

1 Introduction
Over the last few decades, homogenous asymmetric catalysis by transition metals has received considerable attention and numerous chiral ligands, and complexes allowing high efficiency reactions have been reported [1]. However, only a few examples have been transferred to industrial processes [2], in many cases because of the expenses associated to catalyst loss. The efficient separation of expensive catalysts and ligands to enable reuse in subsequent cycles is a main challenge that meets both industrial economical needs and new stricter environmental regulations.
Three major strategies have been reported for immobilizing homogeneous catalysts [3,4]. The first one consists of immobilization on solid supports such as polymers [5,6] or inorganic materials [7] (silica, metal oxides, metal phosphates…). At the end of the reaction, the catalyst is easily recovered by filtration, but its activity and selectivity are often lower relative to the corresponding homogeneous catalyst. As a second approach, in order to combine the advantages of heterogeneous and homogeneous catalysis, soluble polymers [8] or dendrimers [9] have been used as catalyst support. The reaction occurs then in homogeneous media and the catalyst is finally recovered by precipitation or by filtration through membranes. However, longer synthetic procedures are involved because of the functionalization needed to link the catalyst to the support. The third strategy, involving non conventional liquid media and multiphasic systems with immobilization of the homogeneous catalyst on a “liquid carrier” has attracted increasing interest [10,11]. The functionalized catalyst, with a specifically designed ligand, is isolated in one phase at the end of the reaction, while the product remains in the other phase allowing easy product isolation and catalyst reuse. Among these “liquid carriers”, supercritical carbon dioxide [12], perfluorinated solvents [13], ionic liquids [14,15], water [16,17] and polyethylene glycols (PEG) [18] have emerged as interesting candidates.
We have long been interested in the design and the synthesis of new chiral catalysts for exploring new asymmetric catalytic reactions or for improving existing ones [19–27]. In this area, chiral phosphines have played a significant role. The possibility to easily modify their electronic and steric properties by a judicious choice of their substituents has proven extremely useful to successfully optimized catalytic reactions. Among the numerous phosphine ligands reported to date, ferrocenyl phosphines functionalized by an oxygen atom (PO ferrocenyl phosphines) constitute a distinct class of hemilabile ligands attracting increasing interest [28–32]. We have recently developed promising PO ferrocenyl ligands in an enantiomerically pure form (planar chirality) and started investigating their efficiency in asymmetric catalysis (allylic substitution) [23]. In addition, we recently became interested in improving catalyst recycling using ionic liquid, water or PEG as catalyst “liquid carriers” and in investigating the influence of these media on both the catalytic reaction and the recycling efficiency. As a first part of our project, we have decided to develop simple chiral phosphine-ethers as new ligands for catalysis in unusual solvents. In this manuscript, we will describe the synthesis of the first members of this new family of phosphine-ethers, their use in the Suzuki-Miyura reaction and the synthesis of new PO ferrocenyl ligands bearing charged (imidazolium) or neutral (monomethylether PEG 750, tetraethylbisphosphonate) polar tags [33] for future catalytic applications in non conventional media.
2 Results and discussion
As an entry to new PO ferrocenyl ligands, we have chosen 2-thiodiphenylphosphino(hydroxymethyl)ferrocene, 1 (Scheme 1). This compound can be prepared in multigram quantities and isolated as a racemic mixture or in a pure enantiomeric form, opening direct access to chiral ligands [23]. The phosphino function is protected from oxidation by a sulphur atom allowing working in air. Its functionalization can be performed in a one pot process by successive addition of a strong acid (HBF4), generating probably a ferrocenyl carbocation, and then the nucleophile of choice (alcohol, thiol…) usually leading to the expected substitution product in high yields [21]. In order to obtain a large variety of tagged ligands, we needed a synthetic intermediate which bears both a PO ferrocenyl framework and a function (Y) easily transformed into R′ tags (Scheme 1). For this purpose, we have designed a bifunctional linker (HO-linker-Y). The bromoalcohol p-HOCH2(C6H4)CH2Br appears to be an interesting first candidate to achieve this task. The benzylic bromide may be easily substituted by various R′ substrates which will increase PO ferrocenyl ligand affinity for “liquid carriers” such as water, ionic liquids or PEG. Moreover, the relative para arrangement of the CH2OH and CH2Br groups allows the R′ tag to be located remotely from the ligand coordinating atoms (P,O). However, before embarking in this functionalization work, we first wished to establish the ligand efficiency in the Suzuki-Miyaura reaction with the simplest member (compound 2b) of this new family of phosphine-ethers (Scheme 2).
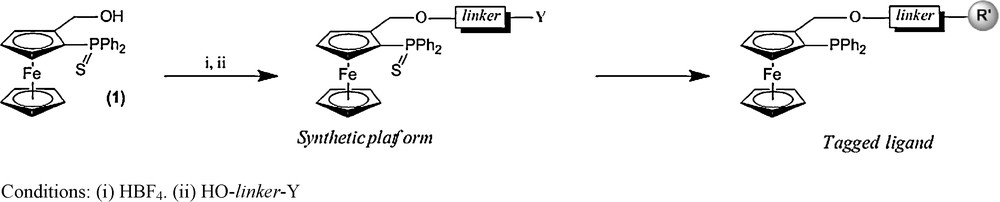
Synthesis of tagged ligands.

Synthesis of ligand 2b.
2.1 Synthesis of ligand 2b
Compound 2b was readily prepared in good yields by addition of an excess of para-methylbenzylalcohol p-HOCH2(C6H4)CH3 to racemic 1 in the presence of tetrafluoroboric acid in dichloromethane, according to the reaction conditions previously optimized for the synthesis of ferrocenyl phosphine thioethers [21]. After purification by flash chromatography on silicagel, compound 2a was obtained as a yellow powder in 45% yield. Desulfurization was achieved with high efficiency using tris(dimethylamino)phosphine in refluxing toluene for 12 h under an argon atmosphere. 2b was recovered as yellow oil after flash chromatography (70% yield). Both compounds have been fully characterized by mass spectrometry and multinuclear NMR. Selected NMR values are reported in Table 1.
31P and 13C (CH2R’) NMR data (CDCl3, δ in ppm).
| compound | R′ | 31P{1H} | 13C{1H} | |
| 43.1 | 21.3 | - | ||
| 2b | H | −21.9 | 22.0 | - |
| 3 | Br | 41.8 | 33.6 | - |
| 4a | CH[PO(OEt)2]2 | 43.0 | 31.6 | |
| 24.3 | dd | JPC = 4.3Hz | ||
| 4b | CH[PO(OEt)2]2 | 23.1 | 30.9 | |
| −22.1 | dd | JPC = 4.3Hz | ||
| 4c | C[PO(OEt)2]2 | 43.1 | 37.3 | |
| 25.7 | dd | JPC = 4.2Hz | ||
| 5a | Imidazolium | 43.0 | 53.2 | - |
| 5b | Imidazolium | −22.4 | 53.4 | - |
| 6a | PEG 750 | 43.0 | 73.0 | - |
| 6b | PEG 750 | −22.0 | 73.1 | - |
2.2 Preliminary evaluation of P,O ferrocenyl ligand efficiency in a Suzuki catalytic reaction
Ligand 2b was tested in the coupling reaction of phenylboronic acid with bromobenzene in the presence of Pd2(dba)3 (1.0% molar of Pd) as catalyst and Cs2CO3 as base (Scheme 3). A control experiment was also carried out under the same conditions but without ligand 2b. Fig. 1 reports the phenyl bromide consumption (%) and the biphenyl formation (%) versus time for both runs. Whereas the reaction is complete within 2 h when using ligand 2b at 70 °C, the conversion remained very low (less than 10%) for the control experiment without ligand 2b, even after 7 h. Comparisons to recently published P,O ferrocenyl ligands underline the efficiency of ligand 2b in the Suzuki-Miyaura reaction [29,32]. For example, Yu et al. [32] obtained a 96% isolated yield of biphenyl in 6 h at 110 °C using a polymer-supported PO ferrocenyl ligand (cf. with a 98% conversion after only 2 h at 70 °C using 2b). Using a similar molar ratio, Hor et al. [29] reported isolated yields of 80–90% at 60 °C in THF but only after 5 h of reaction. These results, obtained under non optimized conditions, confirm the interest of such P,O ligands in Suzuki-Miyaura reactions.
