

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Le syndrome d’Aicardi-Goutières est une encéphalopathie provoquée par des mutations de différents enzymes du métabolisme des acides nucléiques et une production chronique d’interféron de type I [8]. L’étude de certaines protéines à l’origine de cette maladie a montré que la présence anormale d’acides nucléiques dans le cytosol de ces cellules entraîne la production d’interférons. SAMHD1 (Sterile Alpha Motif and HD Domain-containing protein 1) est fréquemment muté dans le syndrome d’Aicardi-Goutières [17], mais son rôle dans l’étiologie de cette maladie était jusqu’à présent inconnu.
Avant de se diviser, chaque cellule doit dupliquer fidèlement son génome afin de le transmettre aux cellules filles. L’intégralité du matériel génétique de chaque cellule est ainsi répliquée par des ADN polymérases pendant la phase S du cycle cellulaire. Cette synthèse est assurée par un complexe macromoléculaire, le réplisome, agissant au niveau de structures appelées fourches de réplication. La progression des fourches est fréquemment ralentie ou bloquée par des obstacles d’origine endogène ou exogène [15]. Ce stress réplicatif est une source majeure d’instabilité génétique chez l’homme [13].
Le stress réplicatif est signalé par ATR et Chk1, les principales kinases du checkpoint de phase S. L’activation de cette voie de surveillance bloque la progression du cycle cellulaire et assure le redémarrage des fourches de réplication. La kinase ATR détecte l’accumulation d’ADN simple-brin recouvert par RPA au niveau des fourches bloqués [6]. On considère généralement que cet ADN simple brin résulte du découplage entre hélicases et polymérases [3], mais des données récentes indiquent qu’il peut aussi être produit par dégradation nucléolytique de l’ADN néosynthétisé. En particulier, il a été montré que la nucléase MRE11 dégrade l’ADN néorépliqué au niveau des fourches bloquées afin de permettre leur redémarrage [11]. Cette dégradation nucléolytique est contrôlée par BRCA2, qui charge la recombinase Rad51 sur l’ADN simple-brin afin de limiter l’activité de MRE11 [18]. La balance entre dégradation et protection des brins naissants est donc un élément déterminant de la physiologie des cellules en réponse au stress réplicatif [5].
La voie interféron est un mécanisme de défense contre les agents pathogènes qui détecte la présence d’acides nucléiques étrangers dans le cytoplasme et active la réponse immunitaire innée. L’ADN cytosolique est ainsi détecté par la protéine cGAS qui, en réaction, synthétise le second messager cGAMP. Cette molécule active à son tour la protéine STING qui déclenche, entre autres, la production d’interférons de type I [4]. D’autre part, il a été montré que des fragments d’ADN issus du métabolisme de l’ADN génomique (réparation, rétrotransposition…) peuvent diffuser dans le cytoplasme et activer la voie interféron [1, 19].
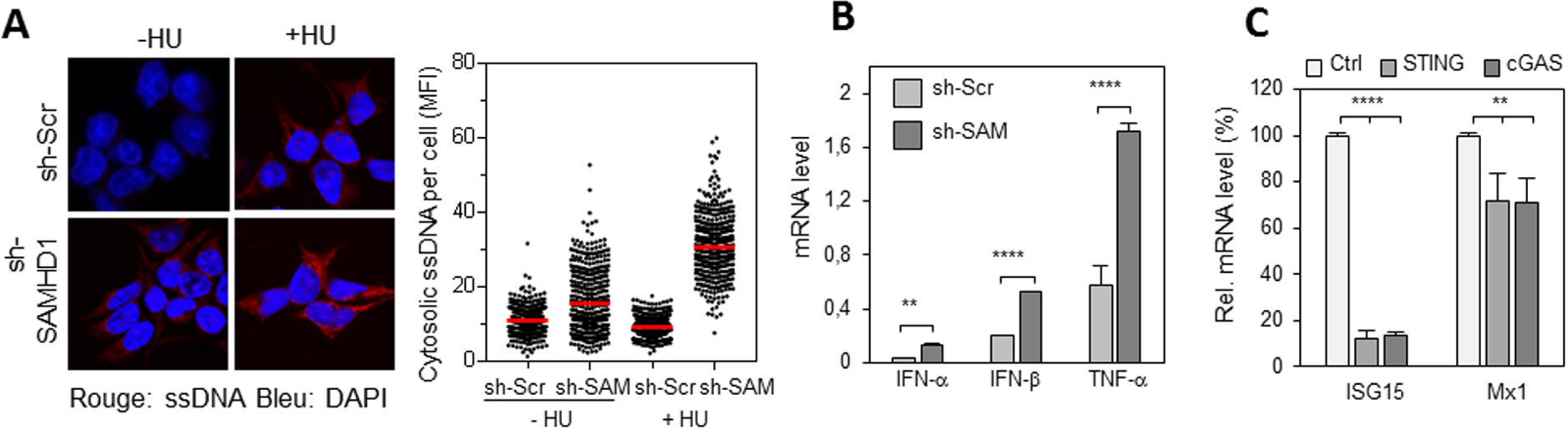
SAMHD1 prévient l’accumulation d’ADN cytosolique et la production d’interférons de type 1 induite par la voie cGAS/STING. (A) Accumulation d’ADN simple brin (ssDNA) dans les cellules déplétées pour SAMHD1. Les cellules HEK293T sh-Scr et sh-SAMHD1 sont traitées ou non avec de l’HU pendant 2 heures, puis fixées et perméabilisées. L’ADN simple brin est détecté avec un anticorps spécifique et un anticorps secondaire (rouge). Le noyau est marqué au DAPI (bleu). L’intensité de fluorescence moyenne de l’anti ADN est quantifiée avec CellProfiler. (B) La déplétion de SAMHD1 entraine une surexpression de gènes pro-inflammatoires. Expression d’IFN-α, β et TNF-α dans des cellules exprimant un shRNA contre SAMHD1 ou un shRNA contrôle et exposées à l’HU. (C) La surexpression des gènes pro-inflammatoire dépend de la voie cGAS/STING. Expression des gènes pro-inflammatoires ISG15 et Mx1 dans des cellules KO pour STING, KO pour cGAS ou contrôle et transfectées avec un siRNA contre SAMHD1. Masquer
SAMHD1 prévient l’accumulation d’ADN cytosolique et la production d’interférons de type 1 induite par la voie cGAS/STING. (A) Accumulation d’ADN simple brin (ssDNA) dans les cellules déplétées pour SAMHD1. Les cellules HEK293T sh-Scr et sh-SAMHD1 sont traitées ou non avec ... Lire la suite
En situation de stress réplicatif, la réponse interféron pourrait ainsi signaler la présence anormale de produits du métabolisme des fourches de réplication, ce qui en ferait une extension du checkpoint de phase S. L’existence d’un tel lien entre stress réplicatif et inflammation reste cependant à démontrer. Une manifestation pathologique de ce processus est le syndrome d’Aicardi-Goutières, une maladie rare caractérisée par une inflammation chronique générant des problèmes neurodégénératifs et développementaux. Dans le cadre de cette encéphalopathie, il a été suggéré que la réplication de l’ADN pouvait générer des fragments d’ADN cytosoliques [20].
