

1 Introduction
Malaria, an infectious disease transmitted by mosquitoes and caused by Plasmodium parasites is responsible for the contamination of more than 300 million people and the death of more than one million people each year 〚1〛. Although the disease has been successfully contented after World War II because of the easy access to a cheap insecticide like DDT and a readily available drug such as chloroquine, its incidence is now dramatically increasing, mainly because of the widespread resistance of many Plasmodium falciparum strains to widely used drugs 〚2〛. Given this loss of activity of almost all classical antimalarial, there is an urgent need for a highly effective, well-tolerated and affordable drug.
As recommended by the WHO, the combination of at least two drugs having different modes of action must be used to prevent the development of drug resistance 〚3〛. We therefore adopted a covalent bitherapy strategy by preparing new molecules named trioxaquines by covalently attaching a trioxane, the moiety responsible for the activity of artemisinin, to a 4-aminoquinoline entity, a key constituent of chloroquine 〚4〛. DU-1102, the first trioxaquine 1 (Fig. 1), proved very active in vitro on chloroquine sensitive and chloroquine resistant Plasmodium falciparum, both on laboratory strains 〚5〛 and on human isolates 〚6〛. DU-1102 is a modular molecule that can be prepared through a convergent synthesis 〚5〛. Many modulations are easy to do at the different steps of the synthesis, leading to a large variety of trioxaquines.
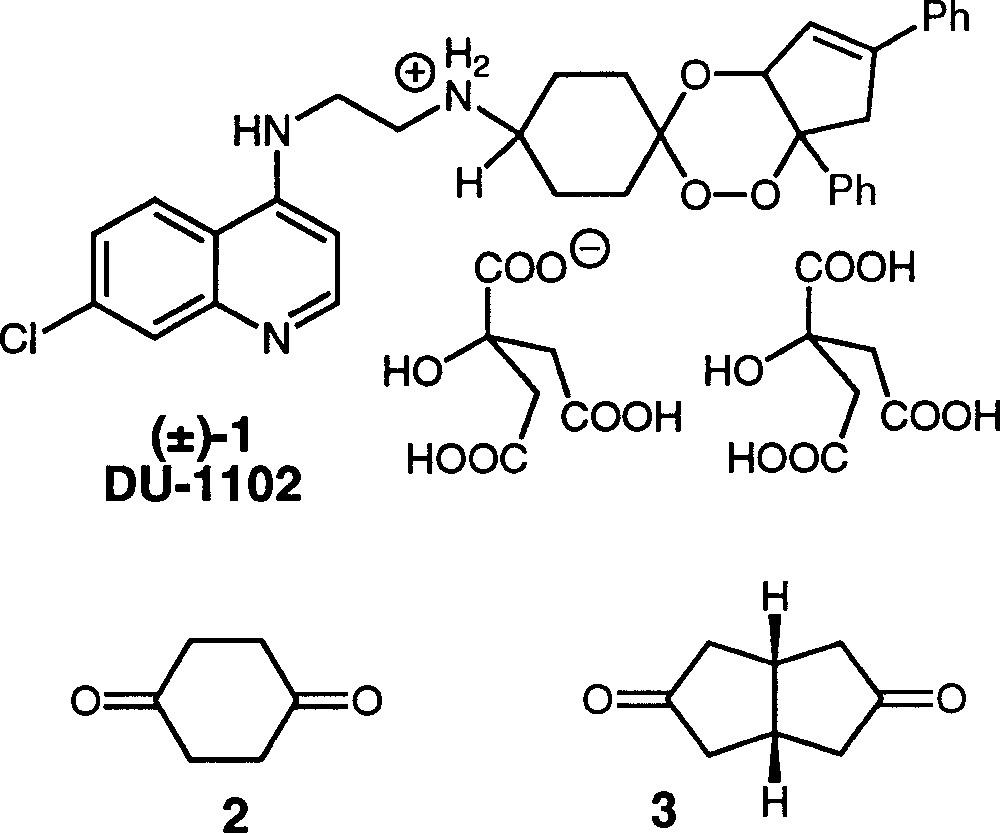
Structures of trioxaquine DU-1102 (±)-1, 1,4-cyclohexanedione 2 and cis-bicyclo〚3.3.0〛octane-3,7-dione 3.
