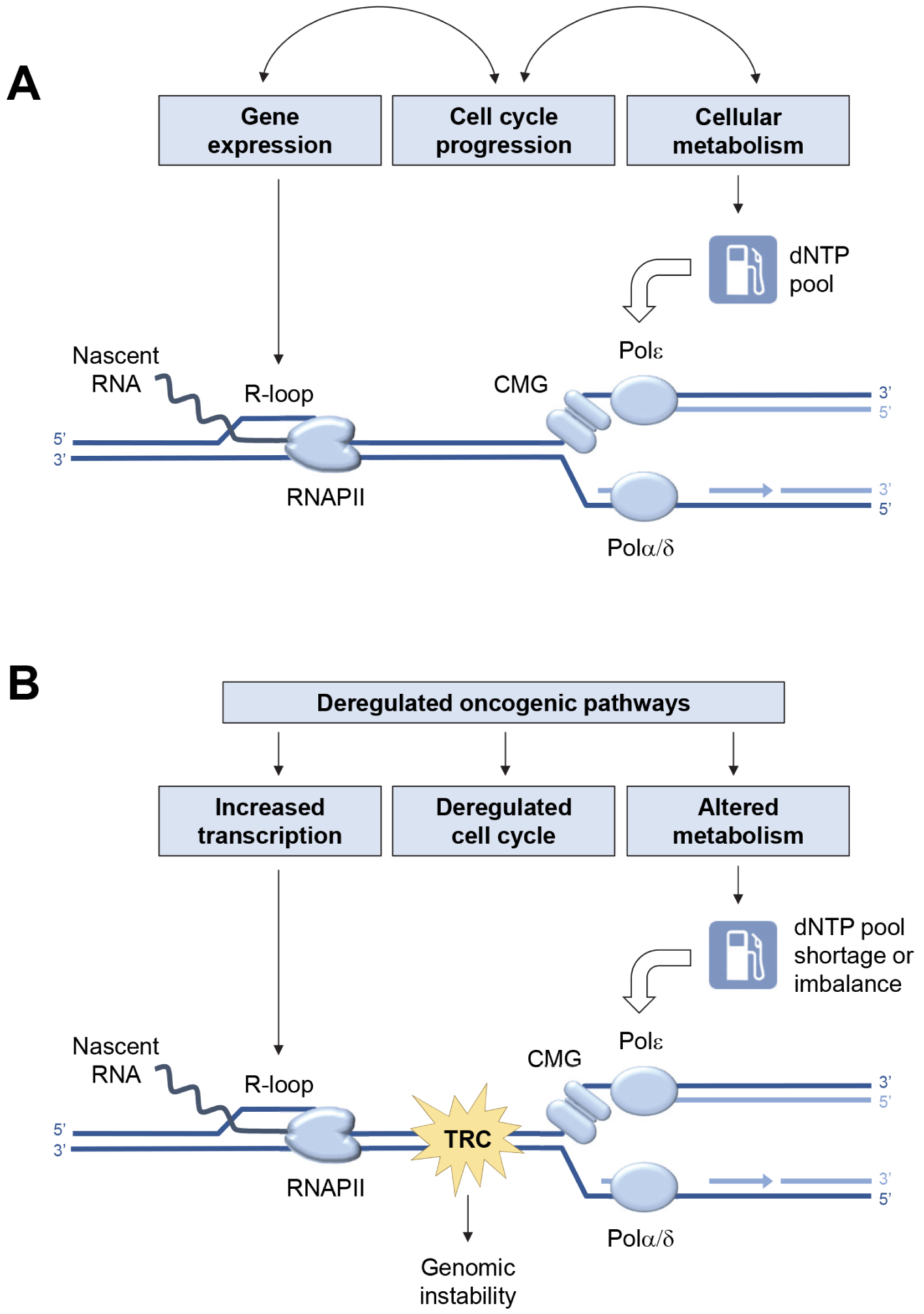
4. Oncogene-induced replication stress
It has been shown that aberrant expression of oncogenes such as Ras, Myc, or CycE can disrupt DNA replication in a variety of ways, for example by deregulating dNTP pools, increasing replication–transcription conflicts, or causing premature entry into S phase [39, 41, 42, 43]. This oncogene-induced replication stress represents a double-edged sword for cancer cells. While it contributes to cancer development by promoting genetic instability, it also slows tumor cell proliferation and activates anticancer barriers leading to apoptosis or senescence [44, 45, 46, 47]. To continue to proliferate, cancer cells must circumvent these barriers, while limiting the deleterious effects of replication stress on DNA replication. To do so, cancer cells become very dependent on the ATR-CHK1 pathway [48, 49, 50]. They also adapt to oncogene-induced replication stress by overexpressing components of the replication fork protection complex such as Claspin and Timeless, independently of ATR signaling [51]. This overexpression is observed in primary colon, breast and lung carcinomas and is generally associated with poor prognosis. Surprisingly, overexpression of Claspin and Timeless appears spontaneously in immortalized human fibroblasts expressing the H-RasV12 oncogene and allows them to escape replication stress and senescence [51].