Ce facteur de restriction du virus HIV-1 est un bon candidat pour connecter la réponse au stress réplicatif à la voie interféron. En effet, SAMHD1 est fréquemment muté dans le syndrome d’Aicardi-Goutières, ainsi que dans différents cancers, dont la leucémie lymphoïde chronique (CLL) et le cancer du côlon [7, 16]. Dans le cadre d’une étude sur la CLL, notre équipe a montré que SAMHD1 est recruté aux sites de dommages de l’ADN [7]. Depuis, une étude a démontré son implication directe dans la réparation des cassures de l’ADN [9].
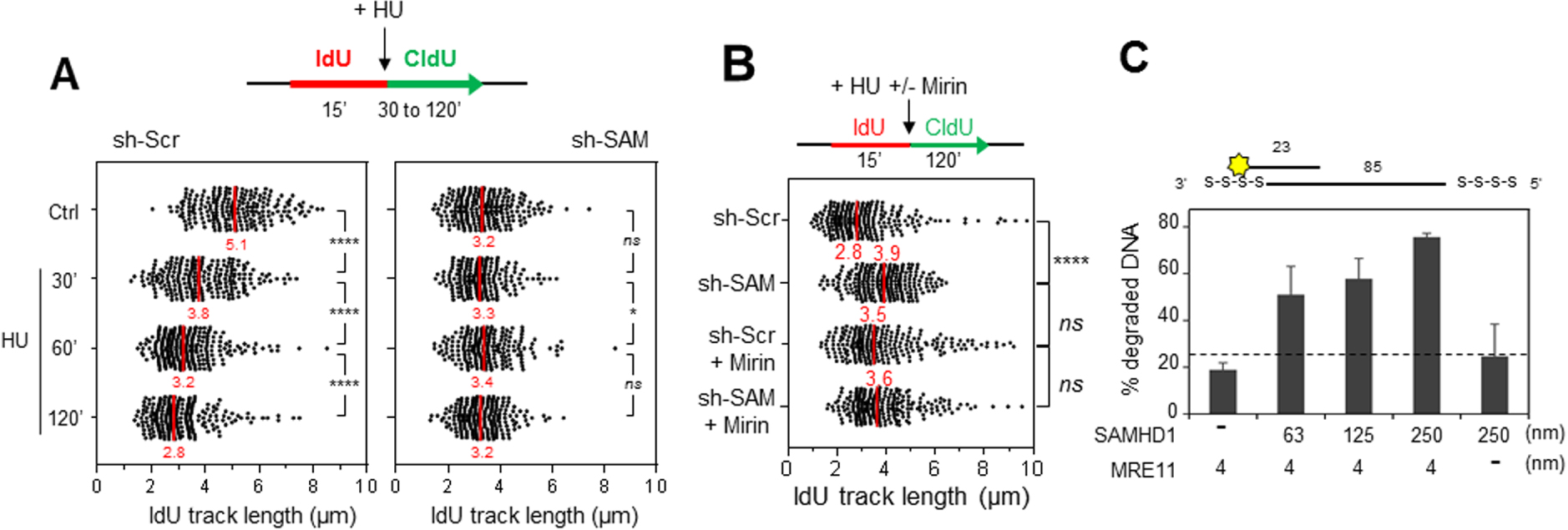
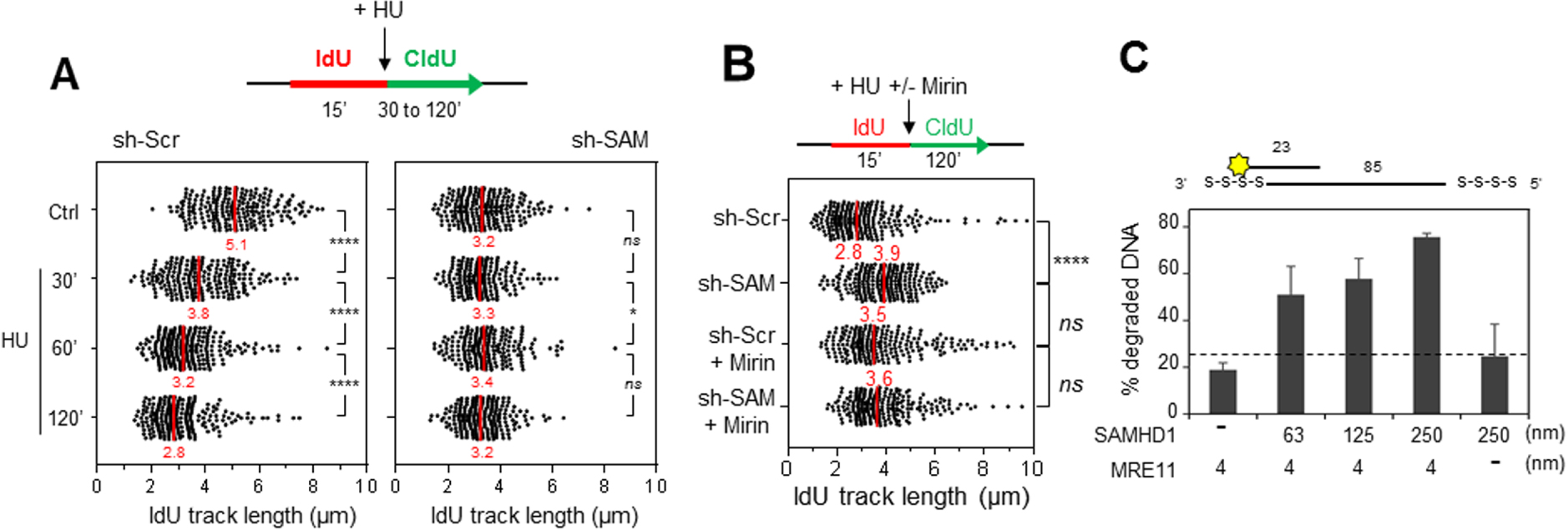
SAMHD1 est nécessaire pour la resection des fourches bloquées par MRE11. (A) La résection des fourches bloquées par l’HU est compromise en absence de SAMHD1. Les cellules ont été marquées par deux pulses successifs d’IdU et de CldU, (analogues de la thymidine) avec ajout d’HU pendant le pulse de CldU. La distribution de la taille des tracks d’IdU est indiquée. Le fragment d’IdU synthétisé avant l’addition d’HU est protégé de la résection en absence de SAMHD1. Par contre, il est dégradé dans les cellules contrôle. (B) SAMHD1 est épistatique de MRE11 pour la résection des fourches de réplication. Les cellules ont été marquées comme dans (A), avec un pré-traitement avec de la mirin (inhibiteur de MRE11). La distribution de la taille des tracks d’IdU est indiquée. Le track d’IdU synthétisé avant l’addition d’HU est protégé de la résection en absence de SAMHD1 et lorsque MRE11 est inhibé dans les cellules contrôle et dans les cellules déficientes pour SAMHD1. (C) SAMHD1 stimule l’activité nucléase de MRE11 in vitro. SAMHD1 et MRE11 ont été exprimées dans des cellules d’insecte (Sf9), purifiées et pré-incubées ensemble. Puis, les enzymes seules ou mélangées sont incubées avec un substrat d’ADN fluorescent. L’activité nucléase de SAMHD1, MRE11 et de la combinaison des deux enzymes est évaluée par quantification de l’ADN dégradé dans chaque réaction. MRE11 et SAMHD1 seuls ont une activité nucléase minimale, par contre, l’activité exonucléase de MRE11 augmente avec la quantité de SAMHD1 utilisée. Masquer
