

1 Introduction
Philosophical arguments over the nature of science have a long history, but during the last decade they have achieved unprecedented visibility in the form of the so-called Science Wars, the most prominent manifestation being the notorious ‘Sokal Hoax’ of 1996 〚1〛. While much of the attention (including the last-mentioned hoax) has focused on literary and cultural theorists and their claims to be able to speak meaningfully about science, recent trends in science studies, particularly sociology of science, have also come under fire 〚2〛. Their focus on the role of human factors in science, and how scientific knowledge is constructed by the operation of these factors, stands in contrast to the more traditional picture of scientific knowledge as determined by objective examination of the natural world. Extreme positions have ranged from the claim that social factors are all that matter, to the accusation that emphasizing the social over the objective amounts to subversively undermining the authority of science. I take a middle ground: while I have disagreed with some of the more sweeping conclusions from science studies 〚3〛, I do believe that the concepts and methods they promulgate are potentially of significant value, to practicing scientists as well as society at large, for understanding and even possibly improving the scientific endeavor 〚4〛.
In this paper I will examine the bond-stretch isomerism controversy – a controversy that involved no more than a dozen or two active participants, elicited interest among inorganic chemists that could be termed significant but well short of dramatic, and was (and remains) virtually unknown to the rest of the chemistry community, let alone the scientific community at large. One might ask, then, why bother? There are two main reasons.
First, the majority of studies of scientific controversy deal with highly visible, even notorious cases. There are obvious reasons for that, not least because those carrying out the study are usually not practicing scientists, and may well not even be aware of the more quiet struggles. But if conclusions based on exceptional cases are to be applied to all of science, there is a clear risk of distortion.
For example, a book 〚5〛 intended to introduce science studies ideas to a lay audience makes a number of claims that have provoked considerable objection, including:
“〚We〛 have shown that scientists at the research front cannot settle their disagreements through better experimentation, more knowledge, more advanced theories, or clearer thinking” (pp. 144–145).
“Experiments in real science hardly ever produce a clear-cut conclusion – that is what we have shown” (p. 149).
But what do ‘at the research front’ and ‘real science’ mean? If they refer simply to cases that provoke prominent and contentious debate (and which are the main subject of the book), then the statements may be unobjectionable – but they are also completely tautological. Hence at least some attention should be devoted to cases that do not reach such heights of visibility, and yet cannot be simply dismissed as ‘textbook science’, the descriptor of choice for science that does not need to be studied from the sociological point of view. The debate over the existence of bond-stretch isomerism, which I have chosen to examine here, appears to satisfy those criteria, and in fact did end with an experiment that produced a clear-cut conclusion. Or did it? We shall see.
On the other hand, many critics of science studies extend their doubts about such grandiose statements to the entire enterprise. If scientific explanations can account satisfactorily for the development and outcome of a given episode, they would ask, why do we need outsiders to come in and tell us how we have gotten it all wrong? A chemist familiar with the course of the bond-stretch isomerism controversy might well summarize it as follows: experiments were done; the wrong conclusions were drawn; an experiment suggested those conclusions were wrong and explained why they were wrong; end of story. But even if such a straightforward version is available, is it nonetheless possible that some of the considerations and questions the science studiers raise are of use, not only for understanding what happened in this particular case but for encouraging valuable reflection on one’s scientific practice?
An additional point is that science studies have been dominated by work on physics, with biology an up-and-coming second; only a few scattered examples from chemistry have received much attention (with the exception of cold fusion, if that is to be counted).
I will first present a brief chronological summary of the case, more or less a ‘straight’ scientific account, and then go back and re-examine the evolution of the controversy, making use of some science studies concepts, and focusing on two central points: the status of X-ray crystallography as an experimental technique; and the nature of closure in this case and the possibility of a ‘decisive experiment’.
2 Chronology
The following is necessarily a severely abridged and highly selective version of the course of the controversy; a much more thorough and detailed account has been provided by Parkin 〚6〛. I have tried to keep from interpreting earlier findings in terms of current knowledge – ‘Whig history’ – to the extent possible without sacrificing clarity and brevity. But it must be acknowledged that the selection of what to mention and what to leave out is, inevitably, highly subject to such influences.