5. Replication–transcription conflicts and cancer
Recent evidence indicates that the replication stress detected in cancer cells is largely due to replication–transcription conflicts [43]. Since transcription and replication complexes progress at roughly the same rate, the most severe conflicts result from head-on collisions [52, 53]. In order to limit the deleterious effects of head-on conflicts, the genome of bacterial species such as Bacillus subtilis has evolved such that most genes are codirectionally oriented with respect to replication forks [54]. In contrast, genes associated with pathogenesis and stress resistance are oriented in the opposite direction to replication, which locally increases DNA damage and promotes targeted evolution of these genes [55, 56]. This codirectional organization of replication and transcription is not found in eukaryotes because unlike bacterial chromosomes, which have only one origin of replication, eukaryotic chromosomes can carry several thousand. Moreover, the diversity of gene expression programs in metazoans does not allow to define a unique organization that would be adapted to all cell types. However, eukaryotes are not helpless against transcription–replication conflicts. Indeed, in the dynamic context of cell differentiation, human cells use a trick of promoting replication initiation upstream of highly transcribed genes [57, 58, 59], allowing these genes to be mostly replicated codirectionally. Thus, the average fork direction profile is positive (codirectional) between the transcriptional start (TSS) and termination (TTS) site in HeLa cells (Figure 2A). Interestingly, the direction of fork progression reverses at the end of the genes, indicating that replication forks initiated downstream of the genes converge with transcription at the TTS [60].
Functional organization of the human genome and replication–transcription conflicts. (A) Distribution of replication fork direction (blue), R-loops (red) and a replication stress marker (p-RPA32-S33, black) at a metagene (16,000 human genes) aligned between transcription start (TSS) and termination (TTS) sites. A positive replication fork direction indicates that the fork is progressing in the same direction as transcription. (B) Schematic representation of the distribution of replication origins and R-loops (1), as well as RNA polymerases prior to replication (2), when replication is initiated upstream of the TSS (3), and when replication and transcription converge at the TTS, inducing replication stress (yellow spark) and accumulation of phospho-RPA32-S33 (4).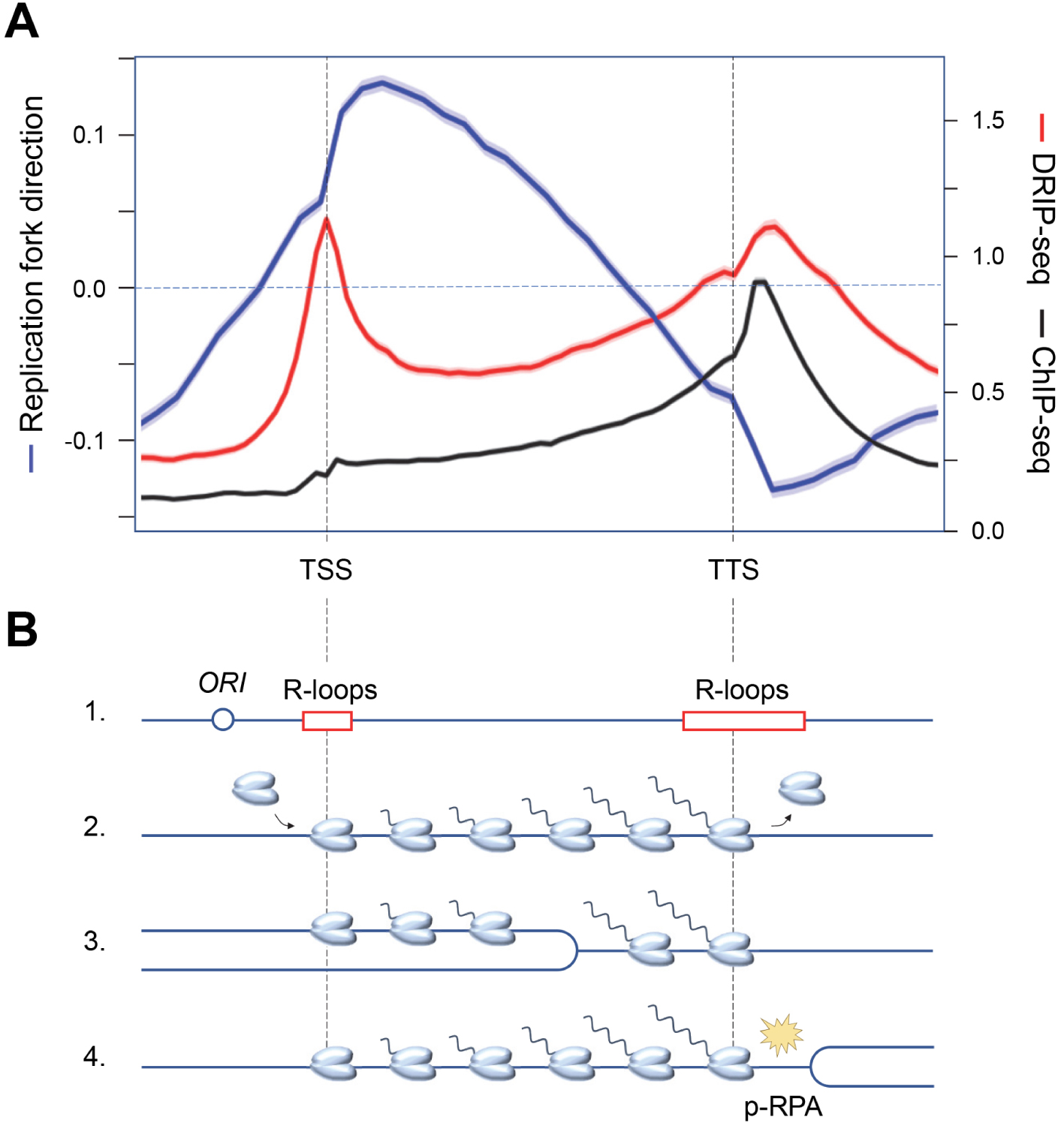
