

1 Introduction
Cyclopentadienyl molybdenum carbonyl halide complexes, CpMo(CO)2(L)X, have been extensively investigated since their first preparation (L = CO) in the early 1950’s 〚1〛. Since then modification of complexes of this type has been achieved by replacing the ring protons with other substituents or modifying the ligand set (CO, L, X), and this has resulted in the synthesis of many hundreds of complexes of this type 〚2,3〛. Extensive studies of these complexes have included thermal 〚4–8〛, photochemical 〚9,10〛 and induced 〚11–13〛 substitution reactions (of CO, L, X), matrix isolation studies 〚14〛, isomerization studies 〚10,15,16〛, etc. A significant breakthrough in this area of chemistry was the physical separation of the pure cis and trans isomers of (η5-C5H5)Mo(CO)2(PPh3)I in 1975 〚17〛.
We recently reported the synthesis of a series of related rhenium piano stool complexes, (η5-C5H4R)Re(CO)(L)X2 (R = Me, Et, iPr, tBu, SiMe3; L = CNC6H3Me2, P(OR)3, PPh3; X = Br, I) 〚18–20〛. Of particular importance was the finding that these complexes underwent isomerization reactions in the solid state. Other than a short comment on the solid-state isomerization reaction of (η5-C5H5)Mo(CO)2(PBu3)I 〚15〛 and a description of the cis/trans interconversion of Cp′M(CO)2(PMe3)GeX3 (Cp′ = C5H5, C5Me5; M = Mo, W) 〚21〛 no other solid-state isomerization reactions of four-legged piano stool complexes have been reported in the literature. Here we report results of the thermal solid-state isomerization reactions of (η5-C5H4Me)Mo(CO)2(P(OiPr)3)I to further explore the generality of the reaction.
2 Experimental section
(η5-C5H4Me)Mo(CO)3I was prepared by the standard procedure 〚22〛. Trimethylamine N-oxide dihydrate (Aldrich) was used as received. All reactions were carried out using standard Schlenk techniques under nitrogen. Solvents were dried by conventional methods, distilled under nitrogen, and used immediately. Melting points were recorded on a Kofler hot stage melting point apparatus. Solution infrared spectra were measured on a Bruker Vector 22 FTIR spectrometer, in KBr cells. NMR spectra were measured on a Bruker AC200 spectrometer operating at 200 MHz. DSC profiles were measured on a Du Pont 911 DSC instrument and TGA data were measured on a Perkin Elmer Pyris 1. Powder X-ray diffraction data were collected on a Phillips PW 1710 diffractometer using a Cu tube anode. Microanalyses were carried out at the CSIR, Pretoria, South Africa.
2.1 Preparation of cis- and trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I
The complex was prepared (70% total yield) by adapting the method developed by Blumer et al. 〚11〛 for the synthesis of the related unsubstituted compounds. Isomer separation was achieved by dissolving the crude material in CH2Cl2 followed by mixing with a small quantity of silica gel. The yellow powder left after removing the CH2Cl2 was chromatographed on a silica gel column (2 × 60 cm) with a 1:10 CH2Cl2/hexane mixture. The isomer ratio and total product yield (both isomers) of the new (η5-complexes obtained are summarized in Table 1. The spectroscopic and analytical data of the molybdenum complexes are given in Table 2.
Solid-state isomerization reactions of cis- and trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I complexes.
| Complex | Yield (%)a | DSC (°C)b | m.p. (°C)c | Reaction temperature and time | (Trans/cis) equilibrium ratio (%)d |
| cis-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I | 70 | 113ex, 124ex,135en | 122–124 | 90 °C, 100 min | 70:30 |
| trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I | 70 | 138en | 126–128 | 90 °C, 100 min | 70:30 |
a Isolated yield.
b Melting point determined by DSC (10 °C min–1): ex = exothermic. en = endothermic.
c Melting point determined by melting point apparatus.
d Determined by 1H NMR spectroscopy.
Spectroscopic and analytical data for (η5-C5H4Me)Mo(CO)2(P(OiPr)3)I complexes.
| Compound | νCO (cm–1)a | 1H NMR (ppm)b | Analysis (%) | ||||
| Cp | Me | Ligand | C | H | |||
| cis-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I | 1964 | 5.26 (m, 1H); 5.22 (m, 1H); 5.14–5.15 (d, 1H, JHH = 2 Hz); 5.09 (m, 1H) | 2.16 (s, 3H) | 4.58–4.68 (septet, 3H); 1.28–1.33 (m, 18H) | calcd | 36.06 | 4.99 |
| 1881 | found | 35.98 | 4.81 | ||||
| trans- (η5-C5H4Me)Mo(CO)2(P(OiPr)3)I | 1966 | 5.14–5.16 (m, 2H); 5.00-5.04 (m, 2H) | 2.25 (s, 3H) | 4.54-4.68 (septet, 3H); 1.31-1.34(d, 18H) | calcd | 36.06 | 4.99 |
| 1884 | found | 36.12 | 5.08 |
a Recorded in CH2Cl2.
b Recorded in CDCl3, relative to TMS: s, singlet; d, doublet; m, multiplet.