2.1 Chatt’s ‘distortional isomers’
In 1970 a full paper reported a series of oxomolybdenum complexes prepared according to equation (1) 〚7〛. Depending on the phosphine ligand L and the halide (or pseudohalide) ligand X, either blue or green products were obtained. In one case, for L = PMe2Ph and X = Cl, both blue and green products could be isolated, depending upon conditions. These were taken to be isomers, both giving acceptable elemental analyses for the formula (calcd C 48.3, H 5.6, Cl 11.9%; found (blue) C 48.5, H 5.9, Cl 12.1%; found (green) C 48.0, H 5.5, Cl 12.7%), and also because solutions of the green compound rapidly changed to blue. The two species in solution exhibited identical electronic and 1H NMR spectra (the 95Mo NMR was later found to be the same for both as well 〚8〛); but did show different IR bands (for crystalline samples as Nujol mulls), at 943 and 954 cm–1 for green and blue respectively. The crystal structure of the blue isomer (cited as a personal communication and subsequently published separately 〚9〛) revealed that it had the cis, mer configuration Ia. Based on the NMR of other green products it was suggested that the green isomer was trans, mer II (equation (1)).
The next year, however, the crystal structure of green MoOCl2(PEt2Ph)3 was determined 〚10〛 (the researchers had been unable to obtain green crystals of the PMe2Ph analog suitable for XRD analysis), and found to have the same cis, mer configuration (Ib) as blue MoOCl2(PMe2Ph)3. Comparing the two molecular structures, they found (i) that the rotational orientation of the organic groups on the phosphine ligands was quite different (Fig. 1), and (ii) that the Mo=O bond distance was considerably shorter for blue Ia than for green Ib – 1.676(7) vs 1.801(9) Å, with smaller differences in the Mo–Cl distances and some bond angles. The authors suggested that green MoOCl2(PMe2Ph)3 would also have structure I, with green Ia differing from blue Ia in the same manner, and that this represented “a new type of isomerism, involving two equilibrium arrangements of ligands, which differ in the distortions of the highly strained co-ordination polyhedron of the metal” – in other words, that non-bonded interactions involving the phosphine ligand substituents were primarily responsible – for which they proposed the term ‘distortional isomerism’.
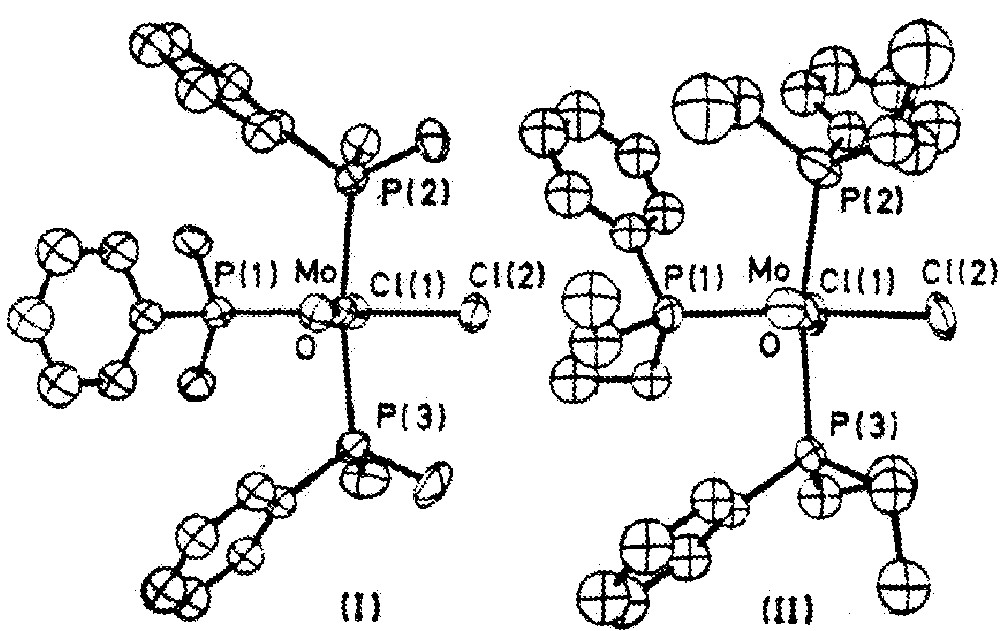
Crystallographic structures of blue MoOCl2(PMe2Ph)3 (I) and green MoOCl2(PEt2Ph)3 (II). From 〚10〛, reproduced by permission of the Royal Society of Chemistry.
In a subsequent full paper 〚11〛 it was noted that spectroscopic considerations suggested that the green isomers of both Ia and Ib should have the same configuration, which cast some doubt on the possible role of ligand steric effects, and that a crystal structure analysis of green Ia was in progress; but no report appeared. A number of years later Haymore was able to get crystals of green Ia. However, the resulting structure was of low quality, and was not published, but reported only as a poster at a conference (the abstract was published 〚12〛), where it drew considerable interest 〚13〛. It did confirm a long Mo=O distance (1.80(2) Å) as in Ib.
