

1 Introduction
In many cases, when good catalytic enantioselectivities are achieved with a C2-symmetric bis-phosphine, a 1,1’-ferrocenylene-bridged analogue is investigated. Sometimes this analogue displays an enhanced efficiency. Two recent examples will illustrate this point. The famous DUPHOS (1) of Burk 〚1〛 has been transposed into the ferrocenylene-bridged bis-phospholane (2) 〚2〛 with rather disappointing results. On the contrary, the bis-phosphetane (3) 〚3〛 has been transposed into (4) 〚4,5〛, with substantial gains in enantioselectivity.
Having recently developed the BIPNOR (5), an efficient P-chiral bis-phosphine for asymmetric catalysis 〚6〛, it seemed logical to test the ferrocenylene analogue (6), even though the connection between the two sub-units of (6) is geometrically different from those found in (2) and (4). This is the subject of the following report.
2 Results and discussion
The synthesis of (6) relies on the usual approach 〚7〛. The necessary bis-phosphole (7) was first synthesized from 1,1’-dilithioferrocene and 1-cyano–3,4-dimethylphosphole 〚8〛 as shown in equation (1).
The crude phosphole (7) contains ca 10% of monophosphole (δ31P –13.4 vs –14.3 for (7) in CDCl3), which are removed by crystallisation from ethanol. The classical 〚1,5〛 shift of the ferrocenylene substituent around the two phosphole rings takes place at ca 170 °C and the resulting bis–2H-phosphole can be trapped by an excess of tolan to give the expected bis–1-phosphanorbornadiene (6) as a 50/50 mixture of the meso and racemic products (equation (2)).
The two diastereomers (δ31P –13.2 and –13.7 in CDCl3) were separated by chromatography and recrystallisation from hexane/dichloromethane (90/10). The X-ray crystal structure of the product that resonates at –13.7 (Fig. 1) identified it as the racemic mixture. The resolution was carried out with the enantiopure palladium complex of Roberts and Wild 〚9〛 (equation (3)).
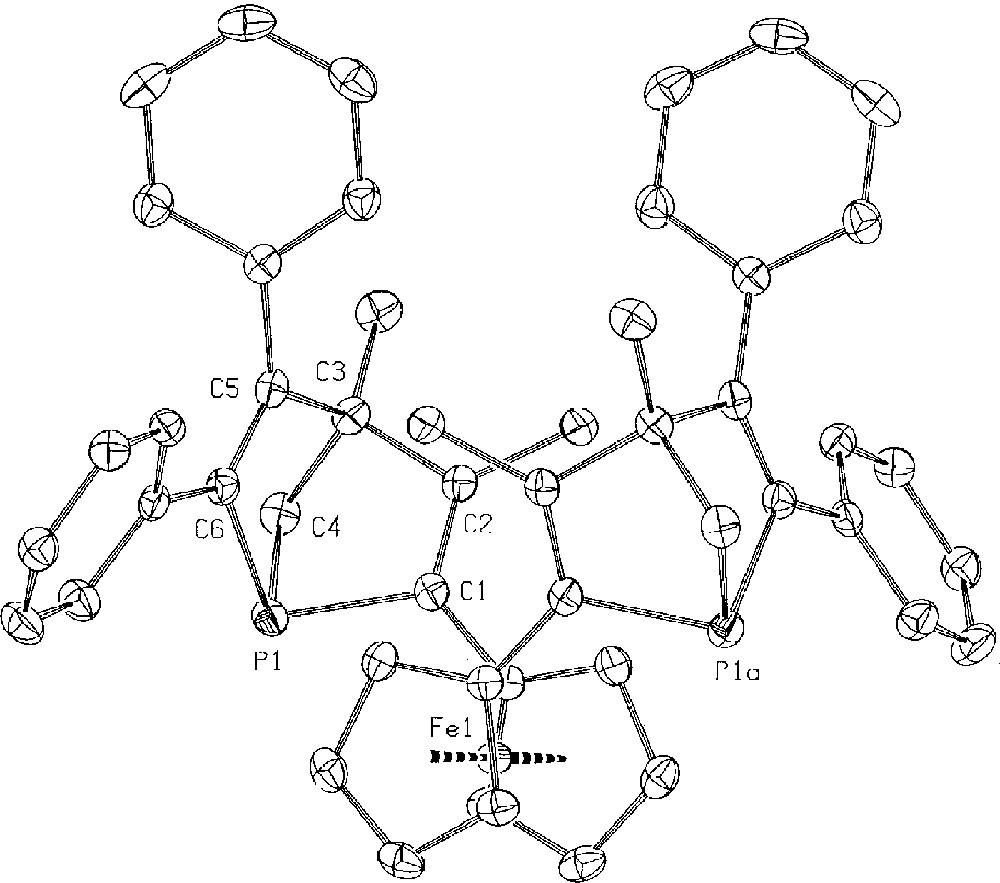
Molecular structure of rac–6, as determined by a single crystal X-ray diffraction study.