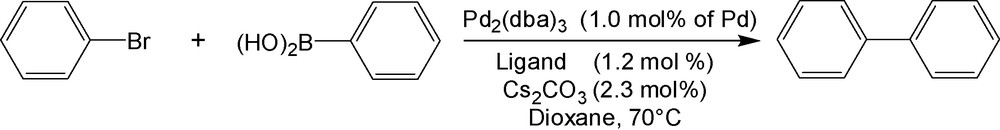
Reagents and conditions for Suzuki-Miyaura reaction.
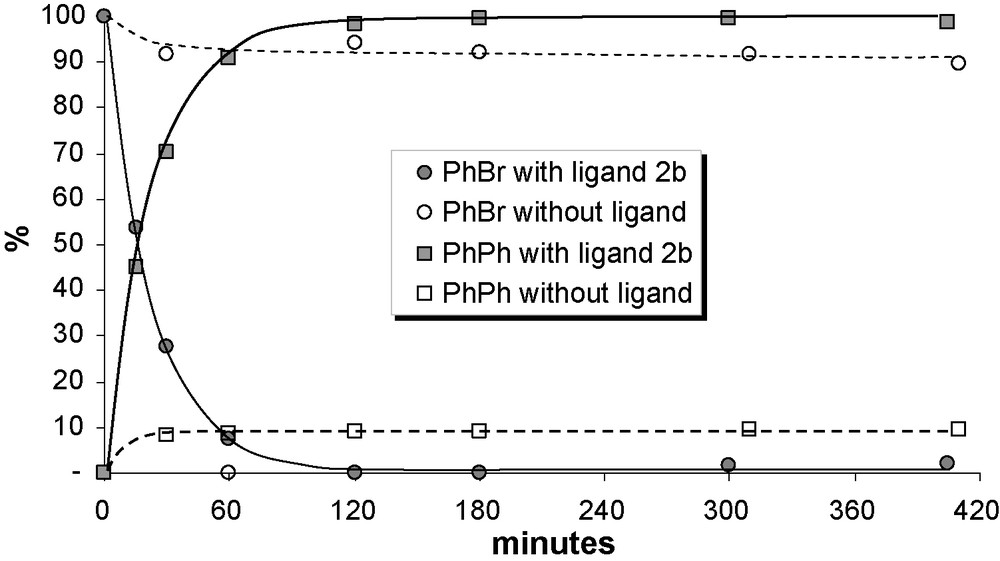
Suzuki-Miyaura reaction of PhBr with PhB(OH)2 in the presence of Pd2(dba)3 with and without ligand 2 (reaction conditions: dioxane, 70 °C, PhBr/Pd/ligand/Cs2CO3 = 100:1:1.2:2.3).
2.3 Synthesis of tagged ligands
Our next goal was to develop recyclable catalytic systems using water, RT ionic liquids or PEG as “liquid carriers”, in particular for Suzuki reactions which have already been efficiently carried out in such solvents [34]. Therefore, we synthesized analogues of ligand 2b bearing various substituents in order to increase their solubility in the “liquid carrier”. All of these tagged ligands have been prepared from the same synthetic intermediate, compound 3 (Scheme 4).
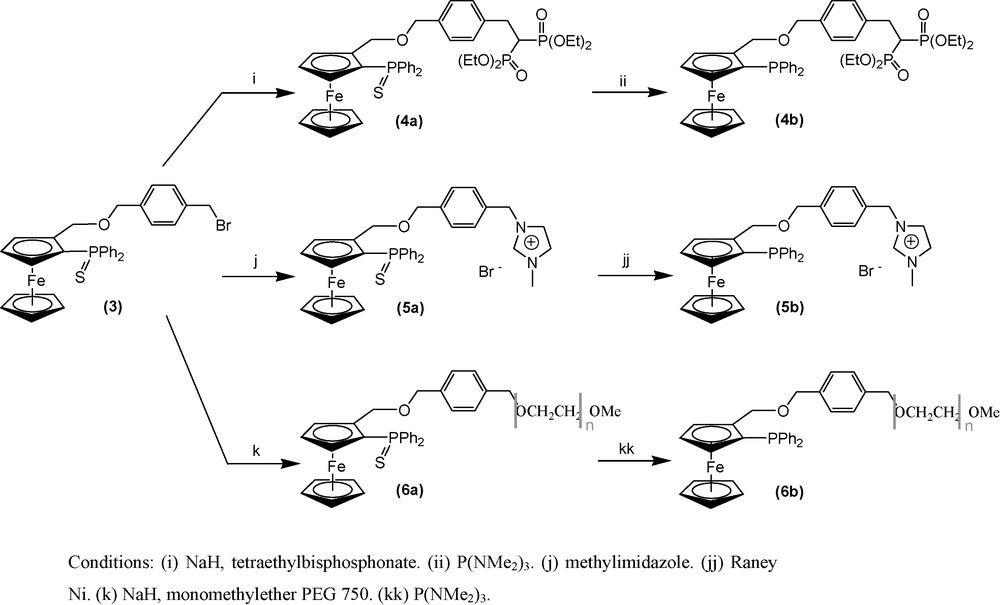
Synthesis of compounds 4 to 6.
2.3.1 Preparation of synthetic intermediate 3
Like 2a, compound 3 was readily prepared in good yields by addition of an excess of p-HOCH2(C6H4)CH2Br to 1 in the presence of tetrafluoroboric acid in dichloromethane. After purification by flash chromatography on silicagel, compound 3 was obtained as a yellow powder with an overall yield of 70%. Its structure was confirmed by NMR, mass spectrometry and X-ray diffraction analyses of the racemic mixture (Table 2). A view of the compound is shown in Fig. 2. As observed in related ferrocenyl derivatives [20,22–24,26,35], the sulfur is endo with respect to the Cp ring, being away from the ring plane by 0.808(6) Å, whereas the oxygen is exo by 1.352(5) Å. The two Cp rings are nearly parallel (dihedral angle of 2.6(2)°) and roughly eclipsed (twist angle of 7.7(2)°). The C2-C21-O2-C22 atoms are nearly planar with the largest deviations from the mean plane being 0.085(2) Å for C22 and this plane is nearly perpendicular to the phenyl ring, with which it has a dihedral angle of 89.5(1)°. The O2 and Br1 atoms are anti with respect to the C22---C23 axis. All bonds lengths and angles in this structure are within expected range (Table 3). NMR data are in agreement with the expected values. The C23 atom (bonded to bromide) gives a signal around δ = 35 ppm in the 13C NMR (CDCl3), upfield relative to the C atoms of the other CH2 groups (δ > 70). This signal is an efficient probe to follow the bromide substitution by the various tags. Selected values are reported in Table 1.
