

1 Results and discussion
1.1 Introduction
In the last few years, dendrimers have generated an exponentially growing interest because of the unique and fascinating properties of these highly branched and well-defined macromolecules 〚1–4〛. Amongst the numerous dendrimers that have already been synthesised, structures bearing organometallic centres attract particular attention owing to the specific physical properties of metals 〚5–10〛. Ferrocenyl dendrimers appear to be especially interesting, because of the unique features of ferrocene, like its high thermal stability or its well-defined electrochemistry 〚11–12〛. Furthermore, since the discovery of ferrocene, 50 years ago, progresses in synthetic procedures gave way to a very large variety of ferrocenyl derivatives. A very large range of substituents can be now selectively introduced on the desired position on ferrocene 〚13–16〛. Consequently, ferrocenes found many applications in various fields, especially materials chemistry and asymmetric catalysis.
One ferrocenyl layer has been efficiently grafted on the surface of various kinds of dendrimers like silicon-based dendrimers 〚11, 12, 17, 18〛, polyamine dendrimers 〚11, 12, 19〛, polyarylether dendrimers 〚20, 21〛, polybenzyl dendrimers 〚22〛, small dendrimers with a polyester 〚23〛, an adamantane 〚24〛, a cyclophosphazene 〚25〛 or a tetraphenylmethane core 〚26〛. Even organometallic species like an organoplatinum dendrimer 〚27〛 or ferrocene itself 〚28〛 have been covered by a ferrocenyl layer. These dendrimers are often rather small, but up to 243 ferrocenes could be attached 〚21〛. Some of these dendrimers have been deposited onto electrode surfaces or used in carbon paste electrodes as mediators for amperometric biosensors 〚11, 12〛. Some have been used as CO gas sensors 〚18〛. Cholesteric first generation dendrimers have been found to be liquid crystals 〚23〛. Some polybenzyl dendrimers are suitable to recognise small inorganic anions 〚22, 29, 30〛 and chiral dendrimers proved to be excellent catalysts for asymmetric hydrogenation 〚24, 25〛.
Recently, we also synthesised new phosphorus-containing dendrimers with various ferrocenyl units (eventually chiral) on the periphery 〚31, 32〛. The 11th generation was reached meaning a theoretical number of ferrocenes up to 6144! The ferrocenyl units were easily grafted on the surface of the dendrimers by nucleophilic substitution of the phenol function borne by the ferrocenyl building block on P–Cl bonds of the dendrimers following a methodology developed by some of us 〚33–39〛.
In contrast to the grafting of ferrocenyl units on the periphery of dendrimers, which has been so often studied, very few publications deal with the presence of ferrocene only at the core of a dendrimer. A third generation dendron has been reported 〚40, 41〛. Some carbosilane dendrimers of various sizes (up to the fourth generation) build up around a 1,1’-diphenylphosphinoferrocene (dppf) core were also described and successfully used for palladium-catalysed allylic substitution 〚42〛. We also described a first generation phosphorus-containing dendrimer built from 1,1’-diphenylphosphinoferrocene 〚43〛 and a family of dendrimers built from 1,1’-ferrocenecarboxaldehyde up to the fourth generation 〚31〛.
Moreover, we described the only example of dendrimers having ferrocenes at each layer within the branches up to the second generation 〚31〛 as well as dendrimers having a single ferrocenyl layer located at a precise level within the dendritic structure 〚44〛. The synthesis was based on the use of a ferrocenyl building block bearing a phenol function and a formyl group, namely 1’-(4-hydroxyphenyl)ferrocenecarboxaldehyde. We want now to disclose our results using a new building block E-4 (Fig. 1) where the hydroxyphenyl part is tethered to the ferrocenecarboxaldehyde part by a vinyl link.
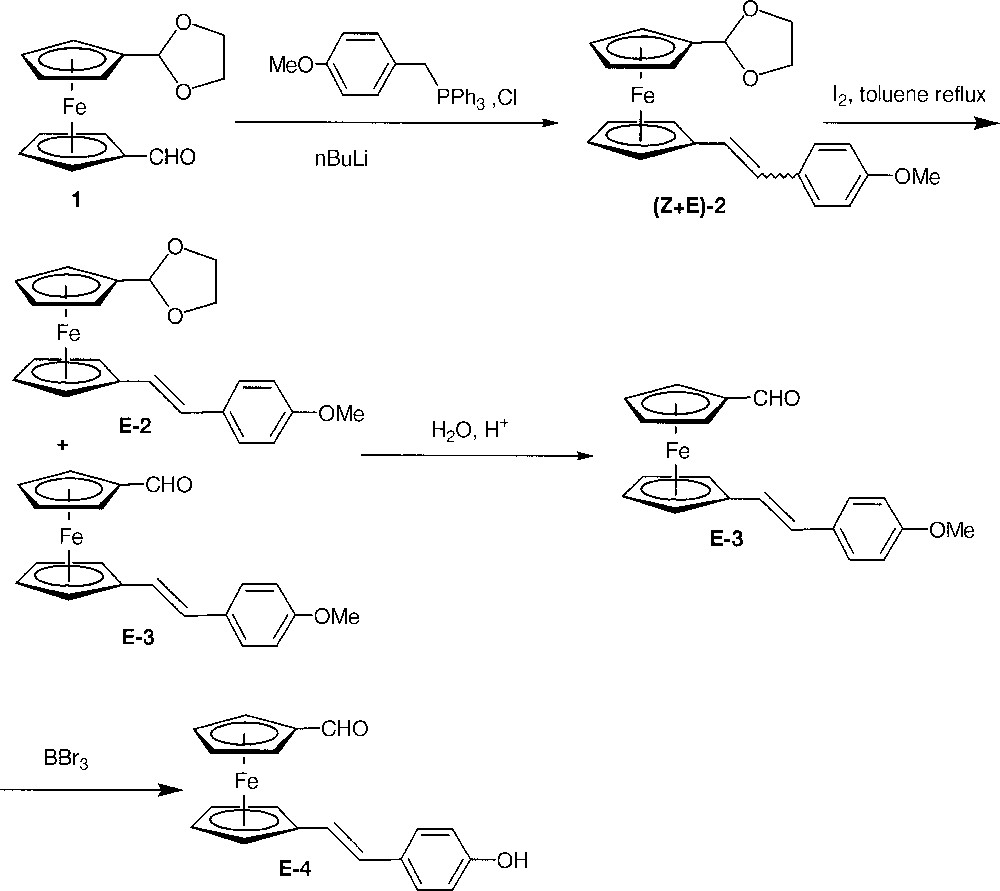
Synthesis of the building block E-4.