The two binuclear enantiopure diastereomeric complexes were separated by chromatography on silica gel with toluene/ethyl acetate (80:20) and recrystallised from Et2O/CH2Cl2 (95:5) at –20 °C. The absolute configuration of 8b was established by X-ray analysis 〚Flack enantiopole = 0.05(4)〛 (Fig. 2). In complex 8b, the ligand 6 has the (S,S) configuration. The enantiopure (R,R)–6 and (S,S)–6 were respectively recovered from 8a and 8b by quantitative decomplexation with sodium cyanide. A series of preliminary catalytic tests were carried out with the two enantiomers. They are summarised in Fig. 3.
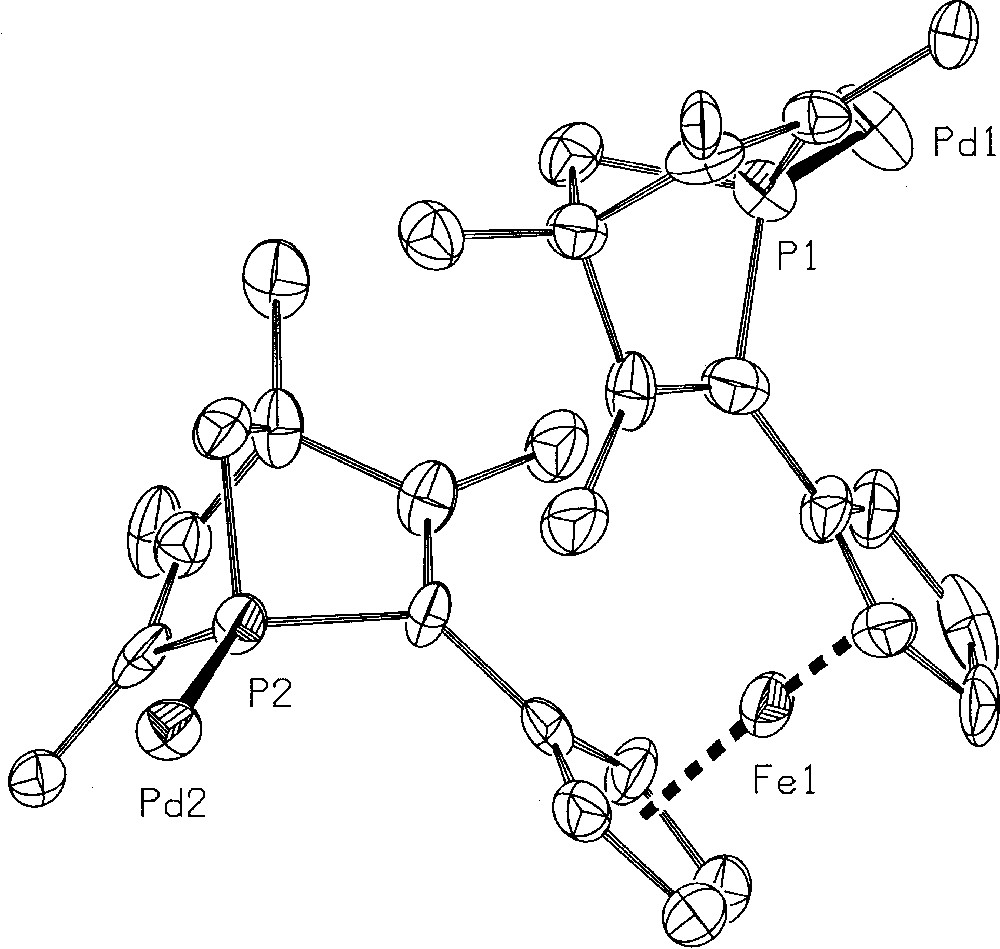
Absolute configuration of complex 8b. The phenyl substituents and the ancillary optically active amine have been removed for clarity.