Chatt’s finding was noted in a number of surveys, but the reaction was mostly rather muted. A typical example stated 〚14〛:
“Due to the differences 〚...〛 which can be mainly ascribed to ligand repulsions, Chatt et al. have suggested that the blue-green isomerization be called ‘distortional isomerism.’ Whether this type of isomerism is confined to these examples or is more universal in nature awaits further structural and chemical investigations.”
Only one review suggested that this might be novel and highly significant 〚15〛:
“It is hardly possible to overestimate the importance of this result; two isomeric forms were discovered which have the same ligand arrangement in the complex but completely different equilibrium internuclear distances and thus different spectroscopic properties 〚...〛”
2.2 Wieghardt’s 〚CnWOCl2〛+ cations, and other examples
While a few more examples that seemed to fall into the class of distortional isomers appeared over the next few years, it was a 1985 paper from Wieghardt’s group 〚6〛 that provoked really serious interest. Salts of the cationic complex 〚CnWOCl2〛+ (Cn = κ3-N,N’,N”-trimethyl-1,4,7-triazacyclonanone) could be crystallized in two different forms, again one blue and one green. In contrast to MoOCl2(PMe2Ph)3, the two forms each appeared to be indefinitely stable in solution (acetonitrile), although addition of water to the green solution caused an immediate color change to blue. The EPR and, particularly, the electronic spectra (Fig. 2) were reported to be significantly different; the IR spectra differed only in the position of the peak assigned to W=O stretching (980 and 960 cm–1 for blue and green respectively). The elemental analyses agreed well with each other and the nominal formula.
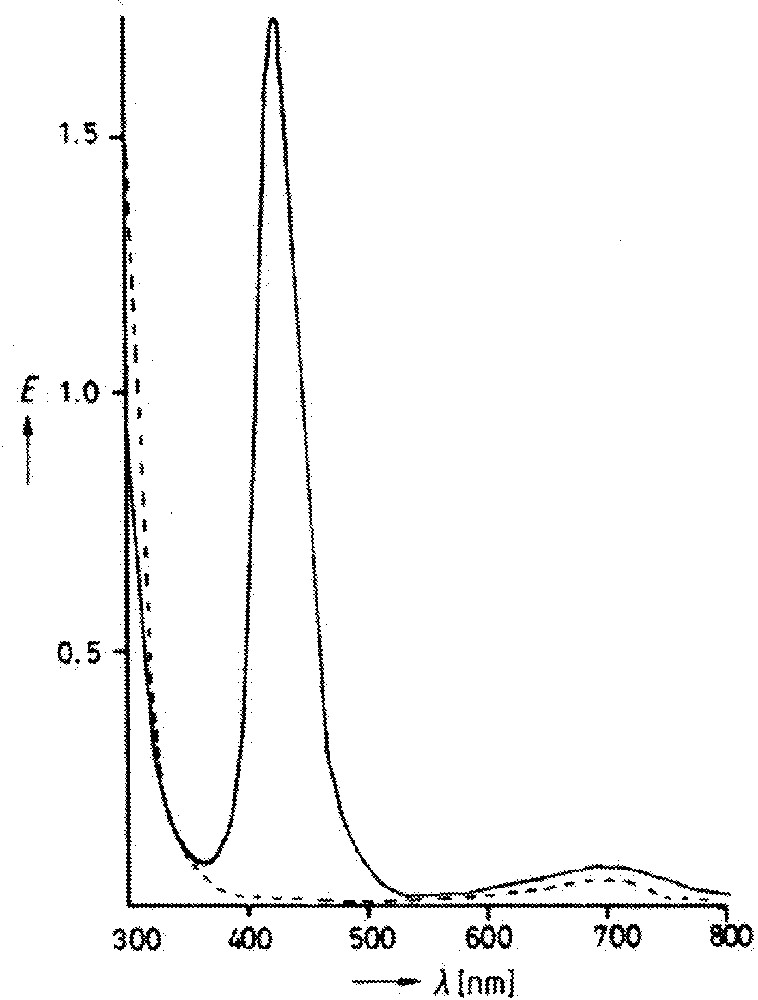
Electronic spectra for the blue (dotted) and green (solid) isomers of 〚CnWOCl2〛+PF6–. From 〚16〛, reprinted by permission of Wiley-VCH and the author.