1.2 Synthesis of the ferrocenyl building block
Our synthetic target was compound E-4 (Fig. 1). If the synthesis of symmetrically 1,1’-disubstituted ferrocenes is well documented and often easy by electrophilic trapping of the readily available 1,1’-dilithioferrocene 〚45〛, access to non-symmetrically 1,1’-disubstituted ferrocenes is trickier. Amongst the different methods described in the literature 〚15〛, we chose to use compound 1, which allowed us to synthesise successfully similar 1’-styrylferrocenecarboxaldehyde 〚46〛. The action of 4-methoxybenzyltriphenylphosphonium chloride in the presence of n-BuLi yielded quantitatively compound 2 as a mixture of diastereoisomer (E/Z = 1) by a Wittig reaction. This mixture of E-2 and Z-2 was treated with I2, following a procedure already successfully applied to the (Z+E→E) isomerisation of several styrylferrocenes 〚47, 48〛. In these conditions, although the isomerisation was complete, the acetal ring has been partly hydrolysed into the corresponding aldehyde E-3 (Fig. 1). The mixture of E-2 and E-3 was completely converted to E-3 with an excellent yield (98% from the (E+Z)-2 mixture) by acidic hydrolysis. The expected phenol E-4 was finally synthesised by deprotection of the methoxy group of E-3 with BBr3 〚49〛. In conclusion, E-4 could be obtained conveniently and efficiently in four steps with a really good overall yield (83% from 1).
Crystals of E-2 suitable for X-ray analysis were obtained by slow diffusion of hexane into a dichloromethane solution of E-2. The X-ray structure confirms the nature of this intermediate. In particular, the E configuration of the ethylenic fragment, which was deduced from the 1H–1H NMR coupling constants, was directly proved. A molecular view of compound E-2 is shown in Fig. 2 with an atom-labelling scheme, whereas selected bond lengths and angles are given in Table 1. The (4-methoxyphenyl)ethenyl part is slightly out of plane to the ferrocenyl group resulting in a slightly twisted conformation with a dihedral angle of 7.9°. Such twisted conformation seems to be general for all previously reported complexes with values ranging from 4.8° to 48.1 〚46–48〛. The value observed for E-2 indicates occurrence of a strong conjugation in the π system. It is worth pointing out that the methyl of the methoxy moiety deviates only from the phenyl plane by–0.024 Å, probably resulting in an optimised overlap between the oxygen lone pairs and the aromatic π system. The five-member dioxane ring is twisted with respect to the Cp ring to which it is attached resulting in a dihedral angle of 102.53°. There is no special feature within the ferrocene fragment.

Molecular view of compound E-2 showing atoms labelling scheme. Ellipsoids are drawn at 50% probability. Hydrogens are omitted for clarity.
Bond lengths (Å) and bond angles (°) for E-2. E.s.d.s in parentheses refers to the last significant digit.