SAMHD1 est nécessaire pour la resection des fourches bloquées par MRE11. (A) La résection des fourches bloquées par l’HU est compromise en absence de SAMHD1. Les cellules ont été marquées par deux pulses successifs d’IdU et de CldU, (analogues de ... Lire la suite
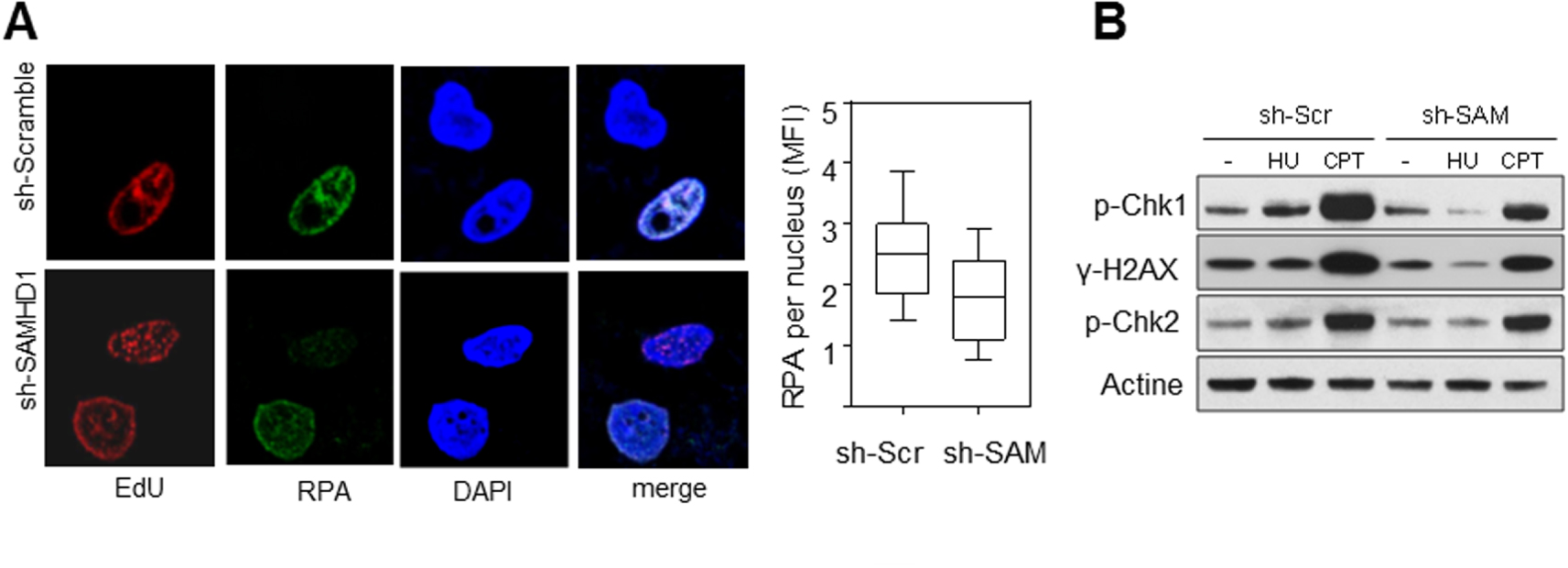
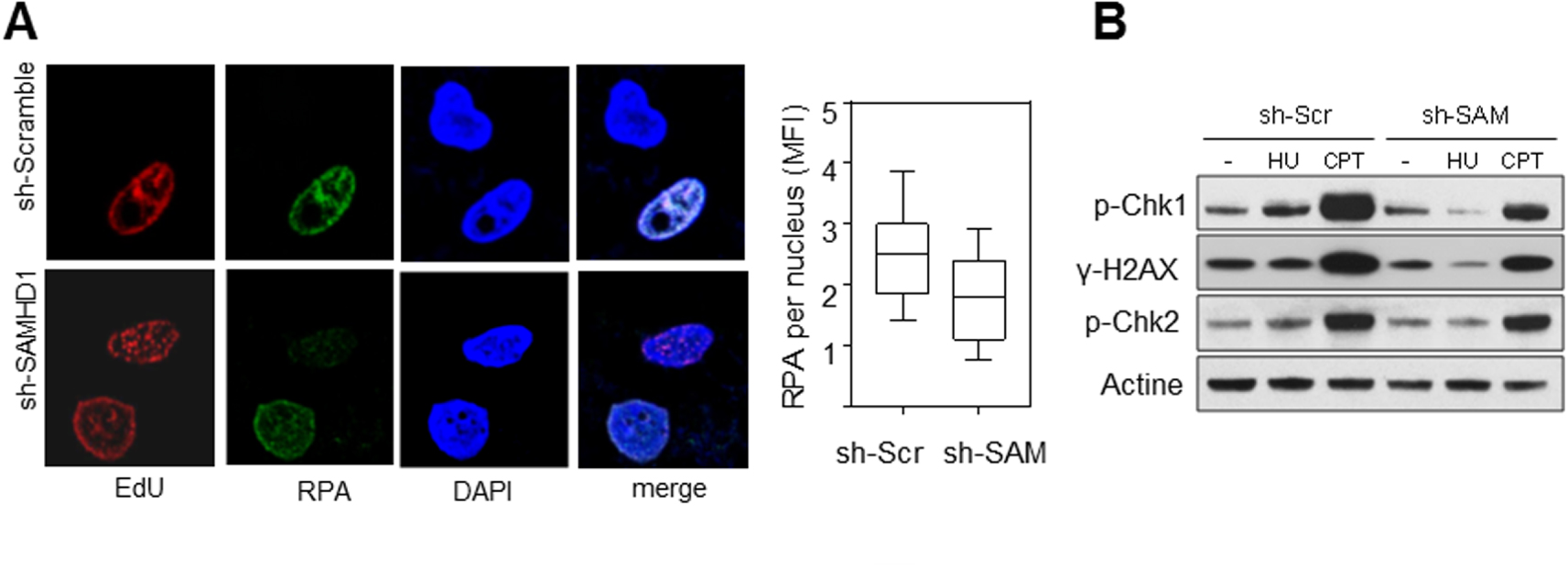
SAMHD1 est nécessaire pour l’activation du checkpoint de réplication. (A) L’accumulation de RPA sur la chromatine en réponse au stress réplicatif induit par l’HU est compromise en absence de SAMHD1. Les foyers de réplication sont marqués à l’EdU (rouge) et RPA est détecté avec un anti-RPA (vert). L’ADN est coloré au DAPI (bleu). L’intensité moyenne de fluorescence (MFI) de RPA est quantifiée avec le logiciel CellProfiler. (B) SAMHD1 est nécessaire pour l’activation du checkpoint de réplication. L’analyse par western blot montre que la phosphorylation de Chk1 et H2AX induite par l’HU et la camptothécine (CPT) est fortement réduite en absence de SAMHD1. Par contre, l’activation de Chk2 spécifique de la voie ATM n’est pas affectée. Masquer
SAMHD1 est nécessaire pour l’activation du checkpoint de réplication. (A) L’accumulation de RPA sur la chromatine en réponse au stress réplicatif induit par l’HU est compromise en absence de SAMHD1. Les foyers de réplication sont marqués à l’EdU (rouge) et ... Lire la suite
SAMHD1 possède une activité dNTPase impliquée dans la régulation des pools de nucléotides [10], ainsi qu’une activité 3′–5′ exonucléase qui est, encore aujourd’hui, controversée [2]. Enfin, une étude récente a montré que la voie de détection de l’ADN cytosolique cGAS/STING est anormalement activée dans les cellules déficientes pour SAMHD1 [14]. Ces résultats suggèrent que SAMHD1 serait important pour limiter la génération de cet ADN cytosolique.