Crystal data and structure refinement parameters for compound 3.
| Identification code | 3 |
| Empirical formula | C31H28BrFeOPS, 1/2 (CH2Cl2) |
| Formula weight | 657.79 |
| Temperature, K | 180(2) |
| Crystal system | 0.71073 |
| Space group | Monoclinic |
| a, Å | C2/c |
| b, Å | 34.2054(8) |
| c, Å | 9.1162(2) |
| β,° | 114.370(3) |
| V, Å3 | 5643.5(2) |
| Z | 8 |
| Dcalc, Mg/m3 | 1.548 |
| μ, mm−1 | 2.200 |
| F(000) | 2680 |
| Crystal size, mm3 | 0.31 × 0.29 × 0.03 |
| θ°, range | 2.08 to 29.07 |
| Reflections collected | 69,915 |
| Unique reflections [Rint] | 7516 [0.133] |
| Obsd reflections [I > 2σ(I)] | 4731 |
| Completeness (%) | 99.5 |
| Absorption correction | Multi-scan |
| Tmin/Tmax | 1.0 and 0.761 |
| Refinement | F2 |
| Parameters | 339 |
| GOF on F2 | 1.022 |
| 0.0472, 0.1173 | |
| 0.0912, 0.1339 | |
| Δρmin, Δρmax (e Å−3) | 1.008/−1.123 |
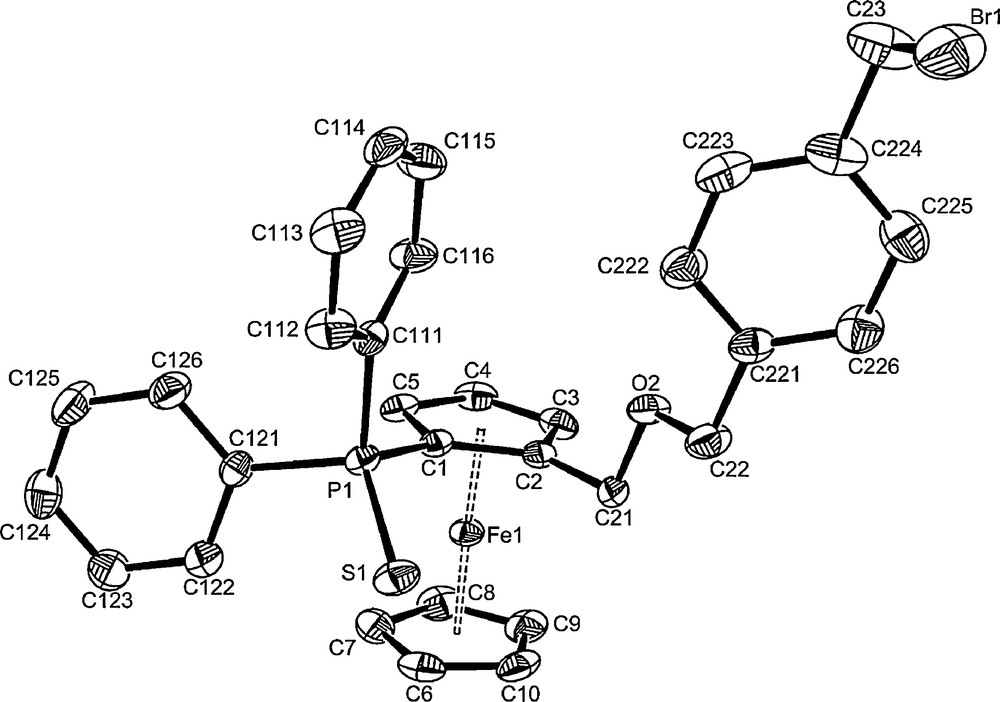
An ORTEP view of the structure of compound 3. Ellipsoids are drawn at the 30% probability level. The dichloromethane solvate and H atoms have been omitted for clarity.
Bond distances (Å) and bond angles (°) with e.s.d.s in parentheses for the structure of compound 3.
| Br(1)-C(23) | 1.964(4) | P(1)-C(111) | 1.816(3) |
| P(1)-C(1) | 1.790(3) | P(1)-S(1) | 1.9623(11) |
| P(1)-C(121) | 1.808(3) | O(2)-C(22) | 1.419(4) |
| O(2)-C(21) | 1.428(3) | C(22)-C(221) | 1.493(4) |
| C(2)-C(21) | 1.488(4) | C(23)-C(224) | 1.487(5) |
| C(1)-P(1)-C(121) | 106.09(14) | C(1)-P(1)-S(1) | 116.62(10) |
| C(1)-P(1)-C(111) | 104.45(13) | C(121)-P(1)-S(1) | 110.49(11) |
| C(121)-P(1)-C(111) | 105.87(13) | C(111)-P(1)-S(1) | 112.53(10) |
| C(22)-O(2)-C(21) | 111.6(2) | O(2)-C(21)-C(2) | 108.0(2) |
| C(2)-C(1)-P(1) | 128.1(2) | C(5)-C(1)-P(1) | 123.8(2) |
| C(3)-C(2)-C(21) | 125.8(3) | C(1)-C(2)-C(21) | 127.3(3) |
| O(2)-C(22)-C(221) | 108.6(2) | C(224)-C(23)-Br(1) | 111.0(2) |
| C(116)-C(111)-P(1) | 122.8(2) | C(126)-C(121)-P(1) | 122.7(2) |
| C(112)-C(111)-P(1) | 118.2(2) | C(122)-C(121)-P(1) | 118.8(2) |
| C(222)-C(221)-C(22) | 120.7(3) | C(226)-C(221)-C(22) | 121.2(3) |
| C(223)-C(224)-C(23) | 120.5(3) | C(225)-C(224)-C(23) | 120.5(4) |
2.3.2 Synthesis of tagged ligands
The last step of our synthetic work consists in increasing the affinity of the ligands with water, ionic liquid or PEG solvents by introducing carefully chosen R′ fragments (Scheme 1). Three different polar tags have been selected to accomplish this task (Scheme 4). The ligand polarity may be increased by introducing the tetraethylbisphosphonate fragment, already used previously to synthesize water soluble phosphine [36], to generate compound 4b. Bisphosphonate esters are also potential precursors of highly charged bisphosphonate anions, R2C(PO32-)2, which may increase water solubility, although they may also compete with the PO donating atoms of the core ligand for metal coordination. Such anionic ligands could be future synthetic targets. We also imagine increasing the ligand solubility by grafting an imidazolium moiety (compound 5b), a cation frequently found in ionic liquids. This cation is specifically designed for use in water or ionic liquid media. The third considered fragment is a monomethylether PEG750 (compound 6b), in order to increase the ligand solubility in water or PEG media.