We now report the synthesis and the NMR study of new trioxaquines prepared from the cis-bicyclo〚3.3.0〛octane-3,7-dione 3 (Fig. 1) in place of the 1,4–cyclohexanedione 2 used to prepare DU-1102 1.
2 Results and discussion
Cis-bicyclo〚3.3.0〛octane-3,7-dione 3 is a cyclic symmetric diketone constituted of two fused cyclopentanones, and hence is in a fixed conformation, unlike 1,4–cyclohexanedione 2, which undergoes rapid conformational changes between two chair-conformations. This diketone 3 reacted with the endoperoxide of 1,4-diphenyl-1,3–cyclopentadiene 5, in dichloromethane in the presence of Me3SiOTf as Lewis acid catalyst, to provide two diastereomeric trioxane-ketones (±)-6a and (±)-6b (Fig. 2). As previously reported for the condensation of the same endoperoxide 5 but with cyclopentanone 〚7〛, the trioxane and the cyclopentene cycles were cis-fused, H5 and phenyl-C9 being contiguous, giving rise only to (5R, 9R) and (5S, 9S) configurations. In addition, a pseudo-asymmetric centre was created at the spiro carbon C3. As a result, four stereoisomers were obtained as two diastereomeric racemates (±)-6a and (±)-6b. The compound (±)-6a has the endoperoxide function on the same side of the mean plane defined by C10–C12–C14–C16 than the apical hydrogen atoms H11 and H15, and is named exo racemate (by extrapolation of the endo/exo nomenclature). The endo racemate (±)-6b has the endoperoxide on the opposite side than H11 and H15 with respect to the C10–C12–C14–C16 plane.
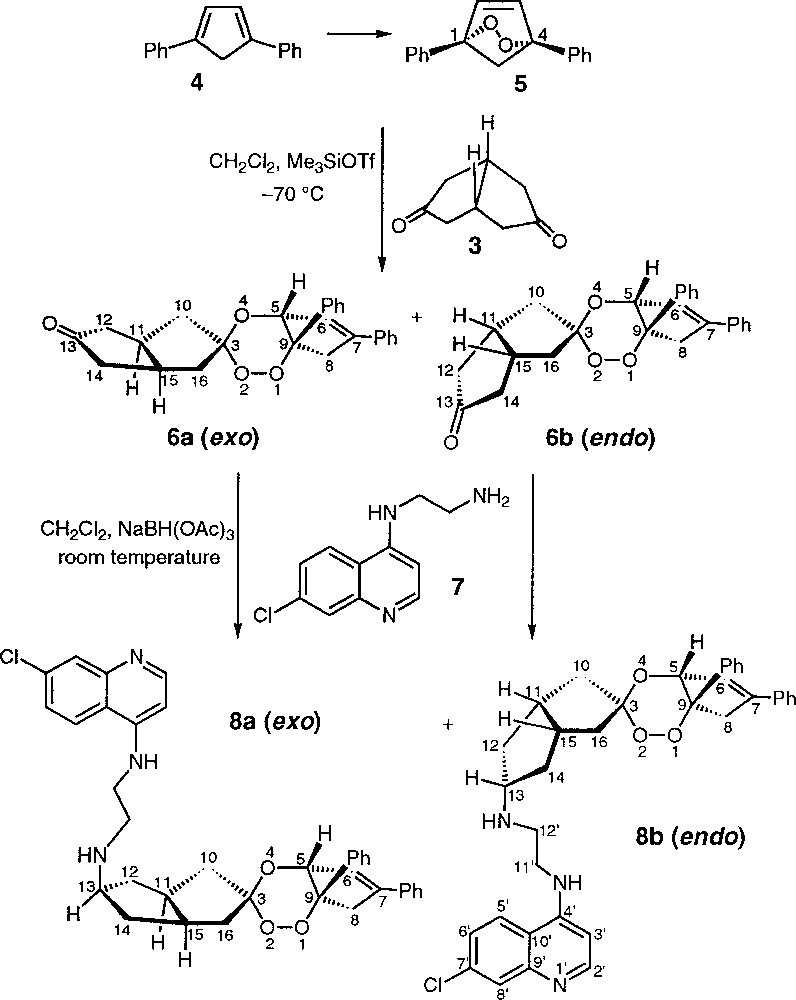
Synthesis of the two diastereomeric racemates of the trioxane-ketones 6a and 6b, and of the two diastereomeric racemates of the trioxaquines 8a, DU-1111, and 8b, DU-1113 – only the enantiomers with configuration (5R, 9R) are depicted.