2.2 Solid-state isomerization of cis- and trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I
2.2.1 Reactions in NMR tubes
Reactions were carried out using 5–20 mg of the isomers packed into sealed NMR tubes under nitrogen and immersed in an oil bath at a pre-set temperature. Care was taken to ensure that the reactant was fully covered by the oil. Temperatures between 80 and 100 °C were chosen and the reaction was monitored at 20 min intervals. The product was analyzed by TLC, solution IR and 1H NMR spectroscopy. Because both isomers had similar red colors, the solid-state isomerization reactions of the complexes could not be monitored visually.
NMR spectroscopy was used to monitor the reaction. An example of the NMR spectra obtained for the cis and trans isomers and the equilibrium mixtures (from solid state reactions) of the isomers are shown in Fig. 1 and the intensity data as a function of time (Me resonance) plotted in Fig. 2.
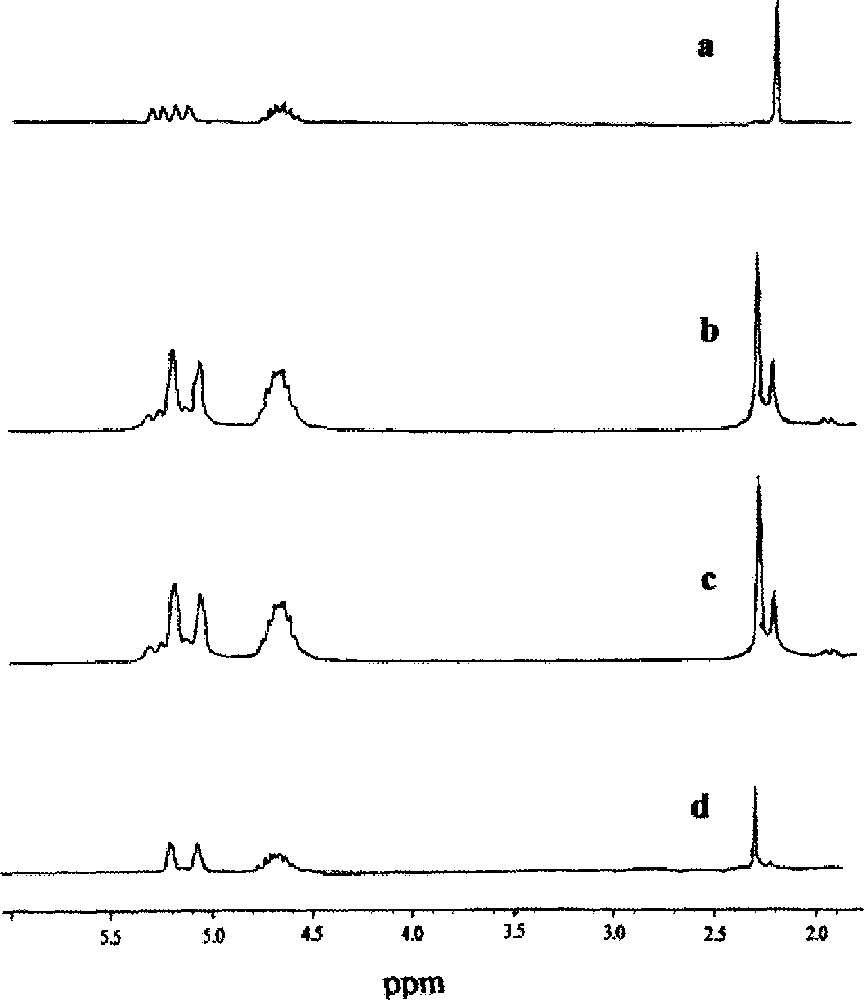
1H NMR spectra of MeCpMo(CO)2P(OiPr)3I after reaction in the solid state at 90 °C. Spectra of the cis isomer at (a) t = 0 min, (b) t = 100 min. Spectra of the trans isomer at (c) t = 100 min, (d) t = 0 min.