| O(1)–C(124) | 1.366(4) | C(6)–C(61) | 1.501(4) |
| O(1)–C(127) | 1.420(4) | C(7)–C(8) | 1.411(4) |
| O(2)–C(61) | 1.412(4) | C(8)–C(9) | 1.416(4) |
| O(2)–C(63) | 1.414(5) | C(9)–C(10) | 1.428(4) |
| O(3)–C(61) | 1.419(4) | C(11)–C(12) | 1.298(5) |
| O(3)–C(62) | 1.419(4) | C(12)–C(121) | 1.495(5) |
| C(1)–C(2) | 1.414(5) | C(62)–C(63) | 1.498(6) |
| C(1)–C(5) | 1.425(5) | C(122)–C(121) | 1.369(5) |
| C(1)–C(11) | 1.469(4) | C(122)–C(123) | 1.388(4) |
| C(2)–C(3) | 1.399(5) | C(125)–C(124) | 1.388(5) |
| C(3)–C(4) | 1.398(6) | C(125)–C(126) | 1.384(4) |
| C(4)–C(5) | 1.413(5) | C(124)–C(123) | 1.382(4) |
| C(6)–C(7) | 1.419(4) | C(126)–C(121) | 1.394(5) |
| C(6)–C(10) | 1.417(4) | ||
| C(124)–O(1)–C(127) | 117.8(2) | C(1)–C(11)–C(12) | 123.3(3) |
| C(61)–O(2)–C(63) | 107.4(3) | C(11)–C(12)–C(121) | 127.7(3) |
| C(61)–O(3)–C(62) | 107.0(3) | O(2)–C(61)–O(3) | 107.7(3) |
| C(2)–C(1)–C(5) | 106.9(3) | O(2)–C(61)–C(6) | 112.9(3) |
| C(2)–C(1)–C(11) | 129.5(3) | O(3)–C(61)–C(6) | 110.1(2) |
| C(5)–C(1)–C(11) | 123.5(3) | O(3)–C(62)–C(63) | 103.1(3) |
| C(1)–C(2)–C(3) | 108.6(3) | O(2)–C(63)–C(62) | 102.8(3) |
| C(2)–C(3)–C(4) | 108.6(3) | C(121)–C(122)–C(123) | 122.1(3) |
| C(3)–C(4)–C(5) | 108.0(3) | C(124)–C(125)–C(126) | 120.0(3) |
| C(1)–C(5)–C(4) | 108.0(3) | O(1)–C(124)–C(125) | 116.4(3) |
| C(7)–C(6)–C(10) | 108.3(2) | O(1)–C(124)–C(123) | 124.2(3) |
| C(7)–C(6)–C(61) | 127.2(3) | C(125)–C(124)–C(123) | 119.4(3) |
| C(10)–C(6)–C(61) | 124.4(2) | C(125)–C(126)–C(121) | 121.1(3) |
| C(6)–C(7)–C(8) | 107.8(2) | C(12)–C(121)–C(122) | 117.3(3) |
| C(7)–C(8)–C(9) | 108.6(2) | C(12)–C(121)–C(126) | 124.7(3) |
| C(8)–C(9)–C(10) | 107.6(3) | C(122)–C(121)–C(126) | 117.9(3) |
| C(6)–C(10)–C(9) | 107.7(2) | C(122)–C(123)–C(124) | 119.5(3) |
1.3 Synthesis of the phosphorus-containing dendrimers
The action of a small excess of the sodium salt of E-4 on trichlorophosphane sulphide yields quantitatively the trialdehyde 5-G’0. The formyl groups of 5-G’0 allow the condensation with the –NH2 group of dichlorothiophosphorhydrazide 6 to yield the first generation dendrimer 5-G1 (Fig. 3). The reiteration of the sequence of reactions was attempted on 5-G1. Dendrimer 5-G’1 was obtained in good yield. However, reaction of 5-G’1 with dichlorothiophosphorhydrazide always failed to yield the product of complete condensation, i.e. the second-generation dendrimer. The dendrimer 5-G’1 is only slightly soluble in the solvent of the reaction (chloroform), therefore, the reaction is slow. Furthermore, 5-G’1 is not stable enough in chloroform to sustain a prolonged time of reaction and it is not possible to use another solvent since the dichlorothiophosphorhydrazide 6 is stable only in solution in chloroform. The syntheses of dendrimers 5-G’0, 5-G1 and 5-G’1 are monitored by NMR. The substitution of chlorine by the phenolate induces a shielding of the signal in 31P NMR (from δ = 62.0 ppm for 5-G1 to δ = 61.3 ppm for 5-G’1). An intermediate signal at δ = 67.7 ppm, corresponding to the monosubstitution on each P(S)Cl2 end group, is observed during the course of the reaction and diminishes gradually to disappear totally at the end of the reaction. The condensation step is mainly monitored by 1H NMR and IR, both techniques indicating the disappearance of the signal of the formyl group.