Another important question concerns the impact of R-loops on replication–transcription conflicts. Using methods such as DRIP-seq, which involve immunoprecipitating R-loops with an antibody specifically recognizing DNA:RNA hybrids and sequencing the associated nucleic acids, it has been shown that R-loops are highly abundant and can occupy up to 5% of the human genome [61, 62]. These structures play important physiological roles, such as control of gene expression, recombination of immunoglobulin genes, and mitochondrial DNA replication [39]. The question then arises as to whether all R-loops are inherently difficult to replicate, or whether a particular class of R-loops is particularly toxic to replication forks.
Analysis of the distribution of replication stress markers such as the ATR substrate phospho-RPA32-S33 showed that most R-loops are not associated with replication stress in the human genome [60]. Indeed, cotranscriptional R-loops are mainly located at the transcription initiation (TSS) and termination (TTS) sites, whereas an enrichment of phospho-RPA32-S33 is only detected at TTS (Figure 2A). Since these regions are on average replicated in the opposite direction to the direction of transcription, this means that replication–transcription conflicts occur preferentially at the TTS of highly transcribed genes enriched in R-loops, when the replication and transcription machineries converge (Figure 2B). Importantly, these conflicts do not automatically induce chromosomal breakage. Indeed, these events are only detected in Top1-depleted cells, which fail to manage the topological problems induced by replication and transcription convergence (Figure 3A). Furthermore, the replication stress observed in the absence of Top1 is suppressed by overexpression of RNase H1, an enzyme involved in the degradation of DNA:RNA hybrids, attesting to the fact that R-loops contribute to replication–transcription conflicts [60, 63].