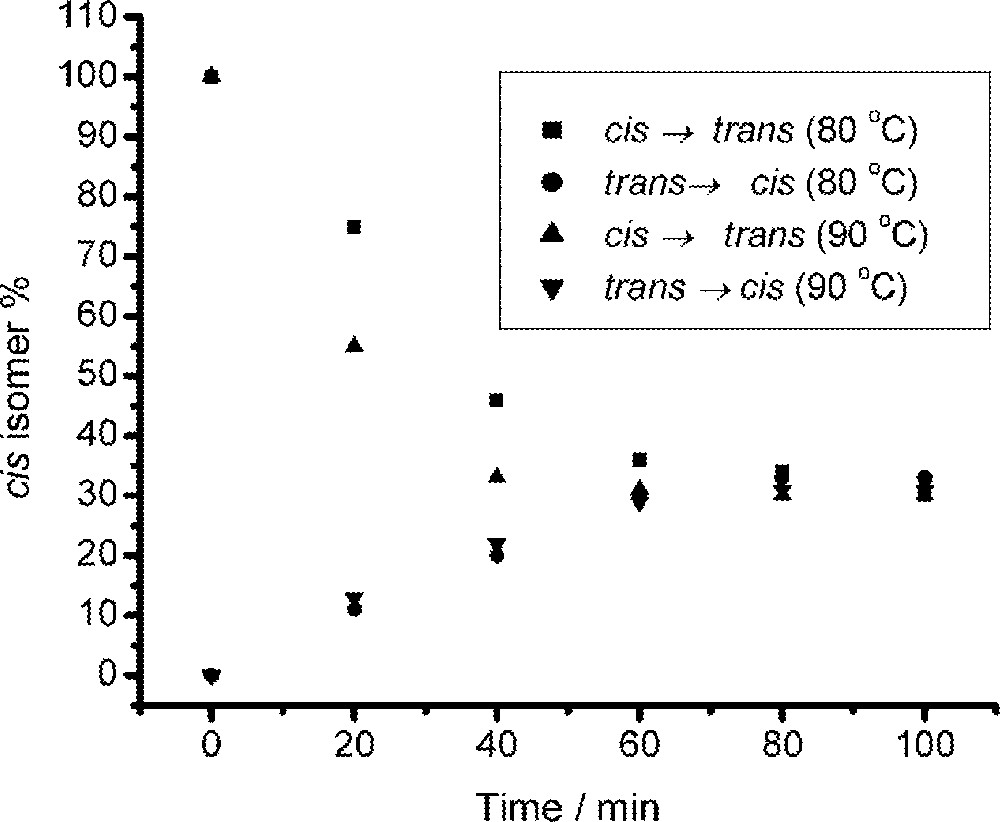
Thermal solid-state isomerization reactions of MeCpMo(CO)2P(OiPr)3I.
2.2.2 Reactions monitored by DSC
The isomers (2–4 mg) were loaded into an aluminum pan and the amorphous reactant and pan were heated under nitrogen in the DSC apparatus. The trans isomer (scan rate, 10 °C min–1) gave a sharp endothermic (melting) peak at 138 °C (Figs. 3 and 4), while the cis isomer gave more complex behavior (Figs. 3 and 5); this will be discussed below.