The two diastereomeric trioxane-ketones (±)-6a and (±)-6b were separated by chromatography on silica gel, using hexane/ethyl acetate (70:30, v/v) as eluent. Compound (±)-6a was eluted first, and then (±)-6b (ratio 6a/6b = 60/40, yield 42 %).
A detailed NMR study of each of the diastereomeric racemates (±)-6a and (±)-6b was carried out to determine their stereochemistry. The case of (±)-6b is presented in Fig. 3. First, the 1H and 13C NMR shifts were assigned by 1H–1H and 1H–13C correlation experiments and the methylene H16 protons were detected at 1.79 and 2.28 ppm (main proton shifts and atom numbering are depicted in Fig. 3A). Then, the stereochemistry HR or HS of H16 observed at 1.79 ppm was established by 1D-NOE selective excitation: this signal was not correlated with the signals of the apical H11 and H15, at 2.94 and 2.87 ppm respectively, suggesting that H16 at 1.79 ppm is HR (Fig. 3B). Finally, selective excitation of the H5 signal at 5.25 ppm produced a more intense NOE on the signal of H16 at 2.28 ppm, then assigned to be HR, than on the HS at 1.79 ppm. These results supported the attribution of (±)-6b as being the endo diastereomeric racemate having the peroxide function on the opposite side than H11 and H15 with respect to the C10–C12–C14–C16 plane (Fig. 3C). A similar study on the (±)-6a isomer should have permitted to confirm this structural proposal: unfortunately, in this case, one of the H10 protons has the same shift that one of the apical H, preventing all NOE experiments from being interpreted. The exo structure for (±)-6a was therefore determined by deduction.
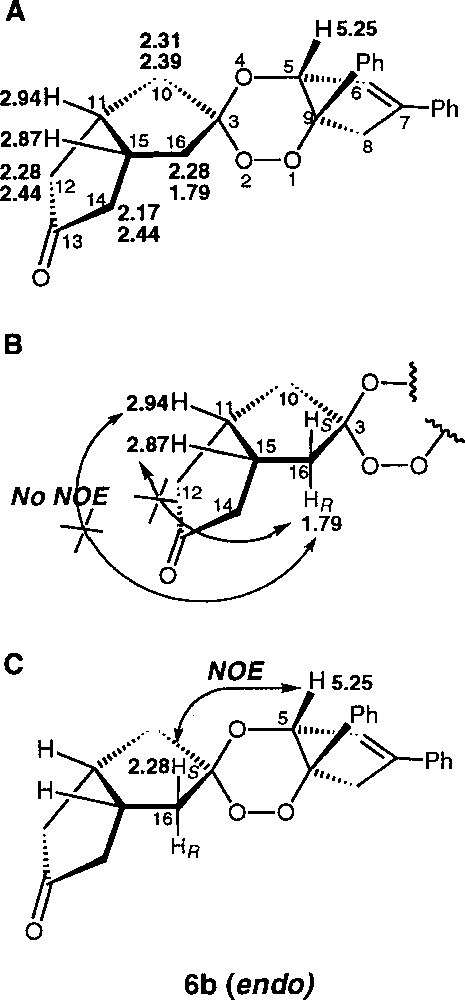
Selected NMR data for trioxane-ketone (±)-6b.
It should be noted that the interpretations of NOE signals are not straightforward because of the fluxionality of the trioxane cycle. Despite attempts in different solvents (dichloromethane, tetrahydrofurane, dimethylformamide, ethyl acetate), it has not been possible to obtain monocrystals of one diastereomer suitable for an X-ray analysis.