Model of replication–transcription conflict involving the formation of post-replicative RNA:DNA hybrids. (A) Convergence of the replisome and RNA polymerases (RNAPII) at transcription termination sites (TTS) induces accumulation of positive DNA supercoiling, which causes replication fork arrest and activation of the ATR kinase. The release of this topological stress is mediated by Top1. (B) ATR promotes the removal of fork-blocking RNA polymerases. At the same time, the DNA:RNA hybrid is degraded by RNase H to remove the R-loop. (C) In the absence of RNase H, the replicative helicase progresses beyond the position of the R-loop, transferring the DNA:RNA hybrid behind the fork and stops at the next RNAPII. (D) RNA:DNA hybrids located behind the fork interfere with post-replicative mechanisms such as resection of nascent DNA and prevent fork restart.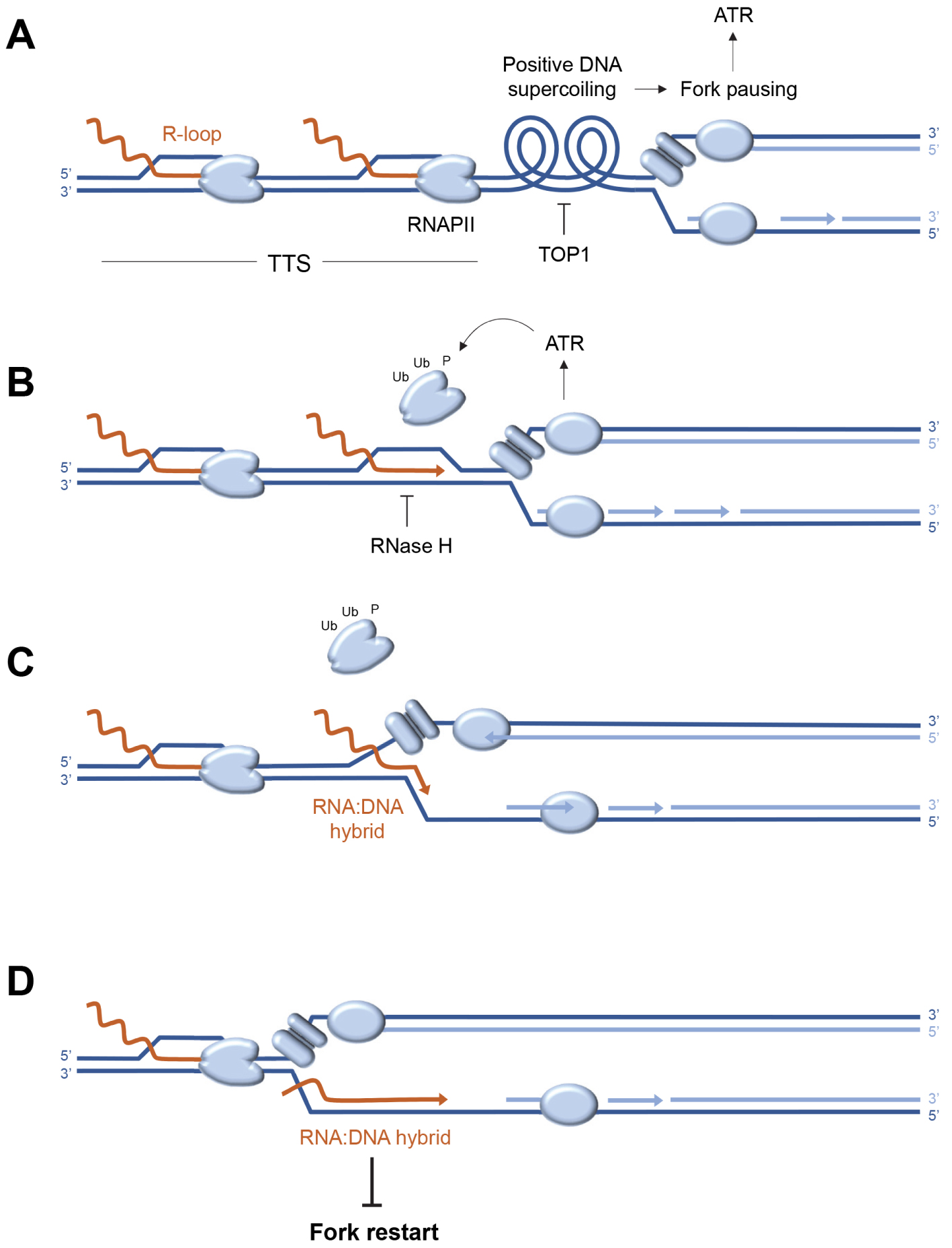
In conclusion, the functional organization of the human genome reduces the genetic instability associated with replication–transcription conflicts by limiting these conflicts to well-defined areas downstream of the genes. However, this regulation cannot operate if the initiation of replication takes place inside the genes, which occurs only very rarely under normal physiological conditions. Interestingly, a new class of replication origins has been identified within genes under oncogenic stress [64]. This intragenic initiation is associated with chromosomal rearrangements in cancers, possibly because it prevents cells from handling replication–transcription conflicts in an optimal manner.
6. R-loops and post-replicative DNA:RNA hybrids
The cell is well equipped to manage conflicts between replication and transcription. On the one hand, the replication fork is able to displace transcription complexes that block its passage (Figure 3B) by mobilizing various factors coordinated by the ATR kinase [65, 66, 67, 68]. On the other hand, the cell has a whole array of enzymes capable of degrading or displacing DNA:RNA hybrids [40]. These include RNases H1 and H2, which specifically degrade the RNA portion of DNA:RNA hybrids [69, 70, 71]. RNase H2 is frequently mutated in a severe interferonopathy called Aicardi–Goutières syndrome [72, 73]. Other key players include helicases such as Senataxin [74, 75] and FANCM [76]. Yet, despite this redundancy of repair mechanisms, some R-loops interfere with replication, following a mechanism that remains to be elucidated.