Most intriguingly, the molecular structures of the two apparent isomers were identical in all respects except for the W=O bond lengths (blue, 1.72(2); green, 1.89(2) Å) and, to a lesser degree (possibly below statistical significance), the W–N distance trans to O (blue, 2.37(2); green, 2.32(2) Å). This finding appeared to exclude the interpretation offered by Chatt and the Muirs, as there are no differences in ligand orientation, steric effects, packing, or anything of that nature – just a difference in bond length.
A number of other examples (see Parkin’s review 〚6〛 for specifics) from several different labs around the same time also suggested that M=O distance, not ligand orientation, was central to this form of isomerism. For example, Cotton found that MoO(NCO)2(PEt2Ph)3 crystallizes with two independent molecules in the asymmetric unit; these have significantly different arrangements of the phosphine ligand substituents but only slightly different Mo=O distances, indicating that Chatt’s proposed distortional effect might not be sufficient to cause substantial bond length perturbation 〚17〛.
2.3 The community takes notice
The flurry of reports in the mid-80s finally caught the attention of the inorganic chemistry community. In particular, several theoretical chemists, who had already been aware of Chatt’s and Haymore’s work, recognized that the new findings, particularly Wieghardt’s, cried out for explanation 〚18〛:
“Chemists appear not to have been very excited over this striking new kind of isomerism 〚...〛 The phenomenon of bond-stretch isomerism is so interesting that it merits analysis and reanalysis by the very best structural techniques available to the profession.”
Two papers by Hoffmann and collaborators appeared in 1988, reporting calculations suggesting that it might indeed be possible to have a double minimum along a coordinate essentially representing one bond distance 〚18, 19〛. They proposed the term ‘bond-stretch isomerism’ as a more appropriate descriptor for this behavior, borrowing from earlier work on quite different classes of molecules 〚20〛.
With a significant number of apparently similar examples as well as a plausible theoretical framework in hand, discussions of the phenomenon of bond-stretch isomerism began to appear in reviews, monographs and even textbooks, although generally hedged with at least some degree of residual doubt. Two typical examples, from a monograph 〚21〛 and a textbook 〚22〛 respectively:
“To our knowledge this is the only suggestion that molecules of any kind have two stable structures with substantially different bond distances. Therefore, if distortional isomerism is a real phenomenon and not an artifact of some kind, it has significant implications beyond metal-ligand multiple bonding.”
“The final form of isomerism to be considered here is quite different than the others. The overall shapes of the molecules are the same, with the difference solely in the length of one or more bonds 〚...〛 The causes and further effects of this type of isomerism are still under investigation; so far, no adequate explanation has been provided.”
During this period there were frequent informal discussions, at ACS meetings and the like, over how this could possibly be right (although nobody was ready to say that it could not be right), and what it might mean. Could it be of some real practical significance, beyond a theoretical curiosity? Connections to catalytic reactions involving molybdenum enzymes or solid molybdenum oxides containing Mo=O bonds were offered as one such possibility 〚23〛.
2.4 The ‘phenomenon’ is explained
In 1991 Ged Parkin published a communication in JACS, which indicated that the crystallographic support for bond-stretch isomerism, at least in the MoOCl2L3 class of complexes, was artifactual 〚24〛. Parkin obtained crystals of green MoOCl2(PMe3)3, which had previously been found in both blue and green forms, and showed that, like Cotton’s previous example, there were two independent molecules in the asymmetric unit. But here the Mo=O bond lengths in the two molecules were in fact quite different, 1.698(8) and 1.866(7) Å. On the other hand, the crystals exhibited only a single IR band in the Mo=O stretching region. Crystallography was repeated on a crystal from a different batch, and again two independent molecules were found, with two more Mo=O distances (1.772(12), 2.154(8) Å). It seemed clear that there could not actually be stable molecules with so many, so widely differing bond lengths, and Parkin was able to explain the anomaly: the samples were contaminated with a different species, MoCl3L3, which can co-crystallize with MoOCl2L3 in almost any proportion. Since substitution of MoCl3L3 for MoOCl2L3 effectively puts a Cl where an O is supposed to be, the calculated structure (based on the assumption that one is dealing with pure MoOCl2L3) will have an apparently long Mo=O distance, as Mo-Cl is longer than Mo=O. Since the electron density of Cl is much higher than that of O, even relatively small amounts of contamination can cause a significant perturbation.