The next step in the trioxaquine synthesis was the reductive amination of the trioxane functionalised by a ketone (±)-6a or (±)-6b with the quinoline-amine 7 (Fig. 2), followed by a protonation of the trioxaquine base forms (±)-8a or (±)-8b with citric acid to produce the corresponding monocitrate salts (±)-9a or (±)-9b, having an enhanced solubility (Fig. 4). The reductive amination of (±)-6 involves the initial formation of an iminium ion, which is subsequently reduced by NaBH(OAc)3 to produce (±)-8 (only the stereoisomers with configuration (5R, 9R) are depicted in Fig. 2〛. In fact, the reaction carried out with each one of the racemates (±)-6a and (±)-6b, separately, showed the same high diastereoselectivity, a single trioxaquine racemate being obtained in each case. The reductive amination of the trioxane-ketone (±)-6a provided the trioxaquine (±)-8a (DU-1111) and the reductive amination of the trioxane-ketone (±)-6b provided the trioxaquine (±)-8b (DU-1113). Compounds (±)-8a and (±)-8b result from an exo attack of the hydride at C13, as it has already been reported for the reductive amination of norcamphor or tropinone by benzylamine 〚8〛. This feature results in an endo stereochemistry of the quinoline-amine substituent with respect to the bis-cyclopentane system. Trioxane-ketones (±)-6a and (±)-6b exhibited a very poor reactivity in this reductive amination step compared to that of the trioxane-ketone prepared from 1,4-cyclohexanedione whose reductive amination was completed in 18 h 〚5〛. In the present case, the reductive amination was accomplished in three weeks for (±)-6a and in one week for (±)-6b. Despite this sluggish reactivity, no side reaction occurred. Then, the reaction mixtures were washed with water, the trioxaquines were recovered from the organic phase, and no further purification was needed. It has been reported that the torsion angle constraint for a cyclopentanone ring increases during the reductive amination reaction, which is unfavourable, whereas the torsion angle constraint for a cyclohexanone ring decreases in the same reaction. Consequently, cyclohexanone was found to be 23 times more reactive than cyclopentanone in reductive amination reactions 〚9〛. Such a phenomenon may explain the poor reactivity of trioxane-ketones (±)-6a and (±)-6b.
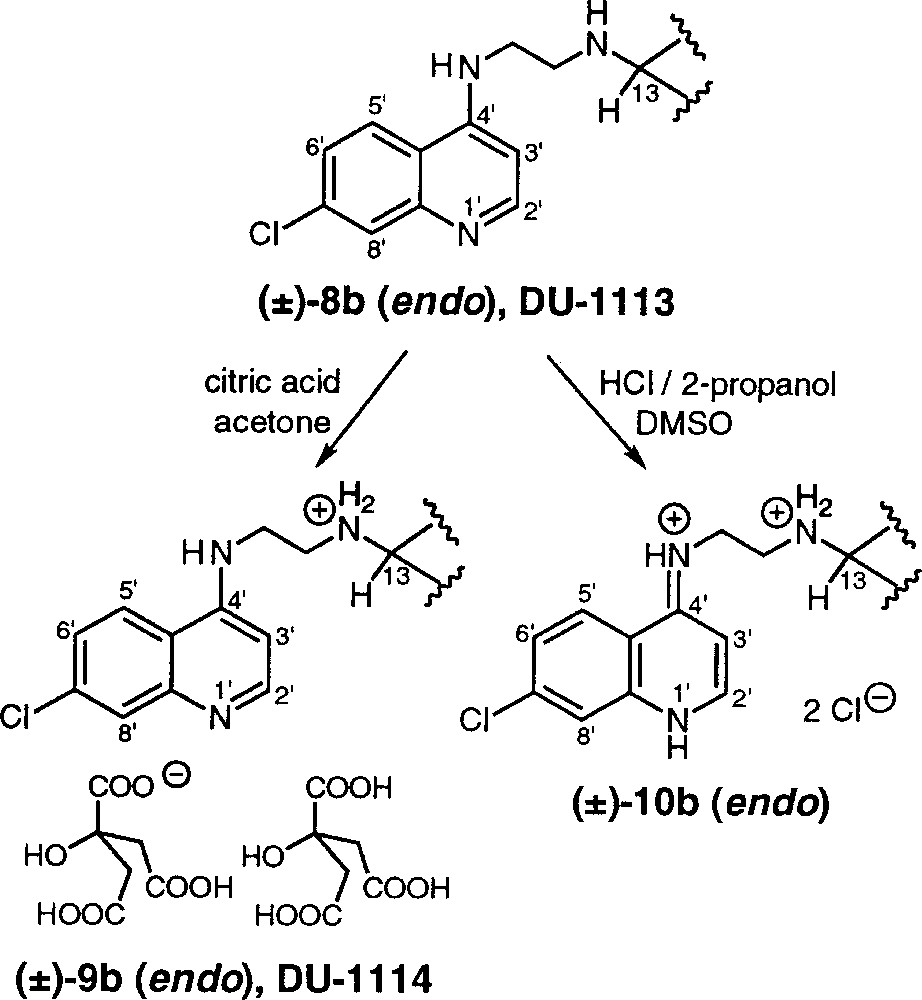
Structures of the trioxaquine citrate (±)-9b, DU-1114, and trioxaquine dihydrochloride (±)-10b.