Ligand 4b was obtained from 3 in a two-step synthesis via compound 4a (Scheme 4). The methodology involved the reactive tetraethyl methylenebisphosphonate carbanion, prepared from the treatment of tetraethyl methylenebisphosphonate with sodium hydride in THF at 0 °C, which efficiently replaced the bromide ion in compound 3. The moderate yield of 40% for this step is partially explained by the subsequent formation of the dialkylated compound 4c (Scheme 5). In agreement with published results [37], this reaction gave mainly monoalkylated product 4a with small amount (60 mg) of dialkylated 4c (12% of the consumed 3) and unreacted starting material. The desulfurization of 4a was achieved with high efficiency using tris(dimethylamino)phosphine in refluxing toluene for 12 h under argon atmosphere. 4b was obtained as an orange oil after flash chromatographic purification (90% overall yield). Compounds 4a, 4b, 4c have been isolated and completely characterized by mass spectrometry and multinuclear NMR. Significant NMR signals are reported in Table 1.
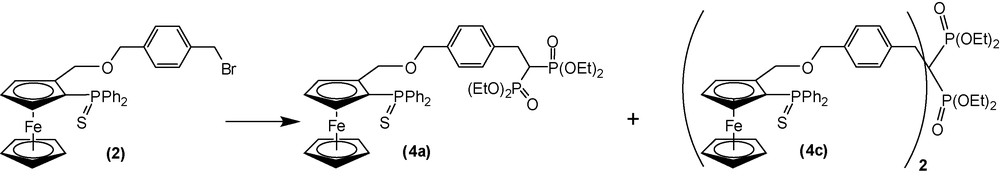
Synthesis of compounds 4a and 4c.
The synthesis of compound 5a was successfully carried out by addition of methylimidazole on compound 3 in refluxing toluene. It was isolated by filtration as a pure yellow powder in a relatively good yield (70%). The same desulfurization procedure used for 4b was not applied because of the high imidazolium affinity to silicagel. Therefore, we turned our attention to a procedure involving heterogeneous conditions [26,38]. Desulfurization was performed by reacting 5a with Raney Ni in acetonitrile under an argon atmosphere at 50 °C. Compound 5b was isolated as a yellow solid in quantitative yield after filtration. Significant NMR signals for both 5a and 5b are reported in Table 1.
The third target is a P,O ferrocenyl ligand bearing a polyethyleneglycol chain. We have chosen PEG 750 monomethylether. 1H NMR integration confirmed the general formula of this PEG: MeO(CH2CH2O)16.3H. Compound 6a was obtained by addition of an excess PEG 750 monomethylether alcoholate, prepared by treatment of PEG 750 monomethylether with sodium hydride, on compound 3 in THF at 0 °C. Compound 6a was isolated as a yellow wax in 60% yield. This low value compared to the crude reaction yield is due to the water solubility of 6a leading to partial loss during the extraction and washing to eliminate the PEG 750 excess. Desulfurization was carried out using tris(dimethylamino)phosphine (Scheme 4) followed by flash chromatographic purification, yielding compound 6b. Compounds 6a and 6b were fully characterized by multinuclear NMR.
2.4 Conclusion
Chiral phosphine-ether 2b has been synthesized and successfully used in a Suzuki-Miyaura coupling reaction. In addition, tagged analogues of 2b bearing various polar substituents (bisphosphonate, imidazolium, PEG750 monomethylether groups) have been prepared from the same synthetic intermediate, compound 3. All of these ligands have been obtained as racemic mixtures but may be easily obtained in an enantiomerically pure form starting from the enantiomerically pure alcohol 1, allowing their use in asymmetric catalysis [39]. The use of this new P,O ligands in a catalytic reaction (the Suzuki reaction…) in non-classical media and the assessment of their efficiency in catalyst recycling is currently underway.
3 Experimental
3.1 General
All reactions were carried out using conventional Schlenk techniques under an inert atmosphere of argon. Thin layer chromatography and flash chromatography were carried out on Merck Kieselgel 60F254 precoated aluminum plates and Merck Kieselgel 60 respectively. Solvents were dried by conventional methods before use. Commercial samples were used as received without further purification (tetrafluoroboric acid diethylether complex, tetraethylmethylenebisphosphonate, N-methylimidazole, Pd2(dba)3, Raney®2800 slurry in H2O and phenylboronic acid from Sigma Aldrich; tris(dimethylamino)phosphine, monomethylether mPEG 750 and diethylene glycol dibutyl ether from Alfa Aesar; bromobenzene from Janssen Chimica). GC analyses were realized with an Agilent 6890 chromatograph equipped with FID detector, automatic sampler and HP5-MS capillary column (30 m × 0.25 mm × 0.25 μm). NMR analyses were performed on Bruker AV500, AV300 and DPX200 instruments. The spectra were referenced internally using the signal from the residual solvent for 1H and 13C, and externally using 85% H3PO4 for 31P. Chemical shifts and coupling constants are given in ppm and Hertz respectively. The following abbreviations are used: s: singulet; d: doublet; t: triplet; quint: quintuplet; m: multiplet; br: broad. Elemental analyses were realized by “Service d’analyse du laboratoire de chimie de coordination”, Toulouse, France. Mass spectral analyses (FAB+ with MNBA matrix; HR MASS) were performed by “Service commun de spectrométrie de masse” of the Université Paul-Sabatier, Toulouse (France).
3.2 Syntheses
Racemic 2-thiodiphenylphosphino(hydroxymethyl)ferrocene, 1, was synthesized according to published procedure [40].
3.2.1 Synthesis and characterization of compound 2a
In a Schlenk tube, 0.75 g of 1 (1.74 mmol) was dissolved in 8 mL of dry dichloromethane. A 54% solution of tetrafluoroboric acid in ether (0.73 mL, 5.30 mmol) was then added. After 1 min stirring, a solution of 2.5 g of 4-methylbenzylalcohol (20.5 mmol) in 8 mL of dry dichloromethane was added. After 1 min of stirring, the crude material was filtered on silica gel with ether as eluent. After evaporation of the solvent, 0.4 g of 2a was obtained as a yellow solid (yield = 43%). 1H NMR (200.1 MHz, CDCl3), δ (ppm): 7.90–7.65 (4H, m: PPh2); 7.52–7.34 (6H, m: PPh2); 7.05 (2H, d, J = 7.9 Hz: Ph); 6.95 (2H, d, J = 7.9 Hz: Ph); 4.89 (1H, d, J = 10.9 Hz: CpCH2); 4.66 (1H, m: subst Cp); 4.45 (1H, d, J = 10.9 Hz: CpCH2); 4.35 (1H, m: subst Cp); 4.33 (5H, s: Cp); 4.29 (2H, d: 2.9 Hz: PhCH2O); 3.85 (1H, m: subst Cp); 2.33 (3H, m: CH3). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 136.8 (s: quat Ph); 135.4 (s: quat Ph); 134.8 (d, JPC = 87.1 Hz: quat PPh2); 133.6 (d, JPC = 86.1 Hz: quat PPh2); 132.2 (d, JPC = 10.7 Hz: PPh2); 132.1 (d, JPC = 10.7 Hz: PPh2); 131.2 (d, JPC = 2.2 Hz: PPh2); 131.1 (d, JPC = 2.2 Hz: PPh2); 128.8 (s: Ph); 128.1 (d, JPC = 12.3 Hz: PPh2); 128.0 (d, JPC = 12.3 Hz: PPh2); 127.7 (s: Ph); 88.3 (d, JPC = 12.1 Hz: quat Cp); 75.2 (d, JPC = 12.5 Hz: subst Cp); 75.6 (d, JPC = 94.7 Hz: quat Cp); 74.5 (d, JPC = 9.4 Hz: subst Cp); 72.4 (s, PhCH2O); 70.7 (s: Cp); 69.4 (d, JPC = 10.4 Hz: subst Cp); 66.7 (s: CpCH2O); 21.3 (s: CH3). 31P NMR (81.0 MHz, CDCl3), δ (ppm): 43.1. HR MS (DCI CH4), C31H29OPSFe, calcd. mass [M]: 536.1026; exp. mass [M]: 536.1039.