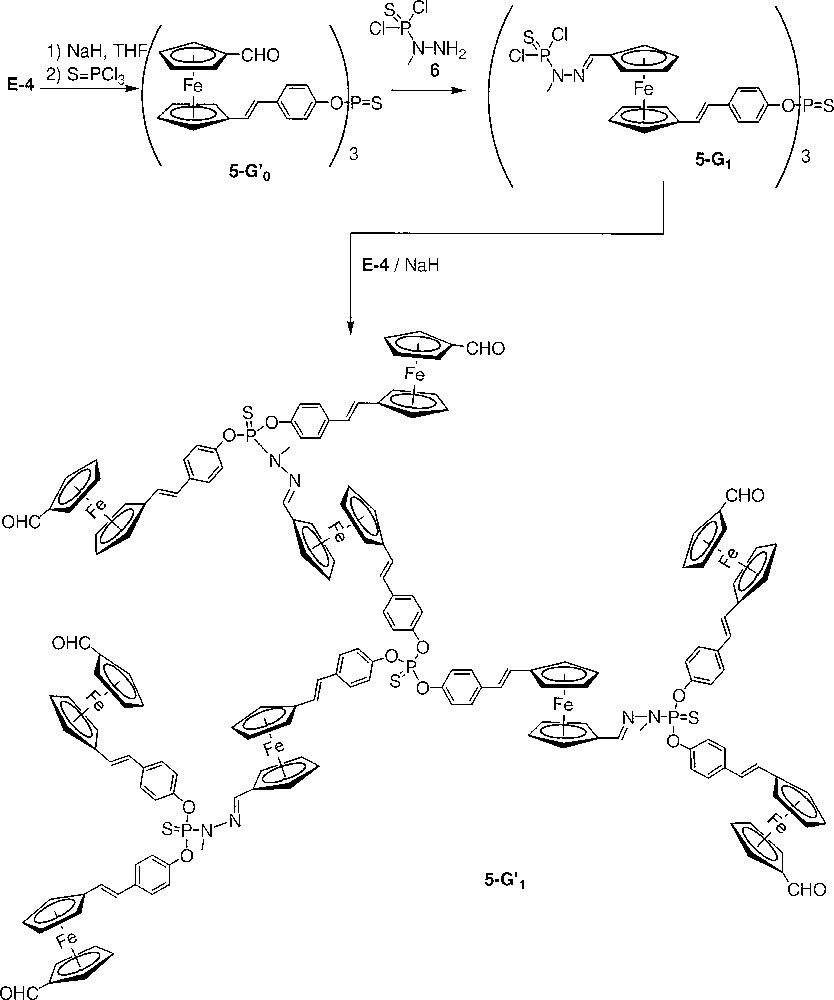
Synthesis of the dendrimers 5-G’0, 5-G1 and 5-G’1.
1.4 Electrochemistry
The cyclic voltammogram of dendrimer 5-G’0 shows that the three ferrocenyl moieties are reversibly oxidised at the same potential (E1/2 = 0.73 V; Ipc/Ipa = 1 and ΔEp = 0.08 V, Table 2 and Fig. 4), indicating the absence of electronic communication. Bulk electrolysis at controlled potential (E = E1/2 + 0.2 V) confirmed the presence of three equivalent electroactive groups, 100% of the theoretical three electrons per molecule being actually transferred at this potential. Such experiment affords a stable and soluble tris ferrocenium dendrimer in the mixture of solvent (THF/acetone, 2:1).
Electrochemical measurements from voltammograms of dendrimers 5-G’0 and 5-G’1.
| Compound | e a | Epc b | Epa b | E | ΔEp c | Ipc/Ipa | Electrolysisd (%) |
| 5-G’0 | 3 | 0.69 | 0.77 | 0.73 | 0.08 | 1 | 100 |
| 5-G’1 first layer | 3 | 0.57 | 0.61 | 0.59 | 0.04 | 1.6 | 92 |
| 5-G’1 second layer | 6 | 0.72 | 0.78 | 0.75 | 0.06 | 1.9 | — |

Cyclic voltammograms (scan rate 100 mV s–1) obtained for compound 5-G’0.
Dendrimer 5-G’0, which possesses two layers of ferrocenyl moieties, exhibits a first wave at E1/2 = 0.59 V and a second one at E1/2 = 0.75 V, with a stripping band (Ipc/Ipa ≈ 2) indicating drastic changes in the solubility properties of the ferroceniums (Fig. 5). The presence of two different waves unambiguously proves that both layers behave independently. The difference between both E1/2 values can be well explained by the difference of electronic properties between formyl and hydrazido groups. Indeed, the replacement of the aldehyde groups by the hydrazido groups increases the electronic density of the first layer’s metallic moieties. Consequently, their potential is shifted from 0.73 V (dendrimer 5-G’0) to 0.59 V (dendrimer 5-G’1), whereas the six electroactive complexes located in the second layer of 5-G’1 are oxidised at 0.75 V. Similar results were already found for similar dendrimers 〚31〛. Bulk electrolysis at controlled potential allowed to count three electrons (with a 95% oxidation ratio) involved in the first transfer: this fact confirms the assignment of this signal to the first layer. Further bulk electrolysis of this dendrimer containing three Fe(III) and six Fe(II) moieties was impossible to perform due to deposition upon the Pt working electrode. Nevertheless, using square wave voltammetry technique, we could assign the second wave to the ferrocenes of the second layer, comparing current peak intensities. As shown in Fig. 6, the second peak is almost twice as high as the second one. The fact that we do not observe a perfect 1/2 ratio for these signals can be explained by a slight overlapping. However, these results appeared reliable enough to assign each wave of the cyclic voltammogram.