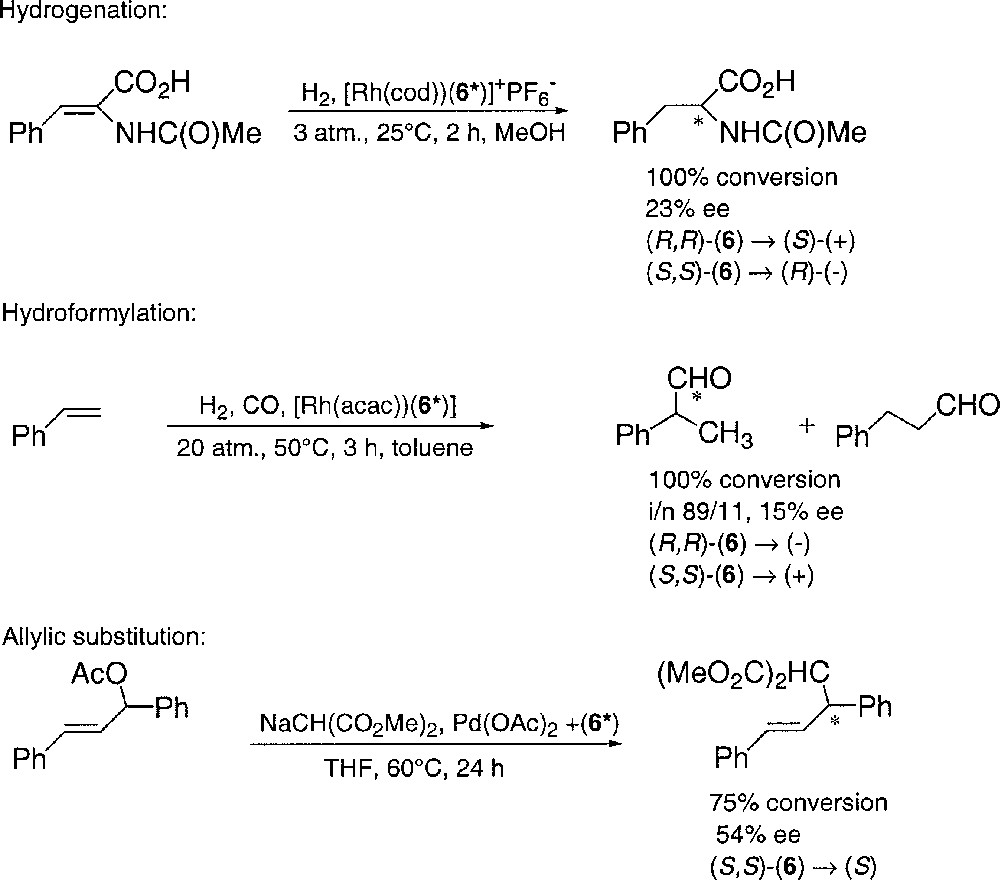
Enantioselective catalysis with (R,R)- and (S,S)–6.
Although these preliminary results are not very encouraging, an optimisation of the 1-phosphanorbornadiene-substitution scheme might lead to efficient catalysts for the enantioselective palladium-catalysed allylic substitutions.