3.2.2 Synthesis and characterization of compound 2b
In a Schlenk tube, 350 mg of 2a (0.22 mmol) were dissolved in 5 mL of dry toluene together with 0.75 mL of tris(dimethylamino)phosphine (3.9 mmol). The solution was kept at reflux overnight under argon. After cooling back to room temperature, the solution was evaporated in vacuo. The crude residue was purified under argon by flash chromatography on silica gel with dichloromethane as eluent. After evaporation of the solvent, 235 mg of 2b were obtained as an orange waxy solid (yield = 70%). 1H NMR (200.1 MHz, CDCl3), δ (ppm): 7.67–7.58 (2H, m: PPh2); 7.48–7.41 (3H, m: PPh2); 7.32–7.29 (5H, m: PPh2); 7.08 (2H, d, J = 8.1 Hz: Ph); 6.97 (2H, d, J = 8.1 Hz: Ph); 4.65 (1H, dd, J = 10.9 Hz, JPH = 2.2 Hz: CpCH2); 4.60 (1H, m: subst Cp); 4.44 (1H, d, J = 10.9 Hz: CpCH2); 4.37 (2H, s: PhCH2O); 4.36 (1H, m: subst Cp); 4.08 (5H, s: Cp); 3.83 (1H, m: subst Cp); 2.37 (3H, m: CH3). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 140.8 (d, JPC = 9.3 Hz: quat PPh2); 138.3 (d, JPC = 8.2 Hz: quat PPh2); 137.6 (s: quat Ph); 136.2 (s: quat Ph); 135.9 (d, JPC = 20.3 Hz: PPh2); 133.2 (d, JPC = 17.9 Hz: PPh2); 129.9 (s: PPh2); 129.6 (s: Ph); 128.9 (d, JPC = 7.5 Hz: PPh2); 128.8 (d, JPC = 5.7 Hz: PPh2); 128.5 (s: PPh2); 128.4 (s: Ph); 90.1 (d, JPC = 24.6 Hz: quat Cp); 77.5 (d, JPC = 8.1 Hz: quat Cp); 73.5 (d, JPC = 3.4 Hz: subst Cp); 72.9 (s, PhCH2O); 72.7 (d, JPC = 3.7 Hz: subst Cp); 70.6 (s: subst Cp); 70.3 (s: Cp); 68.1 (d, JPC = 9.9 Hz: CpCH2); 22.0 (s: CH3). 31P NMR (81.0 MHz, CDCl3), δ (ppm): −22.9 (PPh2). HR MS (DCI CH4), C31H29OPFe, calcd. mass [M + H]: 505.1375; exp. mass [M + H]: 505.1384.
3.2.3 Synthesis and characterization of 4-(bromomethyl)benzyl alcohol
At 0 °C, in a Schlenk tube, 65 mL of BH3.THF (1 M in THF, 65 mmol) were added to 11.6 g of 4-(bromomethyl)benzoic acid (54 mmol) in 20 mL of freshly distilled THF, under argon. After 2 h stirring at room temperature, 20 mL of methanol were added slowly. The clear solution was concentrated. The crude product was solubilized in ether and washed with an aqueous saturated solution of NaHCO3. After concentration, 10.4 g of a white solid were obtained (yield = 96%) without further purification. 1H NMR (200.1 MHz, CDCl3), δ (ppm): 7.46–7.30 (4H, m: Ar); 4.73 (2H, s: CH2Br); 4.54 (2H, s: CH2OH). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 142.0 (s: quat Ar); 138.0 (s: quat Ar); 130.0 (s: Ar); 128.1 (s: Ar); 65.66 (s: CH2OH); 34.01 (s: CH2Br). Anal. Calcd. for C8H9BrO: C, 47.79%; H, 4.51%. Found: C, 47.85%; H, 4.53%.
3.2.4 Synthesis and characterization of compound 3
In a Schlenk tube, 1.5 g of 2-thiodiphenylphosphino(hydroxymethyl)ferrocene 1 (3.47 mmol) was dissolved in 15 mL of dry dichloromethane. Then 1.5 mL of a 54% solution of tetrafluoroboric acid in ether (10.86 mmol) was added. After 1 min stirring, a solution of 5.12 g of 4-(bromomethyl)benzylalcohol in 12 mL of dry dichloromethane was added. After 1 min of stirring, the crude material was filtered on silica gel with ether as an eluent. After evaporation of the solvent, 1.5 g of 3 was obtained as a yellow solid (yield = 70%). 1H NMR (300.1 MHz, CDCl3), δ (ppm): 7.87–7.80 (2H, m: PPh2); 7.72–7.65 (2H, m: PPh2); 7.55–7.40 (4H, m: PPh2); 7.39–7.30 (2H, m: PPh2); 7.23 (2H, d, J = 8.0 Hz: Ph); 6.98 (2H, d, J = 8.0 Hz: Ph); 5.01 (1H, d, J = 10.8 Hz: CpCH2); 4.65 (1H, m: subst Cp); 4.48 (2H, s: CH2Br); 4.44 (1H, d, J = 10.8 Hz: CpCH2); 4.34 (5H, s: Cp); 4.34 (2H, s: PhCH2O); 4.30 (1H, m: subst Cp); 3.84 (1H, m: subst Cp). 13C NMR (75.5 MHz, CDCl3), δ (ppm): 138.9 (s: quat Ph); 136.6 (s: quat Ph); 134.9 (d, JPC = 87.4 Hz: quat PPh2); 133.6 (d, JPC = 86.5 Hz: quat PPh2); 132.2 (d, JPC = 10.0 Hz: PPh2); 132.0 (d, JPC = 10.0 Hz: PPh2); 131.3 (d, JPC = 2.5 Hz: PPh2); 131.1 (d, JPC = 2.5 Hz: PPh2); 128.9 (s: Ph); 127.9 (s: Ph); 128.1 (d, JPC = 12.5 Hz: PPh2); 128.0 (d, JPC = 12.5 Hz: PPh2); 88.0 (d, JPC = 12.0 Hz: quat Cp); 75.3 (d, JPC = 12.6 Hz: subst Cp); 74.7 (d, JPC = 94.9 Hz: quat Cp); 74.6 (d, JPC = 9.3 Hz: subst Cp); 72.0 (s: PhCH2O); 70.7 (s: Cp); 69.5 (d, JPC = 10.0 Hz: subst Cp); 67.1 (s: CpCH2O); 33.6 (s: CH2Br). 31P NMR (121.5 MHz, CDCl3), δ (ppm): 41.8. MS (FAB > 0; MNBA), C31H28BrFeOPS, m/z: 616 ([M]+).