Furthermore, MoCl3L3 happens to be yellow, so its presence admixed with blue MoOCl2L3 will result in a green sample; and it is paramagnetic, so it will not show up in the NMR except as broad, substantially shifted signals – which were observed, once they were looked for. To confirm the interpretation, Parkin prepared a series of samples of MoOCl2L3 deliberately contaminated with increasing amounts of MoCl3L3, and showed that the apparent Mo=O bond distance steadily increased, along with the apparent degree of greenness.
At about the same time, Enemark studied the same system by chromatography and spectroscopy, and found that ‘green MoOCl2(PMe2Ph)3’ could easily be separated into blue and yellow fractions, even though it could be repeatedly recrystallized as green, and that the electronic and NMR spectra revealed evidence for mixtures 〚25〛.
This explanation by contamination was quickly broadcast, by features in Chemical & Engineering News 〚26〛 and Science 〚27〛, and generally accepted, at least for the Mo cases. A detailed chromatographic/spectroscopic reexamination of Wieghardt’s W case followed (the full paper appeared considerably later 〚28〛), showing that a similar situation operated; the electronic spectrum in Fig. 2 above was explained as the superposition of two separate spectra. Theoretical work using higher-level calculatory methodology than that of Hoffmann and collaborators indicated that the proposed mechanism(s) would not produce energy differences, along a single stretching coordinate, sufficiently large to permit two isomers to exist as stable species 〚29〛. And distortional or bond-stretch isomerism soon disappeared from most textbooks (for example the 2nd edition 〚30〛 of the Miessler–Tarr text quoted above contains no mention), unless as a cautionary tale about avoidable error 〚31〛.
3 Another look, with ideas from science studies
It would certainly be possible to stop here, having told a story of error and correction – science working the way it is supposed to! – with perhaps a moral about being more careful with crystallographic results. But there are, I believe, several more general and interesting aspects to this case that may be brought out by reference to some of the ideas promulgated in the field of science studies.
3.1 Black boxing and crystallography
The concept of ‘black boxing’ is by no means unfamiliar to scientists, but those who study science have given it particular attention. Here it is illustrated nicely by a quote from sociologist of science Bruno Latour 〚32〛:
“In 1985, in Paris, John Whittaker obtains ‘nice pictures’ of DNA on a ‘good machine’ 〚...〛 The word black box is used by cyberneticians whenever a piece of machinery or a set of commands is too complex. In its place they draw a little box about which they need to know nothing but its input and output. As far as John Whittaker is concerned the double helix and the machine are two black boxes. That is, no matter how controversial their history, how complex their inner workings, how large the commercial or academic networks that hold them in place, only their input and output count.”
Latour focuses on the stabilization of a technique (or concept): once the ‘inner workings’ are taken for granted, then the results are also taken to be reliable, no longer subject to challenge a priori, without some contradictory result or other force strong enough to compel ‘reopening’ the black box.
I submit that contemporary X-ray crystallography has become a highly ‘black-boxed’ technique. Data collection and analysis are virtually completely automated, to the point where obtaining the crystal structure of a molecule of the size of those discussed above – a project that would have counted as a substantial fraction of a PhD dissertation three decades ago – is now usually a routine procedure taking no more than a matter of days. There are a number of ways of illustrating this change in the status of crystallography; one I find compelling is shown in Table 1. The number of small-molecule crystallographers in the professorial ranks has dropped precipitously over the last 25 years, coinciding with its transition from an intellectually challenging research topic to a routine instrumental method.
Crystallographers in the professorial ranks, US Departments of Chemistry, 1955–1975
| Departments | Faculty | Crystallographersa | Proportion | |
| 1955 | 98 | 1569 | 32 | 1 in 49 |
| 1975 | 187 | 4087 | 101 | 1 in 40 |
| 1995 | 190 | 4459 | 41b | 1 in 109 |
A quote from a professional crystallographer 〚33〛 makes this quite explicit:
“We’ve decided that teaching crystallography is not an absolute necessity anymore because we’ve taken the tedious calculations and put them into computer programs. The ‘lore’ has never been programmed, so the people who are brought up today don’t recognize these problems.”
For clarity I should acknowledge that in the early 1970s, when the first reports appeared, crystallography was by no means as routine as it has become more recently. But this controversy did not really begin in the 1970s. Recognition that there was something sufficiently odd to demand explanation did not become at all general until at least 10–15 years later, by which time crystallography was widely available, quick, cheap and routine. My argument for the central role of black boxing does not center on the original structural determination, but rather on the fact that while many could easily have reexamined the crystallographic findings in either the original or later examples, nobody (until Parkin, who faced a truly glaring anomaly) chose to do so.