The final protonation step was carried out as previously reported for trioxaquine DU-1102 〚5〛: the trioxaquine citrates (±)-9a (DU-1112) and (±)-9b (DU-1114) were prepared from (±)-8a and (±)-8b, respectively, by addition of citric acid (2.5–3 mol equiv) in acetone, and subsequent precipitation of the formed mono-protonated (at –NH–C13 position) trioxaquine derivative (Fig. 4). Under these experimental conditions, the mono-protonated trioxaquine readily co-precipitated with one citric acid molecule, thus preventing di-protonation. The non-protonation of the quinoline nucleus in the citrate salts (±)-9 was attested by 1H NMR in DMSO-d6. For example, the chemical shifts of the quinoline protons of (±)-9b was close to those observed for the trioxaquine (±)-8b: 8.34 compared to 8.22 ppm, 7.95 compared to 7.77 ppm, and 6.73 compared to 6.48 ppm for H5’, H8’ and H3’ respectively (Δδ ≤ +0.25 ppm). On the opposite, by addition of HCl (5 M in 2-propanol) in a solution of trioxaquine (±)-8b in DMSO, the protonation of the quinoline nucleus was evidenced by a more intense downfield shift of H5’, H8’ and H3’ detected at 8.90, 8.18 and 7.00 ppm, respectively (Δδ = +0.68, +0.41 and +0.52 ppm). In addition, the HN–C4’ was detected at 7.20 and 7.51 ppm for the base (±)-8b and the citrate (±)-9b, respectively; whereas HN1’ and HN+=C4’ were at 14.81 and 9.96 ppm, respectively, for the dihydrochloride salt obtained in the presence of HCl in DMSO – the assignment of HN1’ signal in this latter case was confirmed by scalar coupling with H2’ signal at 8.62 ppm (3J = 5 Hz).
3 Conclusion
Modulations in the synthesis of trioxaquines can be done either to improve antimalarial activity, solubility and preparation yield, or to establish a structure–activity relationship.
In the present case, cis-bicyclo〚3.3.0〛octane-3,7-dione 3 is more expensive and less reactive than 1,4-cyclohexanedione 2, and trioxaquines DU-1112 and DU-1114 are unfortunately less soluble in physiological serum than DU-1102 itself. However, the condensation of the endoperoxide derived from 1,4-diphenyl-1,3-cyclopentadiene with cis-bicyclo〚3.3.0〛octane-3,7-dione 3, a cyclic diketone in a fixed conformation, allowed us to separate two diastereomeric racemates of the trioxane-ketones 6, and therefore of the corresponding trioxaquines, unlike it was when the fluxional 1,4-cyclohexadione was used. The comparison of the antimalarial activity of these two trioxaquine diastereomeric racemates DU-1112 and DU-1114 is now under investigation. It will show whether the stereochemistry of the C3 spiro junction influences the biological activity or not.