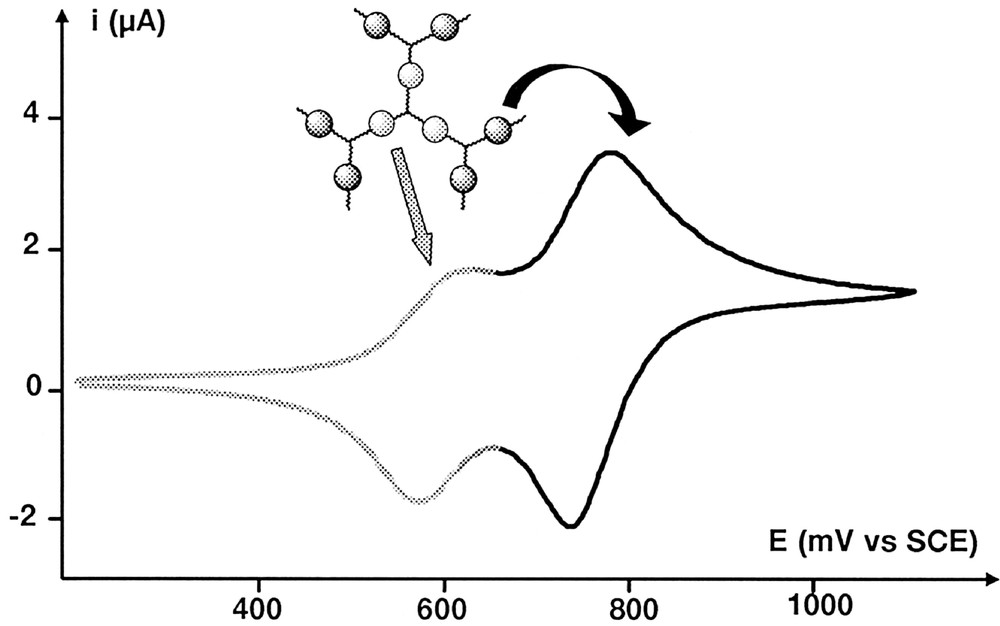
Cyclic voltammograms (scan rate 100 mV s–1) obtained for compound 5-G’1.

Square wave voltammograms (frequency: 50 Hz, potential increment: 10 mV) obtained for compound 5-G’1.
1.5 Conclusion
A new ferrocenyl building block for the synthesis of phosphorus-containing dendrimers bearing ferrocenyl units within the branches was successfully synthesised. The structure of the synthetic intermediate E-2 was determined by X-ray analysis. Two layers of ferrocenes could be introduced on a phosphorus core but any attempts for further growing failed.
Electrochemical studies can differentiate the ferrocenes of the inner layer and of the outer layer which are oxidised at higher potential owing to the presence of electron-withdrawing formyl groups. Surprisingly, oxidation of the inner layers induces drastic changes in the solubility of dendrimers. This behaviour is in marked contrast with most previous observations, indicating that the solubility is mainly governed by the functions located on the surface.
2 Experimental section
2.1 General
All reactions were carried out in the absence of air, using standard Schlenk techniques and vacuum-line manipulations. Commercial samples were used as received. All solvents were dried before use. Thin layer chromatography was carried out on Merck Kieselgel 60F254 precoated silicagel plates. Preparative flash chromatography was performed on Merck Kieselgel. Instrumentation: Bruker AM250 (1H–, 13C– and 31P NMR), Hewlett-Packard HP MSD 7590 (GC/MS), Hewlett-Packard HP 8452A (UV-vis). Elemental analyses were performed by the ‘Service d’analyse’ of the ‘Laboratoire de chimie de coordination’, Toulouse, France. Compounds 1 〚46〛 and 6 〚33–39〛 were synthesised according to published procedures.
2.2 Synthesis of the ferrocenyl building block
2.2.1 2-(1’-(2-(4-methoxyphenyl)ethenyl)ferrocenyl)-1,3-dioxane 2
In a 250-ml two-necked flask under argon, 4.3 g of 4-methoxybenzyltriphenylphosphonium chloride (11 mmol, 1.5 equiv) were suspended in 90 ml of anhydrous THF. The white suspension was cooled down to –78 °C and 6.5 ml (10.4 mmol) of a 1.6 M solution of n-BuLi in pentane was added. After 15 min at –78 °C, the solution was allowed to come back to room temperature and stirred two additional hours at room temperature. The white solution steadily turned bright yellow. The solution was cooled down to 0 °C and then a solution of 1.86 g of 1 (6.52 mmol) in 60 ml of anhydrous THF was added. The reaction mixture was stirred for 18 h at RT. The solution was then diluted by 50 ml of dichloromethane, washed by three 50 ml portions of brine, dried on sodium sulphate and evaporated to yield an orange solid, which was purified by flash chromatography on silicagel. 2.54 g (yield = 99%) of 2 was obtained as a 1:1 mixture of the Z and E isomers.
The E diastereoisomer can be separated by fractional precipitation and characterised.
1H NMR (CD3COCD3): δ (ppm) 7.44 (d, J = 8.8 Hz, 2H, Ar); 6.91 (d, J = 8.8 Hz, 2H, Ar); 6.85 (d (AB), J = 16.2 Hz, 1H, vinyl); 6.75 (d (AB), J = 16.2 Hz, 1H, vinyl), 5.68 (s, 1H, OCHO); 4.53 (t, J = 1.8 Hz, 2H, Cp); 4.30 (t, J = 1.8 Hz, 2H, Cp); 4.26 (t, J = 1.8 Hz, 2H, Cp); 4.14 (t, J = 1.8 Hz, 2H, Cp); 4.0–3.85 (m, 4H, CH2); 3.81 (s, 3H, OCH3). 13C {1H} NMR (CD3COCD3): δ (ppm) 159.3 (quat. Ar); 131.2 (quat. Ar); 127.3 (Ar); 126.2 (vinyl); 124.6 (vinyl); 114.4 (Ar); 102.6 (OCHO); 86.0 (quat. Cp); 85.1 (quat. Cp); 69.8 (Cp); 69.5 (Cp); 68.3 (Cp); 67.7 (Cp); 65.2 (CH2); 55.3 (OCH3). Anal. calcd for C22H22FeO3: C, 67.70; H, 5.70. Found: C, 67.61; H, 5.53. MS (DCI, NH3): m/z = 391 (M+1, 100%).