3.2 Privileging and crystallography
Another point Latour makes that is relevant to the present case is the ‘privileging’ of some experimental methods over others. For example, while spending some time in a biochemistry lab in the 1970s, he noticed that mass spectrometry (not a particularly common tool in that field at that time) seemed to have such a distinction 〚34〛:
“How can inequality be introduced into a set of equally probable statements in such a way that a statement is taken to be more probable than all the alternatives? The technique most frequently used by our scientists was that of increasing the cost for others to raise equally probable alternatives 〚...〛 when Burgus used mass spectrometry to make a point, he made it difficult to raise alternative possibilities because to do so would be to contest the whole of physics 〚...〛 The mass spectrometer is the reified part of a whole field of physics; it is an actual piece of furniture which incorporates the majority of an earlier body of scientific activity.”
In other words, scientists wishing to disagree with the interpretation of the mass spectrometric results but armed only with the more traditional tools of biochemical research found themselves at a disadvantage, because of the association of the former with the ‘harder’, more quantitative world of physics methodology.
A similar situation pertains when crystallography is opposed to other methods of structural determination: it can be very difficult to challenge a crystallographic finding based on any other observation, or even any combination of other observations, that may suggest possible error. Note the following, taken respectively from a book intended for the lay reader 〚35〛 and a monograph on NMR spectroscopy 〚36〛:
“In some cases, when the molecules cooperate, if they crystallize neatly, then, with a machine called an X-ray diffractometer (costing about $100 000) and one week’s work, it is possible to determine the structure of a molecule 〚...〛. All the precious pieces of evidence, all the blips and peaks and valleys provided by hundred-thousand-dollar 〚spectroscopic〛 instruments, none of these alone proves anything. They are but clues 〚...〛.”
“X-ray crystallography is the ultimate arbiter of chemical structure 〚...〛 Spectra and chemical reactions alone can never tell you the structure of a compound. All they can do is give you pieces of information.”
When even a group of spectroscopists make such a strong statement about the relative standing of crystallography and spectroscopy as structure-determining tools, there can be little question that this view is general among chemists.
3.3 Privileging and black boxing: a dangerous combination?
It follows from the above that crystallographic errors are particularly likely to pass through the normal scientific review process unrecognized. They are not readily subject to contest on either intrinsic or extrinsic grounds. One tends to accept the semi-automated process leading to the structure solution without question, in the absence of any obvious difficulty; one also tends to ignore discordant findings from methods considered less reliable, such as spectroscopy, in the face of an ‘ultimate’ answer from crystallography.
Clearly both of these tendencies influenced the course of the bond-stretch story. It is easy to see, in retrospect (perhaps it always is!) that a number of hints that something might be wrong were available from the very beginning. The original Chatt work noted the identical electronic and NMR spectra of the two isomers. It was suggested that, owing to the rapid isomerization of green to blue, only spectra of the latter were observed – a rather facile interpretation. Even the elemental analyses suggest that the two ‘isomers’ might not be strictly isomeric – the Cl content is considerably higher for the green form, although perhaps not outside the range of acceptable experimental error.
For the tungsten case, the electronic spectra in Fig. 2 show an identical position and shape for the visible band of both isomers; the only qualitative difference is the additional band in the UV for the green isomer. The color change on addition of water seems more consistent with a chemical reaction than a simple isomerization.
Nor was Haymore’s finding that green Ia did not yield very good structural results taken as anything more than a problem of disorder in the particular crystals used. John Enemark has commented on the irony that when he (a professional crystallographer) chose to look at this phenomenon, he went at it using spectroscopic and chemical tools rather than reexamining the crystallography; the latter was done by Ged Parkin, a synthetic inorganic chemist 〚23〛. But in fact Parkin did not suspect that anything was wrong with the crystallography until he saw a clear red flag – four different apparent Mo=O bond lengths! – with the PMe3 complex 〚37〛. None of the many lesser anomalies seems to have been sufficient to raise any concern about possible crystallographic errors among interested parties, whether or not they were trained crystallographers.
It is particularly ironic that Enemark had explicitly noted the possibility apparent lengthening of a bond by contamination in an earlier (pre-Chatt!) paper 〚38〛:
“A referee has suggested that the anomalous behavior of the central atom of the azide group might arise from the presence of some of the chloride complex starting material in the crystal 〚...〛 chloride contamination in the bulk sample is very small, probably less than 1%. However, individual crystals could contain larger amounts of chloride complex. Chloride contamination would introduce electron density in the region between N1 and N3. This could affect the N1–N2 distance and the N2 thermal ellipsoid.”