It is generally accepted that DNA:RNA hybrids are inherently difficult to replicate and block fork progression. However, when replication and transcription converge, the DNA:RNA hybrid is positioned on the strand opposite the replicative helicase (CMG), which should not block its progression (Figure 3C). Recent data derived from an in vitro replication system indicate that DNA:RNA hybrids do not block forks in this configuration, unless the displaced DNA strand is capable of forming a G4-like secondary structure [77]. Alternatively, DNA:RNA hybrids could interfere with post-replicative mechanisms acting behind the forks to promote their restart [78, 79]. This model is supported by unpublished data from our team, showing that in the absence of RNase H2, the resection of nascent DNA strands is inhibited by the persistence of undegraded DNA:RNA hybrids. Since fork resection contributes to fork restart [23, 80], these post-replicative DNA:RNA hybrids could induce replication stress not only by blocking forks, but also by preventing their restart (Figure 3D). This model is in agreement with recent data from the team of Massimo Lopes [81], demonstrating by electron microscopy the presence of DNA:RNA hybrids behind the replication forks.
These recent findings led us to propose a model in which R-loops and transcriptional complexes would not only represent an obstacle to the replication fork, but that the limited progression of replicative helicase at the site of conflict could convert the R-loop into a toxic DNA:RNA hybrid, interfering with fork restart if not rapidly degraded by RNase H2 (Figures 3C,D). This model emphasizes the importance for the cell of coordinated removal of the transcription complex and the R-loop to avoid the formation of a post-replicative DNA:RNA hybrid. It is worth noting that cells could be particularly sensitive to transcription–replication conflicts when replication forks are slowed by HU or oxidative stress [29] and that different mechanisms may operate when fork progression is blocked. Indeed, it has been reported that fork recovery at lesions caused by the Top1 poison camptothecin (CPT) involves the MUS81-dependent cleavage of stalled forks in human cells [82]. In fission yeast, fork restart at CPT lesions and at the RTS1 replication fork barrier depends on the degradation by RNase H of RNA primers on Okazaki fragments [83].
7. Conclusion and perspectives
In conclusion, the data gathered by different groups in the last few years shed new light on the molecular mechanisms at the origin of genetic instability in cancers. It now appears that despite optimal functional organization of the genome and redundancy of repair mechanisms, replication–transcription conflicts represent a major source of genetic instability, especially when oncogenic pathways are deregulated. R-loops contribute significantly to these conflicts, but the molecular mechanisms involved are more complex than initially thought. Indeed, these structures would not only act as replication barriers, but also act as postreplicative RNA:DNA hybrids to interfere with the restart of stalled forks. Such postreplicative RNA:DNA hybrids have recently been imaged by electron microscopy [81], but the mechanism of their formation remains unclear. New technologies such as nanopore sequencing could help determine the relative position of nascent DNA and RNA:DNA hybrids at the single molecule level. Another important question that remains to be elucidated is whether these post-replicative hybrids pre-exist before fork passage, as shown in Figure 3, or whether they are synthesized de novo by RNA polymerases acting behind the stalled fork. From this point of view, it is interesting to note that DNA:RNA hybrids are also detected at DNA double-strand breaks and that these structures interfere with DNA end resection, thus affecting homologous recombination repair mechanisms [84]. Finally, it is now well established that the repair mechanisms of forks and chromosomal breaks generate small DNA fragments that accumulate in the cytoplasm, activate the cGAS-STING pathway, and induce an inflammatory response [85, 86]. A very recent study also shows that DNA:RNA hybrids from R-loops after cleavage by repair enzymes can also activate the cGAS-STING pathway [87]. These data indicate that replication stress generated by replication–transcription conflicts and R-loops induces cellular responses acting beyond the boundaries of the cell. This endogenous stress could thus stimulate the elimination of abnormal cells by the immune system, opening promising prospects for the development of new cancer therapies.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgements
The author thanks the members of the team “Maintenance of genome integrity during replication” for their contribution to the work mentioned in this review. Our work is supported by the Agence Nationale de la Recherche (ANR), the Institut National du Cancer (INCa), the Ligue contre le Cancer (labelled team) and the MSDAvenir Fund.