3 Experimental section
All reactions were performed under argon and silica gel (70–230 mesh) was used for chromatographic separations. The solvents were purified, dried and degassed by standard techniques. Diphenylacetylene was purchased from Aldrich; ferrocene, 〚Rh(acac)(CO)2〛, from Strem and used without further purification. 1H, 13C, and 31P NMR spectra were recorded on a Bruker AC 200 SY spectrometer operating at 200.13, 50.32, and 81.01 MHz, respectively, unless stated otherwise. All chemical shifts are reported in ppm downfield from TMS (1H and 13C), and external H3PO4 (31P). Mass spectra (EI) were obtained at 70 eV by the direct inlet method on a Hewlett-Packard (HP) 5989B coupled with a GC HP 5890. Elemental analyses were performed by the ‘Service de microanalyse’, ICSN, CNRS, Gif-sur-Yvette, France.
3.1 Bis-phosphole (7)
In a three-necked flask containing a magnetic stirring bar n-BuLi (27.2 ml, 1.6 M in hexane) was added dropwise to a hexane solution (100 ml) of ferrocene (3.72 g, 20 mmol) at room temperature. Then neat N,N’-tetramethylethylenediamine (TMEDA, 6.35 ml, 42 mmol) was added. The mixture was stirred for 5 min at room temperature, then warmed at 60 °C for 1 h. The solution was allowed to cool to room temperature and dry THF (40 ml) was added before cooling at –40 °C. Cyanophosphole (5.84 g, 42 mmol) in THF (20 ml) was added by portion. The solution was allowed to warm to room temperature and stirring was continued for 15 min. Solvents were evaporated in vacuo and the resulting oil was extracted twice with hexane/CH2Cl2 (150 ml, 90:10). The organic extracts were concentrated in vacuo and EtOH was added (50 ml). The brown solution was kept at –20 °C for one night to give a brown solid (6.0 g, 74%) : 31P NMR (CDCl3) δ –14.3; 1H NMR (CDCl3) δ 2.10 (δ, J = 3.1 Hz, 12H), 4.18 (m, 4H), 4.37 (m, 4H), 6.46 (d, J = 37.7 Hz, 4H); 13C NMR (CDCl3) δ 18.4 (d, J = 4.1 Hz), 72.3 (d, J = 4.3 Hz), 74.1 (d, J = 15.3 Hz),130.8, 149.0 (d, J = 9.2 Hz); mass spectrum m/z (relative intensity) 406 (M+, 66), 358 (M–48, 100). Anal. calcd for C22H24FeP: C, 65.02; H, 5.91; Found: C, 64.32, H, 5.96.
3.2 Bis-phosphanorbornadiene (6)
Bis-phosphole (7) (4.0 g, 10 mmol) and diphenylacetylene (5.4 g, 30 mmol) were heated at 170 °C for 55 h in a sealed tube. The oily product was chromatographed with 85:15 hexane/CH2Cl2 as eluent. The meso isomer was first eluted with some racemic isomer, then the racemic with some meso isomer. Each isomer was recrystallised in 90/10 hexane/CH2Cl2 at –20 °C to give pure products : meso–6 (1.8 g, 24%): 31P NMR (CDCl3) δ –13.2; 1H NMR (CDCl3) δ 1.35 (s, 6H), 2.01 (t, J = 9.8 Hz, 2H), 2.03 (s, 6H), 2.21 (d, J = 9.6 Hz, 2H), 4.0 (m, 4H), 4.27 (m, 2H), 4.39 (s, 2H), 7–7.40 (m, 20H); 13C NMR (CDCl3) δ 16.5, 21.7, 64.5, 69.3 (d, J = 7.5 Hz), 69.9 (d, J = 10.7 Hz), 70.5, 71.1, 72.7 (d, J = 5.3 Hz), 85.4 (d, J = 23.3 Hz), 125–130, 138.9 (d, J = 19.8 Hz),139.7, 146.6 (d, J = 21.5 Hz), 151.1 (d, J = 24.9 Hz), 152.8, 160.9; mass spectrum m/z (relative intensity) 763 (M+, 72), 584 (M–PhCCPh, 84) 406 (M–2 × PhCCPh, 100). Rac–6 (1.6 g, 20%): 31P NMR (CDCl3) δ –13.7; 1H NMR (CDCl3) δ –1.29 (s, 6H), 1.91 (t, J = 9.8 Hz, 2H), 1.98 (d, J = 0.4 Hz, 6H), 2.25 (t, J = 9.3 Hz, 2H), 3.95 (m, 4H), 4.08 (m, 2H), 4.36 (s, 2H), 7–7.40 (m, 20H); 13C NMR (CDCl3) δ 16.5, 21.7, 64.3, 69.7 (m), 70.5, 70.8, 72.8 (d, J = 5.6 Hz), 85.5 (d, J = 21.1 Hz), 125–130, 138.9 (d, J = 19.8 Hz),139.8, 146.6 (d, J = 21.4 Hz), 151.1 (d, J = 24.6 Hz), 152.7, 160.9; mass spectrum m/z (relative intensity) 763 (M+, 74), 584 (M-PhCCPh, 84) 406 (M–2 × PhCCPh, 100).