4 Experimental section
4.1 Synthesis of the trioxanes (±)-6a and (±)-6b
The crude peroxide 5 (0.7 mmol) was solubilised in dichloromethane (5 ml) and the solution cooled to –70 °C, cis-bicyclo〚3.3.0〛octane-3,7-dione 3 (410 mg, 3.0 mmol, 4.2 equiv) and Me3SiOTf (0.05 ml, 0.3 mmol, 0.4 equiv) were added and the reaction was stirred at –70 °C for 2 h. Finally, Et3N (0.1 ml) was added and the solution allowed to warm to ambient temperature. After addition of H2O, the organic phase was separated, dried on MgSO4, and evaporated. The obtained brownish oil was purified by column chromatography (SiO2, hexane/ethyl acetate, 70:30, v/v). Two diastereomeric racemates were separated on the column: (±)-6a was eluted first, and then (±)-6b (6a/6b molar ratio = 60/40, global yield = 42 %). For the numbering of atoms, see Fig. 2.
4.1.1 Trioxane (±)-6a
1H NMR (400 MHz, CDCl3) δ, ppm: 1.76 (m, 1H, H2C10), 1.81 (m, 1H, H2C16), 2.15 (m, 1H, H2C14), 2.23 (m, 2H, 1H H2C12 and 1H H2C16), 2.49 (m, 1H, H2C12), 2.53 (m, 1H, H2C14), 2.72 (m, 1H, HC15), 2.83 (m, 2H, 1H H2C10 and HC11), 3.02 and 3.21 (2 × d, 2 × 1H, 2J = 17.0 Hz, H2C8), 5.27 (br s, 1H, HC5), 6.29 (d, 1H J = 2.0 Hz, HC6), 7.30–7.50 (m, 8H, HCphenyl), 7.62 (m, 2H, HCphenyl).
13C NMR (100.6 MHz, CDCl3) δ, ppm: 36.0 (C15H); 38.0 (C11H), 41.4 (C10H2), 42.7 (C16H2), 44.0 (C12H2), 44.4 (C8H2 and C14H2), 80.1 (C5H), 87.2 (C9), 112.7 (C3), 124.5 (C6H), 125.8, 126.2, 127.9, 128.4, 128.6, and 134.5 (6 × Cphenyl), 141.2 (C7), 219.9 (C13).
MS (DCI/NH3+) m/z (%): 406 (MNH4+, 100), 407 (30), 408 (8).
4.1.2 Trioxane (±)-6b
1H NMR (400 MHz, CDCl3) δ, ppm: 1.79 (m, 1H, H2C16), 2.17 (m, 1H, H2C14), 2.28 (m, 2H, 1H H2C12 and 1H H2C16), 2.31 (m, 1H, H2C10), 2.39 (m, 1H, H2C10), 2.44 (m, 2H, 1H H2C14 and 1H H2C12), 2.87 (m, 1H, HC15), 2.94 (m, 1H, HC11), 3.02 and 3.22 (2 × d, 2 × 1H, 2J = 17.0 Hz, H2C8), 5.25 (br s, 1H, HC5), 6.32 (d, 1H, J = 2.0 Hz, HC6), 7.30–7.50 (m, 8H, HCphenyl), 7.60 (m, 2H, HCphenyl).
13C NMR (100.6 MHz, CDCl3) δ, ppm: 36.2 (C15H), 38.0 (C11H), 41.7 (C10H2), 42.8 (C16H2), 44.0 (C12H2), 44.3 (C8H2), 44.7 (C14H2), 80.1 (C5H), 87.2 (C9), 113.2 (C3), 124.3 (C6H), 125.9, 126.2, 127.9, 128.4, 128.6, 134.5 (6 × Cphenyl), 141.3 (C7), 220.1 (C13).
MS (DCI/NH3+) m/z (%): 404 (3), 405 (3), 406 (MNH4+, 100), 407 (31), 408 (6).