Les données mentionnées ci-dessus indiquent que SAMHD1 est impliqué dans la réponse aux dommages à l’ADN et dans le contrôle de la voie interféron. Si SAMHD1 agit au niveau des fourches arrêtées, on peut supposer que dans des cellules déplétées pour SAMHD1, un métabolisme anormal des fourches pourrait entrainer la génération d’ADN cytosolique et la production d’interférons de type I.
2. Présentation des résultats
2.1. SAMHD1 prévient l’accumulation d’ADN cytosolique et la production d’interférons de type I
Il a tout d’abord été vérifié que les cellules déficientes pour SAMHD1 activent la réponse inflammatoire de manière anormale. Pour cela, la production de facteurs pro-inflammatoires et la présence d’ADN cytosolique dans des cellules HEK293T exprimant un shRNA dirigé contre SAMHD1 (sh-SAM) ou un shRNA contrôle (sh-Scr), ont été étudiées.
La présence d’ADN simple brin dans le cytosol de cellules déplétées pour SAMHD1 a été détectée par immunofluorescence. La quantité d’ADN cytosolique augmente après traitement avec de l’HydroxyuréLe (HU), un inhibiteur de la réplication, (Figure 1A), ce qui suggère qu’il est produit par le stress réplicatif. Une production d’interférons de type I (IFN-α, IFN-β) et de TNF-α plus importante dans les cellules déplétées pour SAMHD1 que dans les cellules contrôle, surtout en présence d’HU (Figure 1B) a été observée. Cette réponse inflammatoire est dépendante de la voie de détection de l’ADN cytosolique cGAS/STING (Figure 1C).
2.2. SAMHD1 est nécessaire pour la résection des fourches bloquées
En présence d’HU, l’ADN néorépliqué est rapidement dégradé d’une manière dépendante de l’activité 3′–5′ exonuclease de MRE11 [11, 18]. En absence de SAMHD1, il a été constaté que cette activité de résection est inhibée (Figure 2A), ce qui suggère que SAMHD1 est nécessaire pour la dégradation nucléolytique de l’ADN néorépliqué. Ce résultat a été confirmé avec d’autres techniques comme celle récemment décrite par l’équipe d’André Nussenzweig [5], ainsi qu’avec différentes lignées cellulaires et plusieurs drogues (HU, aphidicoline). Nous pouvons donc conclure que SAMHD1 joue un rôle clé dans la résection des fourches bloquées.
Il est important de noter que cette activité est régulée par phosphorylation par les CDKs de phase S et qu’elle est indépendante de l’activité dNTPase de SAMHD1. De plus, il a été montré que SAMHD1 est épistatique de MRE11 pour cette fonction (Figure 2B). Nos collaborateurs Vincenzo Costanzo et Lumir Krejci ont confirmé ce résultat en démontrant que SAMHD1 stimule l’activité exonucléase de MRE11 in vitro (Figure 2C).
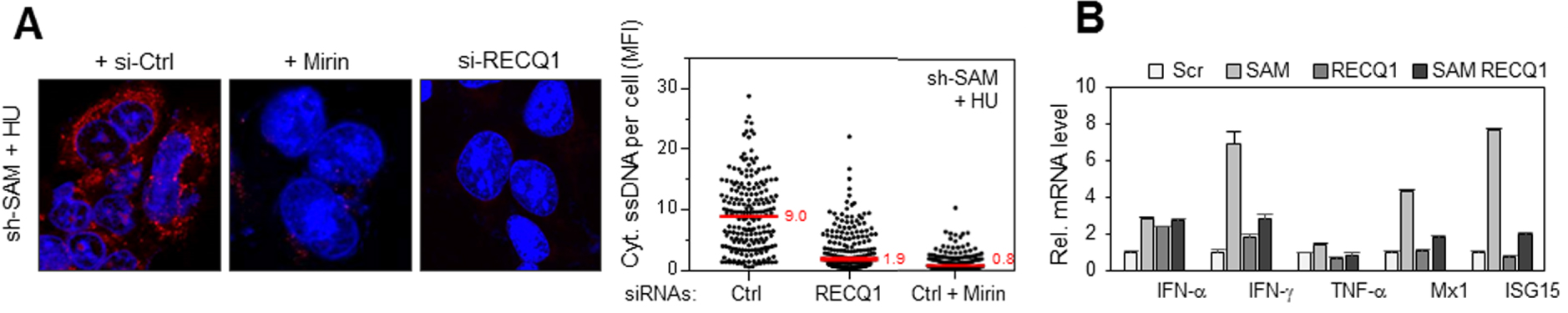
RecQ1 et MRE11 déterminent la réponse inflammatoire induite par la déplétion de SAMHD1. (A) RecQ1 et MRE11 génèrent les fragments d’ADN exportés dans le cytosol en l’absence de SAMHD1. Accumulation d’ADN simple brin dans les cellules déplétées pour SAMHD1 co-transfectées avec un siRNA contrôle (si-Ctrl) ou un siRNA dirigé contre RecQ1 (si-RecQ1) ou traitées avec de la mirin, en présence d’HU. La détection et la quantification de l’ADN simple brin sont réalisées comme dans la Figure 1A. L’intensité du signal d’ADN cytosolique diminue très fortement lorsque MRE11 est inhibé ou RecQ1 déplété dans les cellules déficientes pour SAMHD1. (B) La déplétion de RecQ1 empêche la synthèse de gènes pro-inflammatoires induite par l’absence de SAMHD1. Expression d’IFN-α, 𝛾, de TNF-α et des gènes induits par les interférons Mx1 et ISG15 dans des cellules exprimant un siRNA contre SAMHD1 ou un siRNA contrôle couplé ou non à un siRNA contre RecQ1 et exposées à l’HU. Masquer
RecQ1 et MRE11 déterminent la réponse inflammatoire induite par la déplétion de SAMHD1. (A) RecQ1 et MRE11 génèrent les fragments d’ADN exportés dans le cytosol en l’absence de SAMHD1. Accumulation d’ADN simple brin dans les cellules déplétées pour SAMHD1 co-transfectées ... Lire la suite
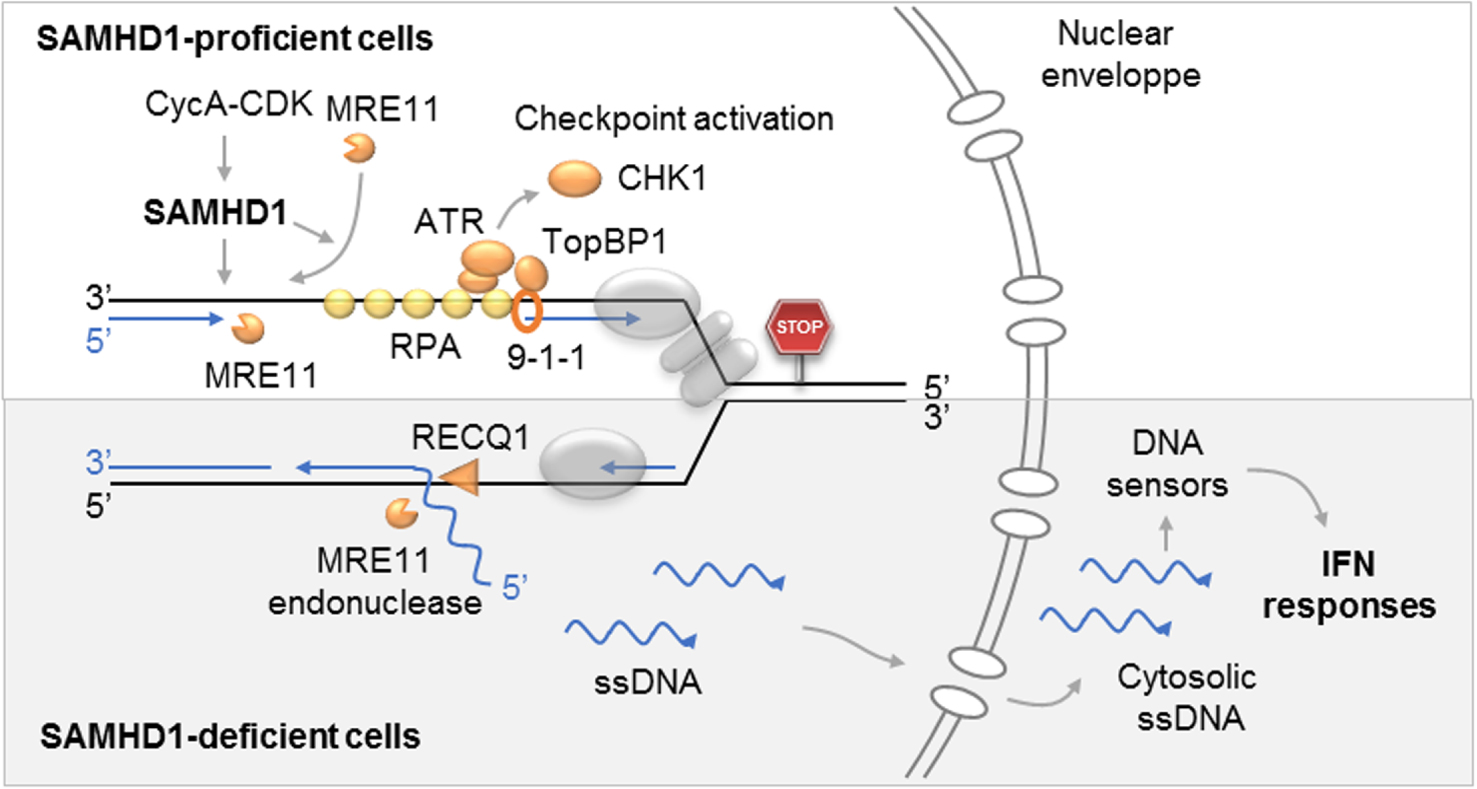