3.2.5 Synthesis and characterization of compounds 4a and 4c
In a Schlenk tube, 250 mg of tetraethylmethylenebisphosphonate (0.87 mmol) were reacted with a suspension of 250 mg of sodium hydride (60% dispersion in mineral oil) in 5 mL of THF. After 1 h at room temperature, the filtered solution was transferred via cannula on 500 mg of compound 3 (0.81 mmol) dissolved in 20 mL of THF. After 1 day, 10 mL of a saturated aqueous solution of ammonium chloride were added and the product was extracted with ethyl acetate. The crude material was purified by flash chromatography on silica gel with ethyl acetate/ethanol (9/1) as eluent. Compounds 3, 4a and 4c were successively recovered. After evaporation of the solvent, 270 mg of 4a was obtained as an orange solid (yield = 40%). 1H NMR (200.1 MHz, CDCl3), δ (ppm): 7.87–7.62 (4H, m: PPh2); 7.50–7.16 (6H, m: PPh2); 7.14 (2H, d, J = 8.1 Hz: Ph); 6.96 (2H, d, J = 8.1 Hz: Ph); 4.38 (1H, d, J = 11.0 Hz: CpCH2). 4.89 (1H, dd, J = 11.0 Hz: CpCH2); 4.63 (1H, m: subst Cp); 4.31 (2H, s: PhCH2O); 4.31 (5H, s: Cp); 4.27 (1H, m: subst Cp); 4.20–4.00 (8H, m: CH2 phosphonates); 3.82 (1H, m: subst Cp); 3.21 (2H, t of d, J = 6.0 Hz, JPH = 16.7 Hz: PhCH2); 2.62 (1H, t of t, J = 6.2 Hz, JPH = 23.9 Hz: CHP2); 1.28 (12H, t of d, J = 7.1 Hz, JPH = 2.6 Hz: CH3 phosphonates). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 135.7 (d, JPC = 87.6 Hz: quat PPh2); 139.4 (dd, JPC = 7.3 Hz: quat Ph); 134.3 (d, JPC = 86.3 Hz: quat PPh2); 137.5 (s: quat Ph); 132.9 (d, JPC = 10.1 Hz: PPh2); 132.8 (d, JPC = 10.1 Hz: PPh2); 132.0 (d, JPC = 2.4 Hz: PPh2); 131.8 (d, JPC = 2.4 Hz: PPh2); 129.4 (s: Ph); 128.9 (d, JPC = 12.8 Hz: PPh2); 128.8 (d, JPC = 12.4 Hz: PPh2); 128.3 (s: Ph); 89.0 (d, JPC = 11.9 Hz: quat Cp); 75.0 (d, JPC = 11.8 Hz: subst Cp); 75.3 (d, JPC = 95.3 Hz: quat Cp); 75.2 (d, JPC = 9.3 Hz: subst Cp); 72.0 (s: PhCH2O); 71.47 (s: Cp); 70.2 (d, JPC = 10.4 Hz: subst Cp); 67.5 (s: CpCH2O); 63.4 (d, JPC = 7.4 Hz: CH2 phosphonates); 63.2 (d, JPC = 7.7 Hz: CH2 phosphonates); 39.8 (t, JPC = 132.5 Hz: CHP2); 31.6 (t, JPC = 4.3 Hz: PhCH2); 17.1 (d, JPC = 6.4 Hz: CH3 phosphonates). 31P NMR (81.0 MHz, CDCl3), δ (ppm): 24.3 (phosphonates); 43.0 (PPh2). MS (FAB > 0; MNBA), C40H49FeO7P3S, m/z: 845 ([M + Na]+), 822 ([M]+).
After evaporation of the solvent, 60 mg of 4c were recovered as a waxy solid. 1H NMR (200.1 MHz, CDCl3), δ (ppm): 7.93–7.68 (8H, m: PPh2); 7.56–7.33 (12H, m: PPh2; 4H, d, J = 7.8 Hz: Ph); 7.02 (4H, d, J = 7.8 Hz: Ph); 4.91 (2H, d, J = 11.0 Hz: CpCH2); 4.69 (2H, m: subst Cp); 4.50 (2H, d, J = 11.0 Hz: CpCH2); 4.37–4.34 (4H, s: PhCH2O; 10H, s: Cp; 2H, m: subst Cp); 4.00–4.10 (8H, m: CH2 phosphonates); 3.88 (2H, m: subst Cp), 3.34 (4H, dd, JPH = 16.1 Hz: PhCH2); 1.19 (12H, dd, J = 7.0 Hz: CH3 phosphonates). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 136.7 (s: quat Ph); 135.6 (dd, JPC = 6.1 Hz: quat Ph); 134.9 (d, JPC = 87.8 Hz: quat PPh2); 133.6 (d, JPC = 85.7 Hz: quat PPh2); 132.2 (d, JPC = 10.0 Hz: PPh2); 132.0 (d, JPC = 10.1 Hz: PPh2); 131.5 (s: Ph); 131.3 (d, JPC = 2.2 Hz: PPh2); 131.1 (d, JPC = 2.2 Hz: PPh2); 128.1 (d, JPC = 12.7 Hz: PPh2); 128.0 (d, JPC = 12.2 Hz: PPh2); 126.6 (s: Ph); 88.3 (d, JPC = 12.1 Hz: quat Cp); 75.2 (d, JPC = 12.3 Hz: subst Cp); 75.5 (d, JPC = 94.8 Hz: quat Cp); 74.4 (d, JPC = 9.4 Hz: subst Cp); 72.3 (s, PhCH2O); 70.7 (s: Cp); 69.4 (d, JPC = 10.3 Hz: subst Cp); 66.7 (s: CpCH2O); 62.3 (d, JPC = 4.6 Hz: CH2 phosphonates); 48.7 (t, JPC = 132.7 Hz: CP2); 37.3 (d, JPC = 4.2 Hz: PhCH2); 16.2 (d, JPC = 4.9 Hz: CH3 phosphonates). 31P NMR (81.0 MHz, CDCl3), δ (ppm): 43.1 (PPh2); 25.7 (phosphonates). MS (FAB > 0; MNBA), C71H76Fe2O8P4S2, m/z: 1379 ([M + Na]+), 1356 ([M]+).