That describes almost precisely what seems to have occurred in the Chatt isomers, but it took almost 20 years for it to be recognized. (Enemark suggests that Chatt’s group wouldn’t have been expected to catch it because the yellow MoCl3L3 complexes were not known at the time of their original work 〚23〛; but the original Butcher and Chatt full paper does in fact mention those species, although they were apparently not isolated in pure form at that time 〚7〛.)
3.4 Decisive experiments and closure
The idea of a decisive experiment – central to the standard conception of the scientific method – is viewed much more skeptically by much of the science studies community. The quotes from The Golem cited in the Introduction above illustrate their position, as does the following, drawn from a sketch of the history of relativity in the same book 〚5〛:
“While all this was going on, there were still other tests of relativity that had the same mutually reinforcing relationship to these tests as they had to each other 〚...〛 No test viewed on its own was decisive or clear cut, but taken together they acted as an overwhelming movement 〚...〛 We have no reason to think that relativity is anything but the truth 〚...〛 but it is a truth which came into being as a result of decisions about how we should live our scientific lives, and how we should license our scientific observations; it was a truth brought about by agreement to agree about new things. It was not a truth forced on us by the inexorable logic of a set of crucial experiments” (pp. 52–54).
In other words, controversial episodes are closed not by a single experiment whose results and implications are so clear-cut as to be irresistible, but rather by achieving consensus within the scientific community – ‘agreeing to agree’. Scientists tend to be rather uncomfortable with this position.
How does it fit with the bond-stretch isomerism case? Parkin’s work certainly looks like a decisive experiment, at least with regard to the carefully defined question: do the MoOCl2L3 complexes represent a novel form of isomerism? It accounts for the difference in apparent Mo=O bond lengths – the central observation – as well as explaining away most of the other observations, including the colors of the two ‘isomers’, the ready ‘isomerization’ of green to blue (attributable to hydrolysis of yellow MoCl3L3), even the (initially unnoticed) differing elemental analytic results. And, as noted earlier, the finding was rapidly accepted by nearly all of the community as ending the controversy, even before Enemark’s concurring chromatographic and spectroscopic demonstrations and Hall’s revised theoretical predictions.
(To be sure, arguments over the reality of the phenomenon continue. But most or all of these deal with cases where the question of how broadly to define bond-stretch isomerism arises. For example, one dispute turns on whether two molecules differing in spin state may be termed bond-stretch isomers 〚39, 40〛. The present article is concerned only with a narrow definition of the term, as represented by the examples discussed above. A recent review entitled ‘Bond-Stretch Isomerism in Strained Inorganic Molecules and in Transition Metal Complexes: A Revival?’ considers different classes of molecules that could be considered under this topic 〚41〛.)
Let us compare this case to that of cold fusion. There too a number of initial claims (very large excess heat, large numbers of neutrons, large amounts of tritium, etc.) were soon demonstrated to be artifacts. But that by no means brought an end to the controversy; a substantial fraction of the community continued to argue that the phenomenon was real, even if the levels of the various measurements needed to be revised to considerably lower values. One could thus argue that the bond-stretch isomerism controversy does represent a disagreement settled through better experimentation, even if cold fusion does not, and that science studies indeed may get a distorted picture by concentrating on the most visible cases.
However, the matter may not be quite so straightforward. At least one anomaly in the MoOCl2L3 story – the differing IR bands assigned to Mo=O stretching – was not accounted for by the contamination explanation, as the contaminant contains no Mo=O bond! Commentators either ignored this 〚24〛 or attributed it to an error in the initial report 〚25〛.
The fact that the community was comfortable with an incompletely resolved discrepancy brings us full circle in the characterization of crystallography as privileged. As discussed above, no contrary evidence from any other source was able to call into question the reliability of crystallographic evidence supporting bond-stretch isomers; the IR findings appeared to be generally consistent with the crystallography. As soon as the crystallography was found to point the other way, the IR evidence was no longer consistent, but it was completely discounted even though unexplained. Enemark raises an interesting question 〚23〛: if his spectroscopic and chromatographic work indicating the presence of mixtures had appeared before Parkin’s crystallography, how much impact would it have had? Indeed, a reviewer of his later paper on the W system 〚28〛 criticized it for not including any crystallography 〚23〛!