Rôle de SAMHD1 aux fourches de réplications bloquées. Haut : la phosphorylation de SAMHD1 par le complexe Cycline A-CDK permet la résection des fourches bloquées par MRE11 et l’activation du checkpoint de réplication en exposant l’ADN simple brin qui est couvert par RPA. Dans les cellules déficientes pour SAMHD1 (bas), l’ADN nouvellement synthétisé est déplacé par RecQ1 et clivé par MRE11. Les fragments d’ADN ainsi générés sont exportés dans le cytosol où ils activent la synthèse d’interférons de type I et d’autres cytokines pro-inflammatoires. Masquer
Rôle de SAMHD1 aux fourches de réplications bloquées. Haut : la phosphorylation de SAMHD1 par le complexe Cycline A-CDK permet la résection des fourches bloquées par MRE11 et l’activation du checkpoint de réplication en exposant l’ADN simple brin qui est ... Lire la suite
2.3. SAMHD1 est nécessaire pour l’activation du checkpoint de réplication
La dégradation des brins naissants derrière les fourches bloquées permet d’exposer de l’ADN simple brin et de RPA (Replication Protein A). RPA est un complexe protéique essentiel pour protéger l’ADN simple brin et pour signaler les problèmes de réplication au checkpoint de phase S. La présence de RPA à la chromatine de cellules en phase S (déterminé par un marquage EdU) a été analysée par immunofluorescence. On observe un défaut de recrutement de RPA après traitement avec l’HU dans les cellules déplétées pour SAMHD1 par rapport aux cellules contrôle (Figure 3A).
Un défaut d’activation du checkpoint de phase S dans les cellules déplétées pour SAMHD1 a également été observé (Figure 3B).
2.4. MRE11 et RecQ1 sont responsable de la réponse pro-inflammatoire induite par l’absence de SAMHD1
La dégradation de l’ADN aux fourches de réplication est réduite dans les cellules déplétées en SAMHD1. Pourtant, de l’ADN simple brin est exporté vers le cytosol, il est donc probablement déplacé puis clivé par une ou des protéines qui fonctionneraient avec SAMHD1 ou la remplaceraient en son absence. Nous avons identifié ces protéines en analysant la quantité d’ADN cytosolique dans les cellules déficientes pour SAMHD1 et co-déplétées avec d’autres facteurs ayant un rôle avéré dans le processing des fourches de réplication. Les enzymes MRE11 et RecQ1 ont été identifiées comme étant les facteurs principaux de la réponse pro-inflammatoire en l’absence de SAMHD1. En effet, la quantité d’ADN cytosolique diminue de manière drastique lorsque RecQ1 est déplété ou MRE11 inhibé dans les cellules déficientes pour SAMHD1 (Figure 4A). Enfin, la transcription de gènes pro-inflammatoires est également fortement réduite lorsque RecQ1 est déplété dans les cellules déficientes pour SAMHD1 (Figure 4B).
En résumé, ces travaux ont montré que SAMHD1 est recrutée au niveau des fourches de réplication bloquées, où elle stimule l’activité exonucléase de MRE11 pour activer le checkpoint de réplication. En l’absence de SAMHD1, des petits fragments d’ADN sont libérés de la fourche et s’accumulent dans le cytoplasme, où ils sont détectés par les mécanismes d’immunité innée et induisent la production d’interférons de type I (Figure 5).
3. Perspectives
Nos travaux mettent en évidence que la réponse au stress réplicatif n’est pas restreinte à l’intérieur du noyau mais s’étend dans le cytosol. Sachant que le stress réplicatif, une caractéristique majeure du cancer, peut être augmenté par la chimiothérapie, nous pouvons maintenant envisager de nouvelles stratégies thérapeutiques combinant chimiothérapie et immunothérapie.
Nos résultats permettent également d’envisager de nouvelles stratégies thérapeutiques pour aider les patients atteints d’AGS, qui ne disposent actuellement d’aucun traitement pour limiter les causes de leur inflammation chronique. En effet, nous montrons que MRE11 et RecQ1 sont responsables de la production des fragments d’ADN qui déclenchent la réponse inflammatoire dans les cellules déficientes pour SAMHD1. Dès lors, nous pouvons imaginer que bloquer l’activité de ces enzymes pourrait diminuer la production des fragments d’ADN et, in fine, l’activation de l’immunité innée dans ces cellules.