3.2.6 Synthesis and characterization of compound 4b
In a Schlenk tube, 180 mg of 4a (0.22 mmol) were dissolved in 5 mL of dry toluene with 0.24 mL of tris(dimethylamino)phosphine (1.3 mmol). The solution was kept at reflux overnight under argon. After cooling back to room temperature, the solution was evaporated in vacuo. The crude residue was purified under argon by flash chromatography on silica gel with dichloromethane as eluent. After solvent evaporation, 120 mg of 4b were obtained as an orange waxy solid (yield = 70%). 1H NMR (300.1 MHz, CDCl3), δ (ppm): 7.60–7.50 (2H, m: PPh2); 7.40–7.38 (3H, m: PPh2); 7.25–7.23 (5H, m: PPh2); 7.13 (2H, d, J = 8.1 Hz: Ph); 6.95 (2H, d, J = 8.1 Hz: Ph); 4.58 (1H, dd, J = 10.8 Hz, JPH = 2.2 Hz: CpCH2); 4.54 (1H, m: subst Cp); 4.38 (1H, d, J = 10.8 Hz: CpCH2); 4.32 (2H, s: PhCH2O); 4.31 (1H, m: subst Cp); 4.11 (8H, quint., JHH = JPH = 6.9 Hz: CH2 phosphonates); 4.02 (5H, s: Cp); 3.79 (1H, m: subst Cp); 3.21 (2H, t of d, J = 16.3 Hz, JPH = 6.2 Hz: PhCH2); 2.62 (1H, t of t, J = 6.2 Hz, JPH = 24.0 Hz: CHP2); 1.28 (12H, t of d, J = 6.9 Hz, JPH = 4.9 Hz: CH3 phosphonates). 13C NMR (75.5 MHz, CDCl3), δ (ppm): 140.0 (d, JPC = 9.6 Hz: quat PPh2); 138.6 (t, JPC = 7.1 Hz: quat Ph); 137.5 (d, JPC = 8.4 Hz: quat PPh2); 136.7 (s: quat Ph); 135.1 (d, JPC = 21.3 Hz: PPh2); 132.4 (d, JPC = 18.1 Hz: PPh2); 129.2 (s: PPh2); 128.7 (s: Ph); 128.1 (d, JPC = 8 Hz: PPh2); 128.0 (d, JPC = 6.2 Hz: PPh2); 127.7 (s: PPh2); 127.4 (s: Ph); 89.2 (d, JPC = 24.6 Hz: quat Cp); 76.6 (d, JPC = 8.0 Hz: quat Cp); 72.6 (d, JPC = 3.5 Hz: subst Cp); 72.0 (s, PhCH2O); 71.9 (d, JPC = 3 Hz: subst Cp); 69.9 (s: subst Cp); 69.6 (s: Cp); 67.4 (d, JPC = 10.0 Hz: CpCH2); 62.6 (d, JPC = 6.7 Hz: CH2 phosphonates); 62.5 (d, JPC = 6.7 Hz: CH2 phosphonates); 39.1 (t, JPC = 132.1 Hz: CHP2); 30.9 (t, JPC = 4.3 Hz: PhCH2); 16.3 (d, JPC = 6.3 Hz: CH3 phosphonates); 31P NMR (121.5 MHz, CDCl3), δ (ppm): 23.1 (phosphonates); -22.1 (PPh2). MS (FAB > 0; MNBA), C40H49FeO7P3, m/z: 829 ([M + K]+), 790 ([M]+).
3.2.7 Synthesis and characterization of compound 5a
In a schlenk tube, 100 mg of 3 were reacted with 100 μL N-methylimidazole in 15 mL of refluxing toluene. After 8 h, the solution was cooled down to room temperature. The precipitate was filtered off and washed with toluene then ether and dried in vacuo. The product 5a was obtained as a yellow solid (yield = 90 mg, 80%). 1H NMR (200.1 MHz, MeOD), δ (ppm): 7.89–7.77 (2H, m: PPh2); 7.69–7.35 (8H, m: PPh2; 2H: imidazolium); 7.28 (2H, d, J = 8.1 Hz: Ph); 7.05 (2H, d, J = 8.1 Hz: Ph); 5.39 (2H, s: PhCH2N); 5.07 (1H, d, J = 10.8 Hz: CpCH2); 4.72 (1H, m: subst Cp); 4.48 (1H, m: subst Cp); 4.39 (1H, d, J = 10.8 Hz: CpCH2); 4.38 (2H, s: PhCH2O); 4.33 (5H, s: Cp); 3.96 (3H, s: CH3 imidazolium); 3.89 (1H, m: subst Cp). Imidazolium NCHN signal is observed in CDCl3 at 10.8 (1H, s). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 140.49 (s: quat Ph); 133.4 (s: quat Ph); 138.0 (s: NCHN imidazolium); 134.2 (d, JPC = 84.9 Hz: quat PPh2); 133.6 (d, JPC = 86.5 Hz: quat PPh2); 132.7 (d, JPC = 8.0 Hz: PPh2); 132.5 (d, JPC = 8.1 Hz: PPh2); 132.0 (d, JPC = 2.6 Hz: PPh2); 131.7 (d, JPC = 2.6 Hz: PPh2); 129.0 (s: Ph); 128.9 (s: Ph); 128.7 (d, JPC = 4.2 Hz: PPh2); 128.4 (d, JPC = 4.2 Hz: PPh2); 124.4 (s: CH imidazolium); 122.9 (s: CH imidazolium); 88.0 (d, JPC = 12.0 Hz: quat Cp); 76.0 (d, JPC = 12.0 Hz: subst Cp); 75.6 (d, JPC = 9.1 Hz: subst Cp); 74.7 (d, JPC = 92.0 Hz: quat Cp); 72.0 (s, PhCH2O); 71.1 (s: Cp); 70.3 (d, JPC = 10.3 Hz: subst Cp); 67.4 (s: CpCH2O); 53.2 (s: PhCH2N); 35.4 (s: CH3 imidazolium). 31P NMR (81.0 MHz, MeOD), δ (ppm): 43.0. MS (FAB > 0; MNBA), C35H34FeN2OPS, m/z: 617 ([M]+).
3.2.8 Synthesis and characterization of compound 5b
In a Schlenk tube, 38 mg of 5a (3.47 mmol) were reacted with 2 g of Raney nickel powder (W.R. Grace and Co. Raney®2800, slurry, in H2O) in 6 mL of acetonitrile at 50 °C. Twenty-four hours later the solution was filtered. After evaporation of the solvent, 5b was obtained as a yellow powder. 1H NMR (200.1 MHz, CDCl3), δ (ppm): 10.7 (1H, s: NCHN imidazolium); 7.61–7.52 (2H, m: PPh2; 1H: imidazolium); 7.43–7.37 (4H, m: PPh2); 7.25–7.22 (4H, m: PPh2; 2H: Ph); 7.11 (1H, br s: imidazolium); 6.99 (2H, d, J = 8.0 Hz: Ph). 5.5 (2H, s: PhCH2N); 4.68 (1H, dd, J = 10.8 Hz, JPH = 2.2 Hz: CpCH2); 4.56 (1H, m: subst Cp); 4.41 (1H, d, J = 10.8 Hz: CpCH2); 4.34 (2H, s: PhCH2O); 4.33 (1H, m: subst Cp); 4.06 (3H, s: CH3 imidazolium); 4.03 (5H, s: Cp); 3.81 (1H, m: subst Cp). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 140.3 (s: quat Ph); 140.1 (d, JPC = 9.5 Hz: quat PPh2); 138.0 (s: NCHN imidazolium); 137.3 (d, JPC = 7.4 Hz: quat PPh2); 135.1 (d, JPC = 21.1 Hz: PPh2); 132.4 (d, JPC = 17.9 Hz: PPh2); 131.3 (s: quat Ph); 129.3 (s: PPh2); 129.0 (s: Ph); 128.5 (s: Ph); 128.2 (d, JPC = 7.2 Hz: PPh2); 128.1 (d, JPC = 5.3 Hz: PPh2); 127.9 (s: PPh2); 123.0 (s: CH imidazolium); 121.5 (s: CH imidazolium); 88.7 (d, JPC = 25.5 Hz: quat Cp); 72.9 (d, JPC = 2.9 Hz: subst Cp); 72.2 (d, JPC = 3.0 Hz: subst Cp); 71.5 (s, PhCH2O); 70.1 (s: subst Cp); 69.6 (s: Cp); 67.9 (d, JPC = 9.7 Hz: CpCH2); 53.4 (s: CH2N); 36.8 (s: CH3 imidazolium). 31P NMR (81.0 MHz, CDCl3), δ (ppm): -22.4.