Isomerisation reaction. Into a one necked round bottom flask equipped with a condenser, were dissolved the mixture of E and Z-2 (1.82 g, 4.66 mmol) in 50 ml of toluene and 10 mol% of I2. The system was purged with argon and the mixture was warmed up to toluene reflux for 30 min. After cooling back to room temperature, the mixture was extracted with methylene chloride, washed with a sodium thiosulfate solution (c = 2 mol l–1), then with brine, dried on sodium sulphate and evaporated. After flash chromatography on silicagel, 1.75 g of a dark red solid containing a mixture of E-2 and E-3 was isolated.
2.2.2 1’-((E)-2-(4-methoxyphenyl)ethenyl)-ferrocenecarboxaldehyde 3
Under argon, the mixture of E-2 and E-3 (1.75 g) was dissolved in 30 ml of dichloromethane. To this solution was added a solution of p-toluenesulfonic acid (3.50 mmol) in 15 ml of water and the reaction mixture was heated up to 60 °C for 90 min. After cooling back to room temperature, the solution was diluted with 100 ml of ether, washed by three portions of brine, dried on sodium sulphate and evaporated to yield a red oil, which was dissolved in dichloromethane and filtered on celite. The celite was thoroughly washed with dichloromethane. After evaporation, 1.59 g of E-4 (98% from the (E+Z)-2 mixture, dark red solid) was obtained.
1H NMR (CDCl3): δ (ppm) 9.89 (s, 1H, CHO); 7.37 (d, J = 8.7 Hz, 2H, Ar); 6.87 (d, J = 8.7 Hz, 2H, Ar); 6.69 (d (AB), J = 16.2 Hz, 1H, vinyl); 6.59 (d (AB), J = 16.2 Hz, 1H, vinyl); 4.74 (t, J = 1.9 Hz, 2H, Cp); 4.52 (m, 4H, Cp); 4.34 (t, J = 1.9 Hz, 2H, Cp); 3.81 (s, 3H, OCH3). 13C {1H} NMR (CDCl3): δ (ppm) 193.8 (CHO); 159.1 (quat. Ar); 130.1 (quat. Ar); 127.7 (vinyl); 127.3 (Ar); 122.5 (vinyl); 114.2 (Ar); 86.0 (quat. Cp); 79.8 (quat. Cp); 74.4 (Cp); 70.6 (Cp); 70.2 (Cp); 67.9 (Cp); 55.3 (OCH3). Anal. calcd for C20H18FeO2: C, 69.40; H, 5.24. Found: C, 69.89; H, 5.52. MS (DCI, NH3): m/z = 364 (M+NH4, 100%), 347 (M+H, 89%).
2.2.3 1’-((E)-2-(4-hydroxyphenyl)ethenyl)-ferrocenecarboxaldehyde 4
In a Schlenk tube under argon, 1.40 g of E-3 (4.04 mmol) was dissolved in 40 ml of dry dichloromethane and the solution was cooled down to –78 °C. 15 ml of a molar solution of boron tribromide in pentane (15 mmol, 3.5 equiv) were then added. The brown solution was allowed to come back to room temperature and kept during 45 min at this temperature. The solution was then thrown in an ice/water bath, stirred during 15 min, washed three times with an aqueous 1 N solution of sodium thiosulfate, then by three portions of brine, dried on sodium sulphate and evaporated to yield a red solid which was purified by flash chromatography on silicagel (eluent = pentane/ether). 1.13 g (yield = 85%) of 4 was obtained as a dark red powder.
1H NMR (CDCl3): δ (ppm) 9.89 (s, 1H, CHO); 7.30 (d, J = 8.5 Hz, 2H, Ar); 6.80 (d, J = 8.5 Hz, 2H, Ar); 6.64 (d (AB), J = 16.2 Hz, 1H, vinyl); 6.57 (d (AB), J = 16.2 Hz, 1H, vinyl), 5.46 (s, 1H, OH); 4.75 (t, J = 1.8 Hz, 2H, Cp); 4.57 (t, J = 1.8 Hz, 2H, Cp); 4.53 (t, J = 1.8 Hz, 2H, Cp); 4.34 (t, J = 1.8 Hz, 2H, Cp). 13C {1H} NMR (CDCl3): δ (ppm) 194.1 (CHO); 155.2 (quat. Ar); 130.1 (quat. Ar); 127.7 (vinyl); 127.4 (Ar); 122.4 (vinyl); 115.6 (Ar); 86.0 (quat. Cp); 79.6 (quat. Cp); 74.1 (Cp); 70.6 (Cp); 70.2 (Cp); 67.9 (Cp). Anal. calcd for C19H16FeO2: C, 68.70; H, 4.85. Found: C, 68.49; H, 4.75. MS (DCI, NH3): m/z = 350 (M+NH4, 100%), 333 (M+H, 73%). IR (KBr pellet): 1646 cm–1.