In fact the IR problem was subsequently resolved for the MoOCl2L3 system, when Gibson 〚42〛 found that blue Ia can be obtained in two different crystalline forms, which exhibit the two different IR stretching bands reported by Chatt. The Mo=O lengths also differ (but by much less than in the comparison of ‘isomers’) as do the orientation of the substituents on the phosphine ligands – consistent with the original idea of distortional isomers! The IR differences (and presumably the others as well) disappear in solution, so this is not an inherent characteristic of the independent molecule, but a consequence (at least in part) of crystal packing – a well-known phenomenon.
But unresolved IR indications for isomerism have been reported for niobium species 〚43〛. The complex NbSCl3(PMe3)3 was obtained in two crystalline forms, orange-yellow and green, exhibiting different apparent Nb=S bond lengths and stretching frequencies. It was found that contamination might well be influencing bond distances, but the IR spectral difference could not be explained on that basis. Furthermore, they persist in solution, apparently ruling out the solid-state effect that accounts for the similar observations in the Mo system 〚44〛. Thus the generally accepted conclusion that all reported cases of bond-stretch isomerism are artifactual appears to leave at least one residual discrepancy unexplained. Nonetheless, nobody seems very interested in cleaning it up, or pursuing its implications. (It is not even mentioned in the recent review cited above 〚41〛.)
4 Conclusions
One question I wanted to examine was whether an outsider’s perspective is of any value to practicing scientists. As noted earlier, one could make this a straightforward story of scientific error and correction, without any reference to science studies. Crystallographer Richard Harlow (citing the bond-stretch case among a number of others) has exhorted his colleagues to reduce the incidence of crystallographic errors by just paying more attention to warning signs 〚45〛:
“With the automated data collection procedures, low-temperature capabilities and computer analysis packages available today, the conversion of diffraction spots into a crystal structure ought to be perfectly straightforward for the average molecular crystal and only somewhat more difficult for inorganic structures where problems with absorption, twinning, pseudosymmetry etc., are more common. As it turns out, however, a fair number of ‘wrong’ structures are being published each year which indicates that the authors, the reviewers, and the editors have all failed to recognize one or more symptoms of incorrect structures.”
Errors persist nonetheless. A notable recent example is a revised structure for a natural product, mischaracterized crystallographically and corrected on the basis of old-fashioned synthetic chemistry 〚46〛!
Harlow argues that the computerization of crystallography provides all the tools one needs to avoid error. In contrast, the earlier discussion of black boxing and privileging suggest that this automation may be responsible in part for a tendency to overlook error! While I wouldn’t insist that all crystallographers should read Latour, it does seem fair to propose that scientists might sometimes profit from a different outlook.
The other main issue was whether science studies gets a distorted picture by focusing on highly visible cases, and whether this look at a quieter controversy might reveal something different about how closure is reached. The bond-stretch isomerism story was settled by Parkin’s and Enemark’s work, which appears to contradict the broad claims quoted earlier about the impossibility of a decisive experiment, or at least to make them sound exaggerated and rhetorically excessive.
On closer examination, though, we can find a few traces of ‘agreeing to agree’ as opposed to ‘inexorable logic’ in the rapid closure of the controversy. Thus the sociological perspective on closure does provide some flashes of illumination that may be applied to the present case. I would like to suggest that more thorough studies (the present one is only a sketch, of course) of other episodes, particularly in areas that have not previously attracted a high level of attention, may be similarly rewarding.
I close with a speculation. I contrasted this case with that of cold fusion on the basis of visibility; but another key difference (obviously not entirely separable from visibility) lies in the stakes involved. After all, nobody had a really compelling interest in proving bond-stretch isomerism correct. What if, for example, defending a patent for a new and commercially valuable catalyst rested on distinguishing it from prior art on the grounds of bond-stretch isomerism? Far-fetched, admittedly, but perhaps not totally inconceivable. Perhaps in that alternate world the story would have gone on quite a bit longer; unexplained anomalies would have been vigorously pursued; Parkin’s and Enemark’s experiments would not have been so widely accepted as decisive – in short, there might well have been more resemblance to the cold fusion saga. Arguments based on contrafactuals are suspect, of course. But perhaps this one lends some support to the contention that to fully understand scientific controversies we may often (always?) need to go beyond what appears to be the straightforward scientific content.
N.B. The evolution of the number of crystallographers in the professorial ranks, US Departments of Chemistry, 1955–1975, is given in Table 1 (based on analysis of data from ACS Directories of Graduate Research).
Acknowledgements
I am indebted to John Enemark, Vern Gibson, Barry Haymore, Roald Hoffmann, Ged Parkin, Karl Wieghardt, Mike Hall and Jeremy Burdett for extensive discussions and/or providing reminiscences and documents.