3.2.9 Synthesis and characterization of compound 6a
In a Schlenk tube, 720 mg of monomethylether mPEG 750 were reacted with a suspension of 210 mg of sodium hydride (60% dispersion in mineral oil) in 6 mL of THF. After 1 h at room temperature, the solution was transferred via cannula on 90 mg of 3 dissolved in 4 mL of THF. Twenty-four hours later, 10 mL of a saturated aqueous solution of ammonium chloride were added and the product was extracted with ethyl acetate and washed with water saturated with NaCl. After solvent evaporation, 110 mg of 6a were obtained as a yellow wax (yield = 60%). 1H NMR (200.1 MHz, CDCl3), δ (ppm): 7.87–7.63 (4H, m: PPh2); 7.55–7.30 (6H, m: PPh2); 7.18 (2H, d, J = 8.0 Hz: Ph); 6.99 (2H, d, J = 8.0 Hz: Ph); 4.92 (1H, d, J = 10.8 Hz: CpCH2); 4.64 (1H, m: subst Cp); 4.52 (2H, s: PhCH2O); 4.42 (1H, d, J = 10.8 Hz: CpCH2); 4.31 (5H, s: Cp); 4.29 (2H, s: PhCH2O); 4.29 (1H, m: subst Cp); 3.81 (1H, m: subst Cp); 3.75–3.55 (64H, m, CH2-PEG); 3.38 (3H, s, CH3O-PEG). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 137.8 (s: quat Ph); 137.1 (s: quat Ph); 134.8 (d, JPC = 87.3 Hz: quat PPh2); 133.6 (d, JPC = 85.5 Hz: quat PPh2); 132.1 (d, JPC = 10.9 Hz: PPh2); 132.0 (d, JPC = 10.9 Hz: PPh2); 131.2 (d, JPC = 2.5 Hz: PPh2); 131.1 (d, JPC = 2.5 Hz: PPh2); 128.1 (d, JPC = 12.4 Hz: PPh2); 128.0 (d, JPC = 12.4 Hz: PPh2); 127.8 (2 C, s: Ph); 88.1 (d, JPC = 12.0 Hz: quat Cp); 75.2 (d, JPC = 12.6 Hz: subst Cp); 74.6 (d, JPC = 95 Hz: quat Cp); 74.5 (d, JPC = 9.3 Hz: subst Cp); 73.0 (s: PhCH2-PEG); 72.2 (s: CH2); 71.9 (s: CH2); 70.5 (s: Cp); 70.5 (s: CH2-PEG); 69.4 (d, JPC = 10.1 Hz: subst Cp); 69.2 (s: CH2-PEG); 66.8 (s: CH2); 59.1 (s: CH3O-PEG). 31P NMR (81.0 MHz, CDCl3), δ (ppm): 43.0.
3.2.10 Synthesis and characterization of compound 6b
In a Schlenk tube, 80 mg of 6a were dissolved in 2 mL of dry toluene with 0.40 mL of tris(dimethylamino)phosphine. The solution was kept at reflux overnight under argon. After cooling back to room temperature, the solution was evaporated in vacuo. The crude residue was purified under argon by flash chromatography on silica gel with dichloromethane as eluent. After evaporation of the solvent, 35 mg of 6b were obtained as an orange waxy solid (yield = 45%). 1H NMR (200.1 MHz, CDCl3), δ (ppm): 7.62–7.52 (2H, m: PPh2); 7.41–7.36 (3H, m: PPh2); 7.25–7.23 (5H, m: PPh2); 7.18 (2H, d, J = 8.1 Hz: Ph); 6.98 (2H, d, J = 8.1 Hz: Ph). 4.60 (1H, dd, J = 10.9 Hz, J = 2.3 Hz: CpCH2); 4.55 (1H, m: subst Cp); 4.52 (2H, s: PhCH2O); 4.38 (1H, d, J = 10.9 Hz: CpCH2); 4.33 (2H, s: PhCH2O); 4.31 (1H, m: subst Cp); 4.02 (5H, s: Cp); 3.78 (1H, m: subst Cp); 3.66–3.65 (64H, m, CH2-PEG); 3.39 (3H, s, CH3O-PEG). 13C NMR (50.3 MHz, CDCl3), δ (ppm): 139.9 (d, JPC = 8.9 Hz: quat PPh2); 137.8 (s: quat Ph); 137.4 (d, JPC = 8.2 Hz: quat PPh2); 137.1 (s: quat Ph); 135.1 (d, JPC = 20.6 Hz: PPh2); 132.4 (d, JPC = 17.9 Hz: PPh2); 129.1 (s: PPh2); 128.1 (d, JPC = 7.4 Hz: PPh2); 128.0 (d, JPC = 5.5 Hz: PPh2); 127.7 (s: PPh2); 127.7 (s: Ph); 127.5 (s: Ph); 89.1 (d, JPC = 25.0 Hz: quat Cp); 76.7 (d, JPC = 8.0 Hz: quat Cp); 73.1 (s: PhCH2-PEG); 72.8 (d, JPC = 3.6 Hz: subst Cp); 72.0 (d, JPC = 3.7 Hz: subst Cp); 72.0 (s: CH2); 70.9 (s: CH2-PEG); 69.8 (s: subst Cp); 69.5 (s: Cp); 67.5 (s, JPC = 9.7 Hz: CpCH2); 59.1 (s: CH3O-PEG). 31P NMR (81.0 MHz, CDCl3), δ (ppm): −22.0.
3.3 General procedure for coupling reaction of aryl bromides with boronic acids
In a typical procedure for coupling reaction (Scheme 2), ligand 2b (6 mg, 0.012 mmol, 1.2 mol %), Pd2(dba)3 (5 mg, 0.005 mmol, 1.1 mol %), phenylboronic acid (146 mg, 1.2 mmol) and cesium carbonate (750 mg, 2.030 mmol, 2.3 mol %) were introduced into a Schlenk tube under an argon stream. A solution of bromobenzene (157 mg, 1.00 mmol) in 10 mL of dioxane was added into the Schlenk tube via a syringe. The reaction mixture was stirred at 70 °C for 8 h under an Ar atmosphere. Aliquots (0.5 mL) of the solution were taken using a polypropylene syringe. One milliliter of water was added in each collected samples. After extraction by two portions of 1 mL of diethylether, the organic phase was then filtered on silica and analysed by GPC using diethylene glycol dibutyl ether as internal standard.
3.4 X-ray diffraction studies
A single crystal of compound 3 was mounted under inert perfluoropolyether at the tip of a glass fibre and cooled in the cryostream of a Bruker APEXII CCD diffractometer. Data were collected using the monochromatic MoKα radiation (λ = 0.71073). The structure was solved by direct methods (SIR97 [41]) and refined by least-squares procedures on F2 using SHELXL-97 [42]. All H atoms attached to carbon were introduced in calculation in idealized positions and treated as riding models. There is half of a dichloromethane solvate per asymmetric unit located around a twofold axis. The drawing of the molecules was realized with the help of ORTEP32 [43]. Crystal data and refinement parameters are shown in Table 1. Crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 745960. Copies of the data can be obtained free of charge on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223 336 033; e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgements
The authors thank LCC CNRS for financial support, the Chemistry Department of “IUT A Paul-Sabatier” for financial support and for offering accesses to some analytical equipment, and L. Orts and T. Scandella for laboratory assistance.