2.3 Synthesis of the phosphorus-containing dendrimers
The numbering scheme used for NMR is depicted in Fig. 7.
Numbering scheme used for NMR.
2.3.1 Compound 5-G’0
To a solution of trichlorophosphane sulphide (32 μL, 0.315 mmol) in THF (10 ml) at 0 °C was added dropwise a slight excess of the sodium salt of E-4 (358 mg, 1.006 mmol) in solution in THF (15 ml). The reaction mixture was stirred overnight at room temperature. After centrifugation and solvent removal under reduced pressure, the crude product was washed with a mixture of pentane and ether (1:1) and then with methanol to afford 5-G’0 as a red powder (yield = 82%).
31P {1H} NMR (CDCl3): δ (ppm) 53.1 (s, P0). 1H NMR (CDCl3). δ (ppm) 9.89 (s, 3H, CHO); 7.45 (d, 3JHH = 8.5 Hz, 6H, C03–H); 7.21 (dd, 3JHH = 8.5 Hz, 4JHP = 1.6 Hz, 6H, C02–H); 6.70 (s, 6H, CH=CH); 4.75 (t, 3JHH = 1.9 Hz, 6H, Cp0–H); 4.54 (m, 12H, Cp0–H); 4,36 (t, 3JHH = 1.9 Hz, 6H, Cp0–H). 13C {1H} NMR (CDCl3): δ (ppm) 193.8 (s, CHO); 149.6 (d, 2JCP = 7.0 Hz, C01); 135.1 (s, C04); 127.2 (s, C03); 126.8 (s, CH=CH); 125.5 (s, CH=CH); 121.4 (d, 3JCP = 4.5 Hz, C02); 85.1 (s, quat –CH=N); 79.8 (s, quat –CH=CH); 74.4 (s, Cp0); 70.6 (s, Cp0); 70.5 (s, Cp0); 68.2 (s, Cp0). Anal. calcd for C57H45Fe3O6PS: C, 64.80; H, 4.29. Found: C, 64.90; H, 4.32. IR (KBr pellets): 1679 cm–1. MS (FAB): m/z = 1056 (〚M+H〛+, 100%).
2.3.2 Compound 5-G1
To a solution of 5-G’0 (270 mg, 0.256 mmol) in THF (20 ml) at 0 °C was added dropwise a slight excess of a freshly prepared solution of dichlorothiophosphorhydrazide 6 in chloroform. The mixture was stirred 4 h at room temperature and after solvent removal under reduced pressure the resulting powder was washed with a mixture of pentane and diethyl oxide (1:1) to afford 5-G1 as a red powder (yield = 78%).
31P {1H} NMR (CDCl3): δ (ppm) 62.0 (s, P1); 52.8 (s, P0). 1H NMR (CDCl3): δ (ppm) 7.1–7.6 (m, 15H, Ar and CH=N); 6.65 (m, 6H, CH=CH); 4.73 (s, 6H, Cp0–H); 4.52 (s, 6H, Cp0–H); 4.34 (s, 6H, Cp0–H); 4.29 (s, 6H, Cp0–H); 2.97 (d, 3JHP = 14 Hz, 9H, N–CH3). 13C {1H} NMR (CDCl3): δ (ppm) 149.3 (d, 2JCP = 7,0 Hz, C01); 142.8 (d, 3JCP = 18.7 Hz, CH=N); 134.9 (s, C04); 126.8 (s, C03); 126.3 (s, CH=CH); 125.4 (s, CH=CH); 121.5 (d, 3JCP = 4.5 Hz, C02); 84.5 (s, quat –CH=N); 79.8 (s, quat –CH=CH); 70.7 (s, Cp0); 69.9 (s, Cp0); 68.6 (s, Cp0); 67.8 (s, Cp0); 31.5 (d, 3JCP = 13.0 Hz, P1–N–CH3). Anal. calcd for C60H54Cl6Fe3N6O3P4S4: C, 46.81; H, 3.53; N, 5.46. Found: C, 46.92; H, 3.59; N, 5.42.
2.3.3 Compound 5-G’1
To a solution of 5-G1 (200 mg, 0.129 mmol) in THF (10 ml) was added dropwise, at 0 °C, a slight excess of the sodium salt of E-4 (302 mg, 0.853 mmol) in THF (15 ml). The reaction mixture was stirred overnight at room temperature. After centrifugation and solvent removal under reduced pressure, the crude product was washed with a mixture of pentane and diethyl oxide (1:1) and then with methanol, to afford 5-G’1 as a red powder (yield = 80%).