English version
1. Introduction
Aicardi-Goutières syndrome is an encephalopathy caused by mutations in different enzymes of nucleic acid metabolism and chronic production of type I interferon [8]. The study of certain proteins that cause this disease has shown that the abnormal presence of nucleic acids in the cytosol of these cells leads to the production of interferons. SAMHD1 (Sterile Alpha Motif and HD Domain-containing protein 1) is frequently mutated in Aicardi-Goutières syndrome [17], but its role in the etiology of this disease was previously unknown.
Before dividing, each cell must faithfully duplicate its genome in order to transmit it to the daughter cells. The entire genetic material of each cell is thus replicated by DNA polymerases during the S phase of the cell cycle. This synthesis is ensured by a macromolecular complex, the replisome, acting at the level of structures called replication forks. The progression of the forks is frequently slowed down or blocked by obstacles of endogenous or exogenous origin [15]. This replication stress is a major source of genetic instability in humans [13].
Replicative stress is reported by ATR and Chk1, the main kinases of the phase S checkpoint. Activation of this monitoring pathway blocks cell cycle progression and ensures the restarting of replication forks. The ATR kinase detects the accumulation of RPA-coated single-stranded DNA at the blocked forks [6]. This single-stranded DNA is generally considered to result from decoupling between helicases and polymerases [3], but recent data indicate that it can also be produced by nucleolytic degradation of neosynthetisized DNA. In particular, the MRE11 nuclease has been shown to degrade neosynthetisized DNA at blocked forks to allow them to restart [11]. This nucleolytic degradation is controlled by BRCA2, which loads Rad51 recombinase on single-stranded DNA to limit the activity of MRE11 [18] . The balance between degradation and protection of the nascent strands is therefore a key determinant of cell physiology in response to replicative stress [5].
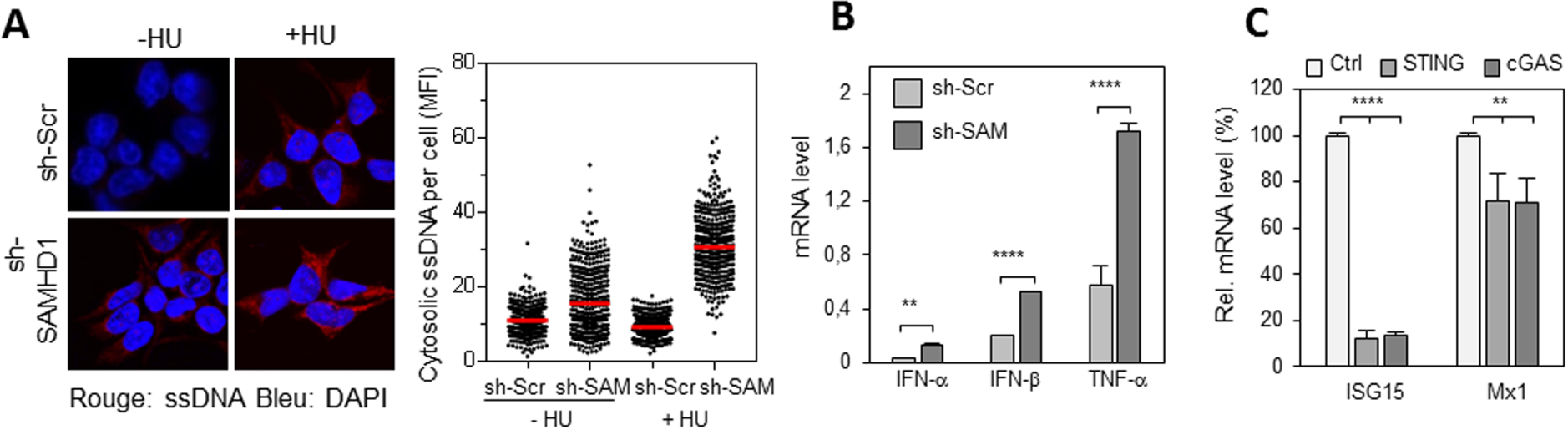
SAMHD1 prevents cGAS/STING-induced cytosolic DNA accumulation and type 1 interferon production. (A) Accumulation of single-stranded DNA (ssDNA) in cells depleted for SAMHD1. HEK293T sh-Scr and sh-SAMHD1 cells are treated or untreated with HU for 2 hours and then fixed and permeabilized. Single-stranded DNA is detected with a specific antibody and a secondary (red) antibody. The nucleus is labelled with DAPI (blue). The average fluorescence intensity of the DNA signal is quantified with CellProfiler. (B) Depletion of SAMHD1 leads to overexpression of pro-inflammatory genes. Expression of IFN-α, β and TNF-α in cells expressing shRNA against SAMHD1 or shRNA control and exposed to HU. (C) Overexpression of pro-inflammatory genes is dependent on the cGAS/STING pathway. Expression of ISG15 and Mx1 pro-inflammatory genes in KO for STING, KO for cGAS or control cells and transfected with siRNA against SAMHD1. Masquer
SAMHD1 prevents cGAS/STING-induced cytosolic DNA accumulation and type 1 interferon production. (A) Accumulation of single-stranded DNA (ssDNA) in cells depleted for SAMHD1. HEK293T sh-Scr and sh-SAMHD1 cells are treated or untreated with HU for 2 hours and then fixed and ... Lire la suite
The interferon pathway is a defence mechanism against pathogens that detects the presence of foreign nucleic acids in the cytoplasm and activates the innate immune response. Cytosolic DNA is detected by the cGAS protein which, in response, synthesizes the second messenger cGAMP. This molecule in turn activates the STING protein which, among other things, triggers the production of type I interferons [4]. It has also been shown that DNA fragments resulting from genomic DNA metabolism (repair, retrotransposition, etc.) can diffuse into the cytoplasm and activate the interferon pathway [1, 19].
In a situation of replicative stress, the interferon response could thus signal the abnormal presence of products of the replication fork metabolism, making it an extension of the phase S checkpoint. However, the existence of such a link between replicative stress and inflammation remains to be demonstrated. A pathological manifestation of this process is the Aicardi-Goutières syndrome, a rare disease characterized by chronic inflammation leading to neurodegenerative and developmental problems. In this encephalopathy, it has been suggested that DNA replication can generate cytosolic DNA fragments [20].
This restriction factor of the HIV-1 virus is a good candidate to connect the replicative stress response to the interferon pathway. Indeed, SAMHD1 is frequently mutated in Aicardi-Goutières syndrome, as well as in various cancers, including chronic lymphocytic leukemia (CLL) and colon cancer [7, 16]. In a study of CLL, our team has shown that SAMHD1 is recruited at sites of DNA damage [7]. Since then, a study has demonstrated its direct involvement in the repair of DNA breaks [9].