3.3 Resolution of 6
A solution of the d,l isomer (1.52 g, 2 mmol) in CH2Cl2 (5 ml) was added dropwise to a solution of the palladium (–) complex (1.16 g, 2 mmol) in CH2Cl2 (5 ml) in a Schlenk tube. After stirring for 15 min at room temperature, the solvent was evaporated. The diastereomers were separated by chromatography with 80:20 toluene/AcOEt. The isomer 8a was eluted first and then 8b. Each diastereomer was recrystallised in 5:95 CH2Cl2/Et2O in a pure form 8a (1.23 g, 46%): 31P NMR (CDCl3) δ 51.9; 1H NMR (CDCl3) δ 1.44 (s, 6H),1.75 (d, J = 6.5 Hz, 6H), 2.54 (s, 6H), 2.87 (m, 10H), 2.96 (s, 6H), 3.78m, 2H), 3.96 (s, 2H), 4.14 (s, 2H), 4.51 (s, 2H), 5.40 (s, 2H), 6.50–7.30 (m, 28H); 13C NMR (CDCl3) δ 17.1 (d, J = 8.1 Hz), 21.1 (d, J = 9.7), 22.4, 47.2, 51.6, 65.8 (d, J = 2.8 Hz), 69.7–71.9 (m), 75.4, 81.4 (d, J = 19.5 Hz),122.1–130.4 (m), 135.6 (d, J = 12.2 Hz), 136.9 (d, J = 9.0Hz), 139.3, 139.7 (d, J = 12.3 Hz), 145.5 (d, J = 24.0 Hz), 147.6, 153.0, 157.5 (d, J = 6.2 Hz), 163.9 (d, J = 7.0 Hz); 8b (0.94 g, 35%): 31P NMR (CDCl3) δ 55.7; 1H NMR (CDCl3) δ 1.44 (s, 6H),1.70 (d, J = 5.7 Hz, 6H), 2.54 (s, 6H), 2.70 (d, J + 10.0 Hz, 2H), 2.83 (s, 6H), 3.78 (m, 2H), 2.89 (s, 6H), 3.0 (d, J = 10 Hz, 2H), 3.75 (s, 4H), 3.87 (s, 2H), 4.36 (s, 2H), 4.79 (s, 2H), 5.69 (t, J = 7.0 Hz, 2H), 5.82 (t, J = 7.0 Hz, 2H), 6.51 (t, J = 7.3 Hz, 2H), 6.70–7.30 (m, 22H); 13C NMR (CDCl3) δ 16.9 (d, J = 7.1 Hz), 21.0 (d, J = 9.5), 22.1, 46.7, 51.2, 65.9 (d, J = 2.8 Hz), 69.4–71.9 (m), 75.8, 81.2 (d, J = 20.0 Hz),122.1, 123.6,124.9 (d, J = 5.7 Hz), 127.0–130.4 (m), 136.0 (d, J = 12.5 Hz), 136.9 (d, J = 8.8 Hz), 139.0 (d, 9.4 Hz), 139.4 (d, J = 10.3 Hz), 145.8 (d, J = 24.1 Hz), 147.7, 153.0, 157.9 (d, J = 6.2 Hz), 163.4 (d, J = 6.8 Hz).