4.2 Synthesis of the trioxaquines (±)-8a, DU-1111, and (±)-8b, DU-1113
4.2.1 Trioxaquine (±)-8a, DU-1111
The trioxane (±)-6a (163 mg, 0.42 mmol) and the quinoline-amine 7 (120 mg, 0.54 mmol) were dissolved in dichloromethane (15 ml) before addition of sodium triacetoxyborohydride (114 mg, 0.54 mmol). The reaction was stirred at room temperature for three weeks and then washed with distilled water. The organic layer was dried and evaporated under vacuum to dryness (yield = 66 %). For the numbering of atoms, see Fig. 2.
1H NMR (300 MHz, CDCl3) δ, ppm : 1.15 (m, 2H, H2Cbicyclopentyl), 1.75 (m, 2H, H2Cbicyclopentyl), 2.0–2.7 (m, 6H, 4H H2Cbicyclopentyl, HNC13, and HC13), 2.78 (m, 1H, HC15 or HC11), 3.00 (m, 3H, 1H H2C8 and 2H H2C12’), 3.15 (m, 1H, HC11 or HC15), 3.30 (m, 3H, 1H H2C8 and 2H H2C11’), 5.28 (br s, 1H, HC5), 5.96 (br s, 1H, HNC4’), 6.28 (d, 1H, HC6), 6.38 (d, 1H, 3J = 5.4 Hz, HC3’), 7.30–7.65 (m, 11H, 10 HCphenyl and HC6’), 7.75 (d, 1H, 3J = 9.0 Hz, HC5’), 7.95 (d, 1H, 4J = 2.1 Hz, HC8’), 8.51 (d, 1H, 3J = 5.4 Hz, HC2’).
13C NMR (75.4 MHz, CDCl3) δ, ppm: 38.2 and 40.0 (C15H and C11H), 41.1, 41.4, 42.3 and 42.9 (C10H2, C16H2, C12H2, and C14H2), 42.9 (C12’H2), 46.9 (C11’H2), 62.5 (C13H), 80.5 (C5H), 87.5 (C9), 99.6 (C3’H), 117.8 (C10’), 121.9 (C5’H), 125–130 (C6H, C6’H, C8’H, and Cphenyl), 135.1 and 135.2 (Cphenyl and C7’), 141.8 (C7), 149.4 (C9’), 150.3 (C4’), 152.4 (C2’H).
MS (DCI/NH3+) m/z: 594 (MH+).
4.2.2 Trioxaquine (±)-8b, DU-1113
The trioxane-ketone (±)-6b (148 mg, 0.38 mmol) and the quinoline-amine 7 (110 mg, 0.50 mmol) were dissolved in dichloromethane (15 ml) before addition of sodium triacetoxyborohydride (154 mg, 0.73 mmol). The reaction mixture was stirred at room temperature for one week and then washed with distilled water. The organic layer was dried and evaporated under vacuum to dryness (yield = 68%).
1H NMR (250 MHz, CDCl3) δ, ppm: 1.25 (m, 3H, H2Cbicyclopentyl), 1.77 (m, 1H, H2Cbicyclopentyl), 2.0–2.6 (m, 7H, 4H H2Cbicyclopentyl, HNC13, HC13, and 1H HC15 or HC11), 3.00 (m, 4H, 1H H2C8, 2H H2C12’, and 1H HC15 or HC11), 3.25 (m, 3H, 1H H2C8 and 2H H2C11’), 5.25 (br s, 1H, HC5), 6.06 (br s, 1H, HNC4’), 6.27 (m, 2H, HC6 and HC3’), 7.30–7.65 (m, 11H, 10 HCphenyl and HC6’), 7.73 (d, 1H, 3J = 9.0 Hz, HC5’), 7.88 (d, 1H, 4J = 2.1 Hz, HC8’), 8.48 (d, 1H, 3J = 5.4 Hz, HC2’).
1H NMR (250 MHz, DMSO-d6, selected data) δ, ppm: 6.48 (H6 and H3’), 7.20 (HN–C4’), 7.77 (H8’), 8.22 (H5’).
MS (DCI/NH3+) m/z (%): 594 (MH+, 100), 595 (44), 596 (44).