SAMHD1 is required for the resection of forks blocked by MRE11. (A) The resection of HU-blocked forks is compromised in the absence of SAMHD1. The cells were marked by two successive pulses of IdU and CldU, (thymidine analogues) with addition of HU during the CldU pulse. The size distribution of the IdU tracks is shown. The IdU fragment synthesized before the addition of HU is protected from resection in the absence of SAMHD1. On the other hand, it is degraded in the control cells. (B) SAMHD1 is epistatic of MRE11 for the resection of the replication forks. The cells were labelled as in (A), with a pre-treatment with mirin (MRE11 inhibitor). The size distribution of the IdU tracks is shown. The IdU track synthesized before the addition of HU is protected from resection in the absence of SAMHD1 and when MRE11 is inhibited in control cells and in SAMHD1 deficient cells. (C) SAMHD1 stimulates the nuclease activity of MRE11 in vitro. SAMHD1 and MRE11 have been expressed in insect cells (Sf9), purified and pre-incubated together. The enzymes alone or mixed are then incubated with a fluorescent DNA substrate. The nuclease activity of SAMHD1, MRE11 and the combination of the two enzymes is evaluated by quantifying the degraded DNA in each reaction. MRE11 and SAMHD1 alone have minimal nuclease activity, whereas the exonuclease activity of MRE11 increases with the amount of SAMHD1 used. Masquer
SAMHD1 is required for the resection of forks blocked by MRE11. (A) The resection of HU-blocked forks is compromised in the absence of SAMHD1. The cells were marked by two successive pulses of IdU and CldU, (thymidine analogues) with addition ... Lire la suite
SAMHD1 is required to activate the replication checkpoint. (A) Accumulation of RPAs on chromatin in response to HU-induced replicative stress is compromised in the absence of SAMHD1. Replication foci are marked with EdU (red) and RPA is detected with anti-RPA (green). DNA is stained with DAPI (blue). The Mean Fluorescence Intensity (MFI) of RPA is quantified with CellProfiler software. (B) SAMHD1 is required to activate the replication checkpoint. Western blot analysis shows that the phosphorylation of Chk1 and H2AX induced by HU and camptothecin (CPT) is strongly reduced in the absence of SAMHD1. On the other hand, the activation of Chk2 specific to the ATM pathway is not affected. Masquer
SAMHD1 is required to activate the replication checkpoint. (A) Accumulation of RPAs on chromatin in response to HU-induced replicative stress is compromised in the absence of SAMHD1. Replication foci are marked with EdU (red) and RPA is detected with anti-RPA ... Lire la suite
SAMHD1 has a dNTPase activity involved in the regulation of nucleotide pools [10], as well as a 3′–5′ exonuclease activity that is still controversial [2]. Finally, a recent study has shown that the cGAS/STING cytosolic DNA detection pathway is abnormally activated in SAMHD1 deficient cells [14]. These results suggest that SAMHD1 would be important in limiting the generation of this cytosolic DNA.
The above data indicate that SAMHD1 is involved in the response to DNA damage and in the control of the interferon pathway. If SAMHD1 acts at the level of arrested forks, it can be assumed that in cells depleted for SAMHD1, abnormal metabolism of the forks could lead to cytosolic DNA generation and production of type I interferons.
2. Presentation of results
2.1. SAMHD1 prevents the accumulation of cytosolic DNA and the production of type I interferons
It was first verified that cells deficient in SAMHD1 activate the inflammatory response. To this end, the production of pro-inflammatory factors and the presence of cytosolic DNA in HEK293T cells expressing a shRNA directed against SAMHD1 (sh-SAM) or a shRNA control (sh-Scr) were studied.
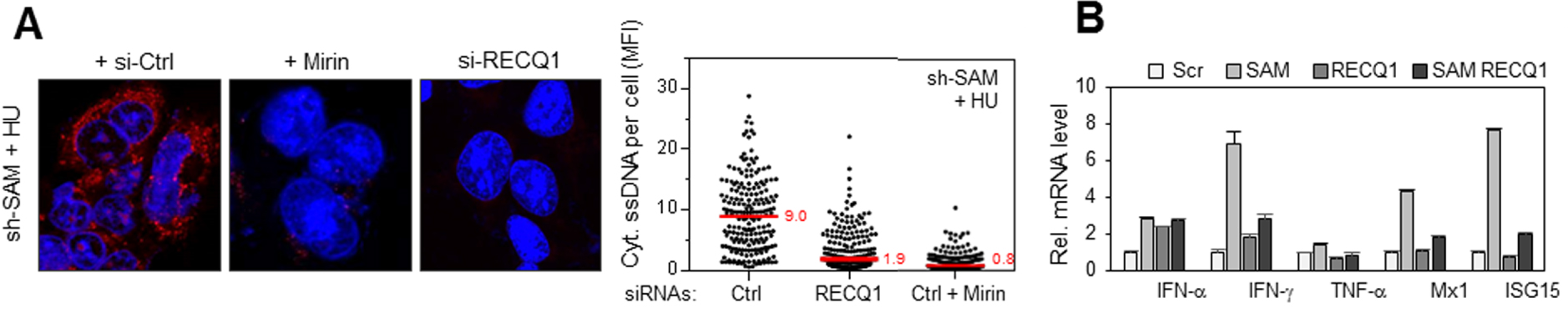
RecQ1 and MRE11 determine the inflammatory response induced by SAMHD1 depletion. (A) RecQ1 and MRE11 generate the DNA fragments exported in the cytosol in the absence of SAMHD1. Accumulation of single-stranded DNA in SAMHD1 depleted cells co-transfected with a siRNA control (si-Ctrl) or a siRNA directed against RecQ1 (si-RecQ1) or treated with mirin, in the presence of HU. Detection and quantification of single-stranded DNA is performed as in Figure 1A. The intensity of the cytosolic DNA signal decreases very strongly when MRE11 is inhibited or RecQ1 is depleted in cells deficient for SAMHD1. (B) Depletion of RecQ1 prevents pro-inflammatory gene synthesis induced by the absence of SAMHD1. Expression of IFN-α, 𝛾, TNF-α and genes induced by interferons Mx1 and ISG15 in cells expressing a siRNA against SAMHD1 or a control siRNA coupled or not coupled to a siRNA against RecQ1 and exposed to HU. Masquer
RecQ1 and MRE11 determine the inflammatory response induced by SAMHD1 depletion. (A) RecQ1 and MRE11 generate the DNA fragments exported in the cytosol in the absence of SAMHD1. Accumulation of single-stranded DNA in SAMHD1 depleted cells co-transfected with a siRNA control ... Lire la suite