3.4 Decomplexation of 8
NaCN (200 mg, 4 mmol) in distilled and degassed water (2 ml) were added to (8) (0.67 g, 0.5 mmol) in CH2Cl2 (4 ml). After vigorous stirring at room temperature for 20 min, water (10 ml) was added. The mixture was then allowed to settle for decantation and the two phases were separated. The organic phase was washed twice with brine and once with water and the aqueous layer with dichloromethane. The combined organic fractions were dried over magnesium sulfate and the solvent evaporated to yield enantiopure (6) (0.37 g, 96%).
3.5 Hydrogenation of α-acetamidocinnamic acid
To a solution of 525 mg (2.5 mmol) of α-acetamidocinnamic acid in freshly degassed methanol (25 ml) was added 1 mol-% of catalyst precursor (prepared from 11.6 mg of 〚Rh(COD)2〛+PF6– and 19.0 mg of (R,R)- or (S,S)–6 in CH2Cl2 (2 ml)). The solution was subsequently transferred by means of a syringe into a 100 ml autoclave previously purged with argon. The solution was stirred under 3 atm of H2 (initial pressure) for 2 h at room temperature. The conversion was determined from 1H–NMR spectra of the crude product in d6-DMSO after evaporation of the solvent in vacuo. A small sample was converted into its methyl ester with trimethylsilyldiazomethane in hexane/2-propanol 90:10 and the enantiomeric excess was determined by HPLC analysis using a Daicel column Chiralcel OD (hexane/2-propanol 90:10, 1 ml min–1; retention times: 9.7 min (R), 12.3 min (S).
3.6 Palladium-catalysed allylic substitution: reaction of 1,3-diphenylprop–2-enyl acetate with NaCH(CO2Me)2 in THF
A mineral oil dispersion of NaH (60% NaH, 1.8×10–3 mol) was washed until free of oil with dry pentane (2 × 5 ml). The oil-free NaH was suspended in THF (4 ml), cooled to 0 °C and dimethyl malonate (0.23 ml, 1.2 equiv) was added dropwise to the stirred suspension. After the reaction was complete, the resulting solution was cannulated into a 50 ml flask containing 1,3-diphenyl–2-propenyl acetate (250 mg, 1×10–3 mol) in 1 ml of THF and the catalytic precursor 〚prepared by mixing Pd(OAc)2 (2.24 mg, 0.01 mmol) and 1 equiv of (6) (7.6 mg, 0.01 mmol) in THF (2 ml)〛. The solution was stirred at 50 °C for 24 h. The reaction mixture was then worked up to give the product as a yellow oil (dilution in AcOH, extraction with Et2O and washing with brine). The conversion was calculated from the crude product by 1H–NMR. Alumina gel chromatography using hexane/ethyl acetate (80:20) afforded the pure allylation product. Enantiomeric excess was determined by HPLC analysis of the purified material using a Daicel column chiralcel OD (200:1 hexane/2-propanol; 1 ml min–1 flow). Rt((R)–1,3-diphenyl–2-propenyl dimethylmalonate) = 25.1 min. Rt((S)–1,3-diphenyl–2-propenyl dimethylmalonate) = 27.7 min.
3.7 Hydroformylation of styrene
The catalyst was prepared in situ from 〚Rh(acac)(CO)2〛 (5.2 mg, 0.02 mmol) and the phosphine (15.2 mg, 0.02 mmol) in toluene (10 ml) containing styrene (420 mg, 4 mmol). The solution was introduced under argon into a stainless-steel autoclave equipped with a magnetic stirring bar . The autoclave was then flushed with CO at 5 bar before pressurising to 20 bar with the syn gas and introduced in a hot bath at the desired temperature. Then the reaction mixture was stirred. After the selected reaction time, the autoclave was cooled to room temperature and depressurised. Conversion was measured by gas chromatography of the crude reaction mixture without evaporation of the solvent. Regioselectivity was determined by 1H–NMR spectroscopy after evaporation of the solvent. After a short-path distillation, optical rotation was used to determine the absolute configuration and the enantiomeric excess.
Supplementary material
Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre: CCDC no. 176584 and 176585.
Acknowledgements
The authors thank Rhodia SA for financial support.