4.3 Synthesis of the trioxaquine citrates (±)-9a, DU-1112, and (±)-9b, DU-1114
4.3.1 Trioxaquine citrate (±)-9a, DU-1112
The trioxaquine (±)-8a (DU-1111, 40 mg, 0.067 mmol) was solubilised in acetone (2 ml) before adding a solution of citric acid (34 mg, 2.6 equiv) in acetone (2 ml). The trioxaquine citrate (±)-9a precipitated spontaneously. After centrifugation, the solid precipitate was washed with diethyl ether and dried under vacuum.
1H NMR (250 MHz, DMSO-d6) δ, ppm: 1.4–1.8 (m, 4H, H2Cbicyclopentyl), 2.0–2.6 (m, 6H, 4H H2Cbicyclopentyl, HC15, and HC11), 2.69 (d, 4H, H2CCOOH citric acid), 2.79 (d, 4H, H2CCOOH citric acid), 3.0–3.8 (m, 7H, H2C8, H2C12’, H2C11’, and HC13), 5.53 (br s, 1H, HC5), 6.59 (m, 1H, HC6), 6.79 (d, 1H, HC3’), 7.54 (m, 6H, HCphenyl), 7.74 (m, 5H, 4 HCphenyl and HC6’), 7.99 (d, 1H, HC8’), 8.42 (d, 1H, HC5’), 8.64 (d, 1H, HC2’).
MS (ES+) m/z: 594.3 (M+).
Anal. calc. for C48H52O17N3Cl, H2O: C 57.86, H 5.46, N 4.22; found: C 57.81, H 5.36, N 4.26.
4.3.2 Trioxaquine citrate (±)-9b, DU-1114
The trioxaquine (±)-8b (DU-1113, 153 mg, 0.26 mmol) was solubilised in acetone (5 ml) before adding a solution of citric acid (150 mg, 3.0 equiv) in acetone (5 ml). The trioxaquine citrate (±)-9b precipitated spontaneously. After centrifugation, the solid precipitate was washed with diethyl ether and dried under vacuum.
1H NMR (250 MHz, DMSO-d6) δ, ppm: 1.4–1.8 (m, 4H, H2Cbicyclopentyl), 2.0–2.6 (m, 6H, 4H H2Cbicyclopentyl, HC15, and HC11), 2.65 (d, 4H, H2CCOOH citric acid), 2.77 (d, 4H, H2CCOOH citric acid), 3.20 (m, 3H, 1H H2C8 and 2H H2C12’), 3.44 (m, 1H, HC13), 3.69 (m, 3H, 1H H2C8 and 2H H2C11’), 5.55 (br s, 1H, HC5), 6.60 (m, 1H, HC6), 6.73 (d, 1H, HC3’), 7.51 (m, 7H, 6H HCphenyl and 1H HN–C4’), 7.73 (m, 5H, 4H HCphenyl and HC6’), 7.95 (d, 1H, HC8’), 8.34 (d, 1H, HC5’), 8.60 (d, 1H, HC2’).
MS (ES+) m/z (%): 594.2 (M+), (ES–) m/z: 190.9 (citrate).
Anal. calc. for C48H52O17N3Cl, H2O: C 57.86, H 5.46, N 4.22; found: C 58.19, H 5.05, N 4.17.
4.3.3 Trioxaquine dihydrochoride (±)-10b
In a DMSO-d6 solution of the trioxaquine (±)-8b, HCl 5M in 2-propanol was added in large excess. The trioxaquine dihydrochloride was not isolated, but characterised by 1H NMR.
1H NMR (250 MHz, DMSO-d6, selected data) δ, ppm: 6.46 (br s, 1H, H6), 7.00 (d, 3J = 7.3 Hz, 1H, H3’), 8.18 (d, 3J = 2.4 Hz, 1H, H8’), 8.62 (dd, 3J = 7.3 and 5 Hz, 1H, H2’), 8.90 (d, 3J = 8.8 Hz, 1H, H5’), 9.96 (dd, 1H, HN+=C4’), 14.81 (br d, 3J = 5 Hz, 1H, HN1).
Acknowledgements
We are grateful to Dr. Y. Coppel (LCC–CNRS, Toulouse) for some NMR experiments and fruitful discussions.