31P {1H} NMR (CDCl3): δ (ppm) 61.3 (s, P1); 53.1 (s, P0). 1H NMR (CDCl3): δ (ppm) 9.90 (s, 6H, CHO); 7.6–7.1 (m, 39H, Ar and CH=N); 6.62 (br s, 18H, CH=CH); 4.7–4.1 (m, 72H, Cp0,1–H); 3.05 (d, 3JHP = 11 Hz, 9H, N–CH3). 13C {1H} NMR (CDCl3): δ (ppm) 193.6 (s, CHO); 149.8 (d, 2JCP = 8.0 Hz, C11); 149.3 (d, 2JCP = 8.0 Hz, C01); 139.8 (d, 3JCP = 14 Hz, CH=N); 135.1 (s, C04); 134.5 (s, C14); 126.9 (br s, Cp0–CH and C0,13); 126.5 (s, Cp1–CH); 125.3 (s, Cp0–CH); 125.0 (s, Cp1–CH); 121.6 (d, 3JCP = 4.0 Hz, C12); 121.4 (d, 3JCP = 4.0 Hz, C02); 85.1 (s, quat –CH=N); 84.3 (s, quat –CH=N); 80.8 (s, quat –CH=CH); 79.6 (s, quat –CH=CH); 74.3 (s, Cp1); 70.7 (s, Cp0); 70.5 (s, Cp1); 70.3 (s, Cp1); 68.3 (s, Cp0); 68.0 (s, Cp1); 67.8 (s, Cp0); 67.7 (s, Cp0); 32.7 (d, 3JCP = 13.7 Hz, P1–N–CH3). Anal. calcd for C174H144Fe9N6O15P4S4: C, 63.07; H, 4.38; N, 2.54. Found: C, 63.12; H, 4.49; N, 2.47. IR (KBr pellets) = 1679 cm–1.
2.4 Electrochemical measurements
Voltammetric and electrolytic measurements were carried out with a home-made potentiostat 〚50〛 using the interrupt method to minimise the uncompensated resistance (Ri) drop; square wave voltammetry measurements were carried out with a PAR model 273. Experiments were performed at room temperature in an airtight three-electrode cell connected to a vacuum/argon line. The reference electrode consisted of a saturated calomel electrode (SCE) separated from the solutions by a bridge compartment. The counter electrode was a spiral of ca 1 cm2 apparent surface, made of a platinum wire 8 cm in length and 0.5 cm in diameter. The working electrode was a Pt electrode (1 mm diameter) for cycling voltammetry and a Pt gauze electrode for bulk electrolysis. E1/2 values were determined as the average of the cathodic and the anodic peak potentials. The supporting electrolyte (nBu4N)〚BF4〛 (Fluka, electrochemical grade) was used as received.
2.5 X-ray crystal structure analysis
Data for E-2 were collected on an Enraf-Nonius CAD4 diffractometer equipped with a graphite oriented monochromator utilising Mo Kα radiation (λ = 0.710 73). The final unit cell parameters were obtained by the least-squares refinement of 25 selected reflections. Only statistical fluctuations were observed in the intensity monitors over the course of the data collection.
The structure was solved by direct methods (SIR92 〚51〛) and refined by least-squares procedures on Fobs. All H atoms attached to carbon were introduced in calculation in idealised positions (d(C–H) = 0.96Å) and their atomic coordinates were recalculated after each cycle. They were given isotropic thermal parameters 20% higher than those of the carbon to which they are attached. Least-squares refinements were carried out by minimising the function , where F0 and Fc are the observed and calculated structure factors. The weighting scheme used in the last refinement cycles was , where with three coefficients Ar for the Chebyshev polynomial Ar Tr(x), where x was Fc/Fc max 〚52〛. Models reached convergence with and , having values listed in Table 3.
Crystal data for compound E-2.
| Crystal parameters | |
| Formula | C22H22O3Fe |
| FW (g) | 390.26 |
| Shape (colour) | plate (yellow) |
| Size (mm) | 1.13, 0.38, 0.05 |
| Crystal system | monoclinic |
| Space group | P21/c |
| a (Å) | 24.421(6) |
| b (Å) | 5.824(2) |
| c (Å) | 12.582(4) |
| β (°) | 93.68(2) |
| V (Å3) | 1786(1) |
| Z | 4 |
| F(000) | 817 |
| ρ (calcd) (g cm–3) | 1.451 |
| μ (MoKα) (cm–1) | 8.63 |
| Data collection | |
| Diffractometer | CAD4 Enraf-Nonius |
| Monochromator | graphite |
| Radiation | MoKα (λ = 0.710 73) |
| Temperature (K) | 293(2) |
| Scan type | ω/2θ |
| Scan range θ (°) | 0.8 + 0.345 tanθ |
| 2 θ range (°) | 3 < 2θ < 50 |
| Absorption method | φ scan |
| Min. and Max. correction | 1.00, 1.139 |
| Number of reflections collected | 3015 |
| Number of independent reflections | 2675 |
| Merging factor R(int) | 0.017 |
| Reflections used, (I > 2 σ(I)) | 1934 |
| Refinement | |
| R | 0.0360 |
| Rw | 0.0422 |
| Weighting scheme | Chebyshev |
| Coefficient Ar | 2.98, 0.723, 2.44 |
| (Δ/σ)max | 0.001 |
| Δρmin/Δρmax | –0.26/0.53 |
| GOF | 1.012 |
| Variable parameters | 235 |
The calculations were carried out with the CRYSTALS package programs 〚53〛. The drawing of the molecules was realised with the help of CAMERON 〚54〛. Full details for data collection and refinement procedures, fractional atomic coordinates, anisotropic thermal parameters for non-hydrogen atoms and atomic coordinates for H atoms have been deposited with the Cambridge Crystallographic Data Centre (CCDC 179305).