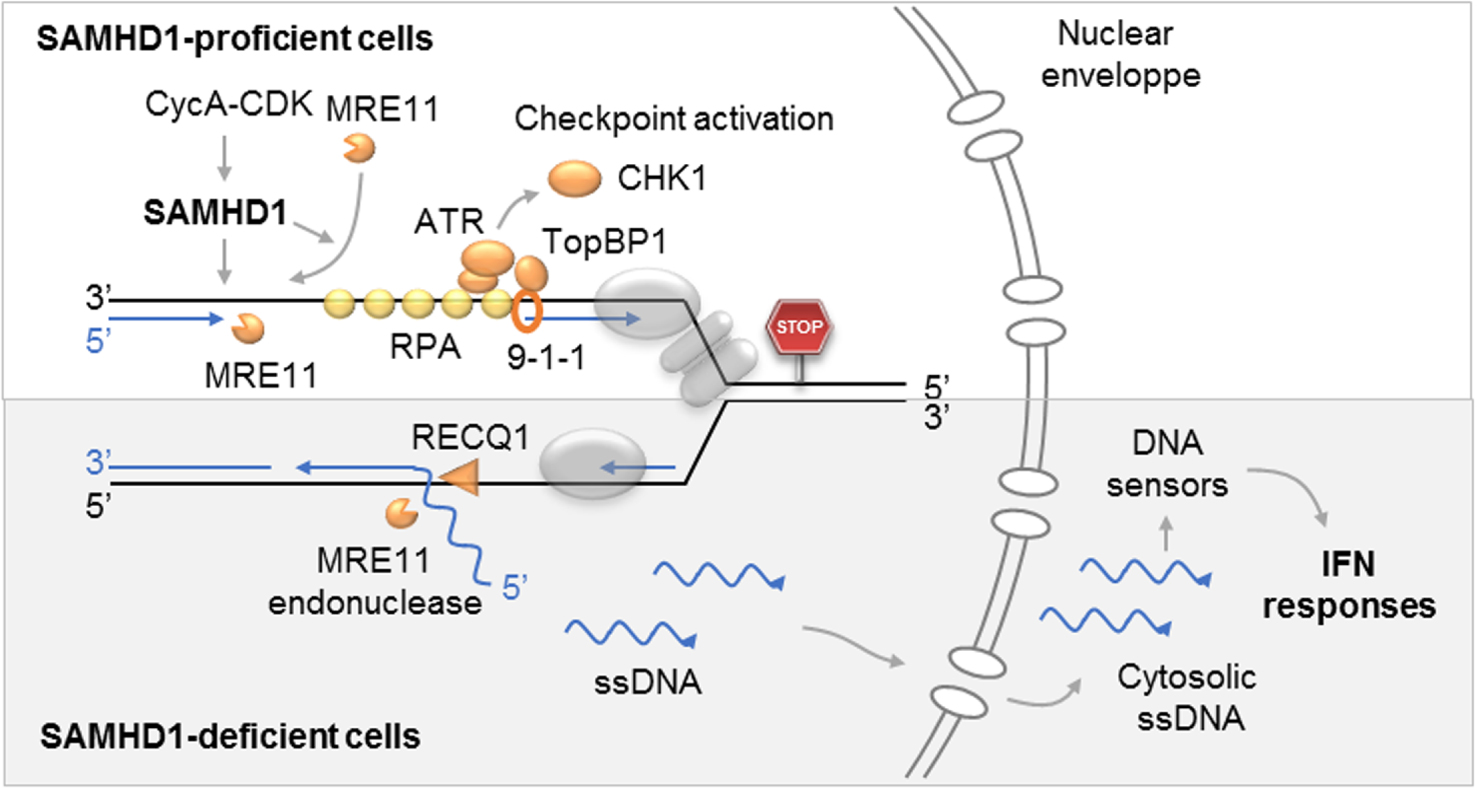
Role of SAMHD1 at blocked replication forks. Top: Phosphorylation of SAMHD1 by Cyclin A-CDK complex allows resection of forks blocked by MRE11 and activation of the replication checkpoint by exposing the single stranded DNA which is covered by RPA. In cells deficient for SAMHD1 (bottom), newly synthesized DNA is displaced by RecQ1 and cleaved by MRE11. The DNA fragments thus generated are exported to the cytosol where they activate the synthesis of type I interferons and other pro-inflammatory cytokines.
Single-stranded DNA in the cytosol of cells depleted for SAMHD1 was detected by immunofluorescence. The amount of cytosolic DNA increases after treatment with Hydroxyurea (HU), a replication inhibitor, (Figure 1A), suggesting that it is produced by replicative stress. A higher production of type I interferons (IFN-α, IFN-β) and TNF-α was observed in SAMHD1 depleted cells than in control cells, especially in the presence of HU (Figure 1B). This inflammatory response is dependent on the cGAS/STING cytosolic DNA detection pathway (Figure 1C).
2.2. SAMHD1 is required for the resection of locked forks
In the presence of HU, the neoreplicated DNA is rapidly degraded by the 3′–5′ exonuclease activity of MRE11 [11, 18]. In the absence of SAMHD1, this resection activity has been found to be inhibited (Figure 2A), suggesting that SAMHD1 is required for the nucleolytic degradation of neo-replicated DNA. This result has been confirmed with other techniques such as the one recently described by André Nussenzweig’s team [5], as well as with different cell lines and several drugs (HU, aphidicolin). We can therefore conclude that SAMHD1 plays a key role in the resection of blocked forks.
It is important to note that this activity is regulated by phosphorylation by S-phase CDKs and is independent of the dNTPase activity of SAMHD1. Furthermore, SAMHD1 has been shown to be epistatic of MRE11 for this function (Figure 2B). Our collaborators Vincenzo Costanzo and Lumir Krejci confirmed this result by demonstrating that SAMHD1 stimulates the exonuclease activity of MRE11 in vitro (Figure 2C).
2.3. SAMHD1 is required to activate the replication checkpoint
The degradation of the nascent strands behind the blocked forks exposes single-stranded DNA and RPA (Replication Protein A). RPA is an essential protein complex to protect single-stranded DNA and to signal replication problems at the S-phase checkpoint. The presence of RPA in the chromatin of S-phase cells (determined by EdU labelling) was analysed by immunofluorescence. There was a defect in RPA recruitment after treatment with HU in cells depleted for SAMHD1 compared to control cells (Figure 3A).
A failure to activate the S-phase checkpoint in SAMHD1 depleted cells was also observed (Figure 3B).
2.4. MRE11 and RecQ1 are responsible for the pro-inflammatory response induced by the absence of SAMHD1
DNA degradation at the replication forks is reduced in cells depleted in SAMHD1. However, single-stranded DNA is exported to the cytosol, so it is likely to be displaced and then cleaved by a protein or proteins that would function with SAMHD1 or replace it in its absence. We identified these proteins by analyzing the amount of cytosolic DNA in cells deficient for SAMHD1 and co-deployed with other factors that have a proven role in the processing of replication forks. The enzymes MRE11 and RecQ1 were identified as the primary factors in the pro-inflammatory response in the absence of SAMHD1. Indeed, the amount of cytosolic DNA decreases drastically when RecQ1 is depleted or MRE11 is inhibited in cells deficient for SAMHD1 (Figure 4A). Finally, transcription of pro-inflammatory genes is also strongly reduced when RecQ1 is depleted in SAMHD1 deficient cells (Figure 4B).
In summary, this work showed that SAMHD1 is recruited at the blocked replication forks, where it stimulates the exonuclease activity of MRE11 to activate the replication checkpoint. In the absence of SAMHD1, small fragments of DNA are released from the fork and accumulate in the cytoplasm, where they are detected by innate immunity mechanisms and induce the production of type I interferons (Figure 5).
3. Perspectives
Our work shows that the response to replicative stress is not restricted within the nucleus but extends into the cytosol. Knowing that replicative stress, a major characteristic of cancer, can be increased by chemotherapy, we can now consider new therapeutic strategies combining chemotherapy and immunotherapy.
Our results also allow us to consider new therapeutic strategies to help Aicardi-Goutières patients, who currently have no treatment to limit the causes of their chronic inflammation. Indeed, we show that MRE11 and RecQ1 are responsible for producing the DNA fragments that trigger the inflammatory response in SAMHD1 deficient cells. Therefore, we can imagine that blocking the activity of these enzymes could decrease the production of DNA fragments and, ultimately, the activation of innate immunity in these cells.