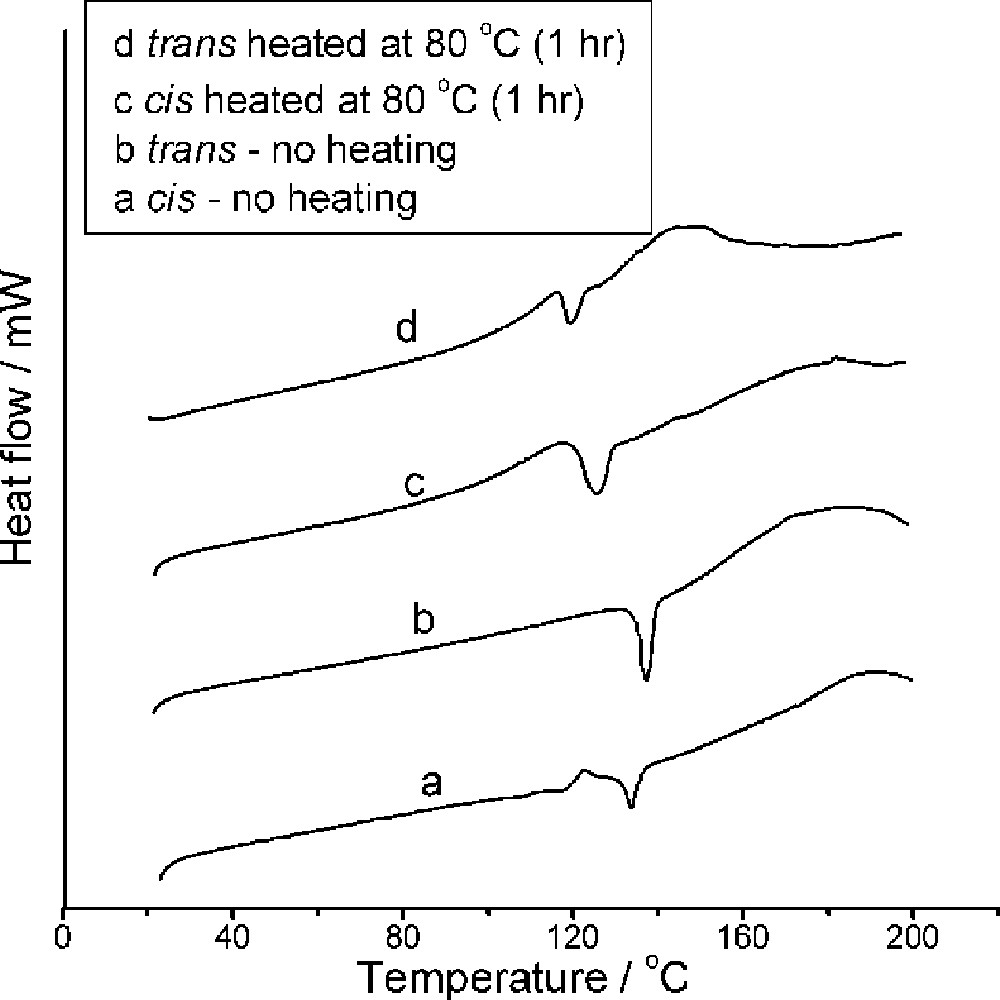
DSC profiles of MeCpMo(CO)2P(OiPr)3I: (a) cis isomer, (b) trans isomer, (c) cis isomer after pre-heating to 80 °C for 1 h, (d) trans isomer after pre-heating to 80 °C for 1 h.
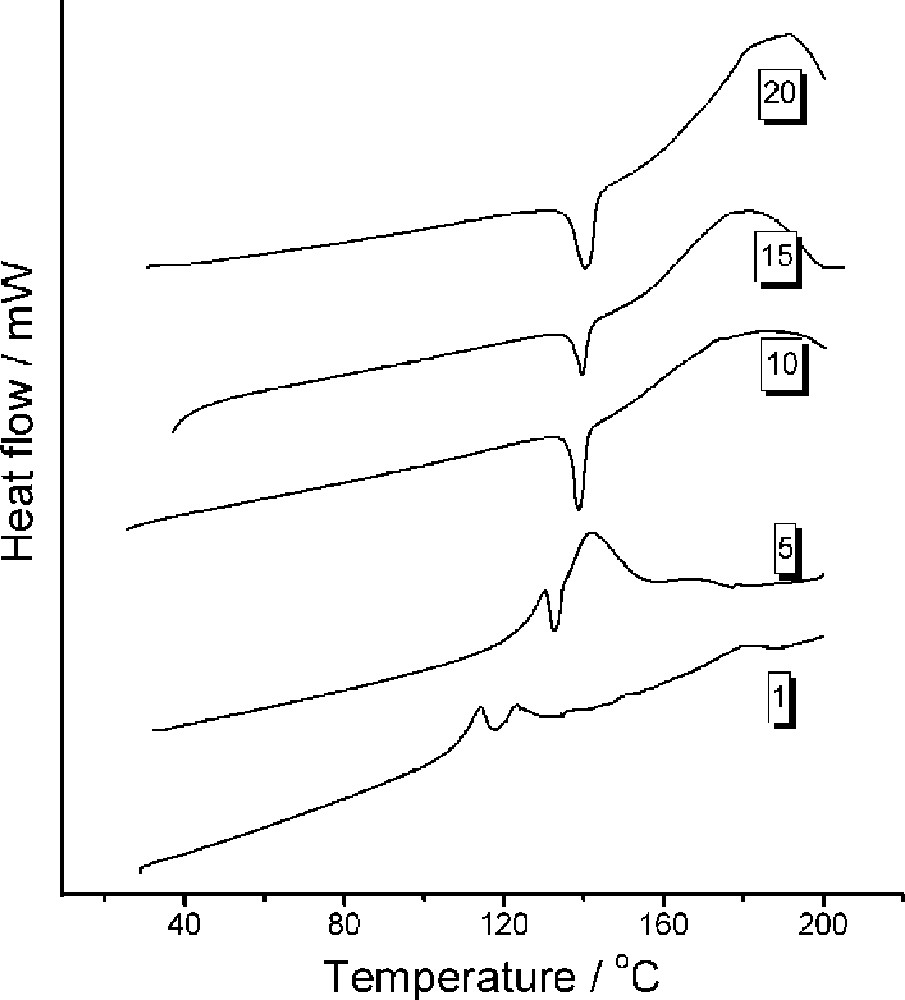
DSC profiles as a function of scanning rate (°C min–1): trans-MeCpMo(CO)2P(OiPr)3I (scanning rates indicated in the figure).
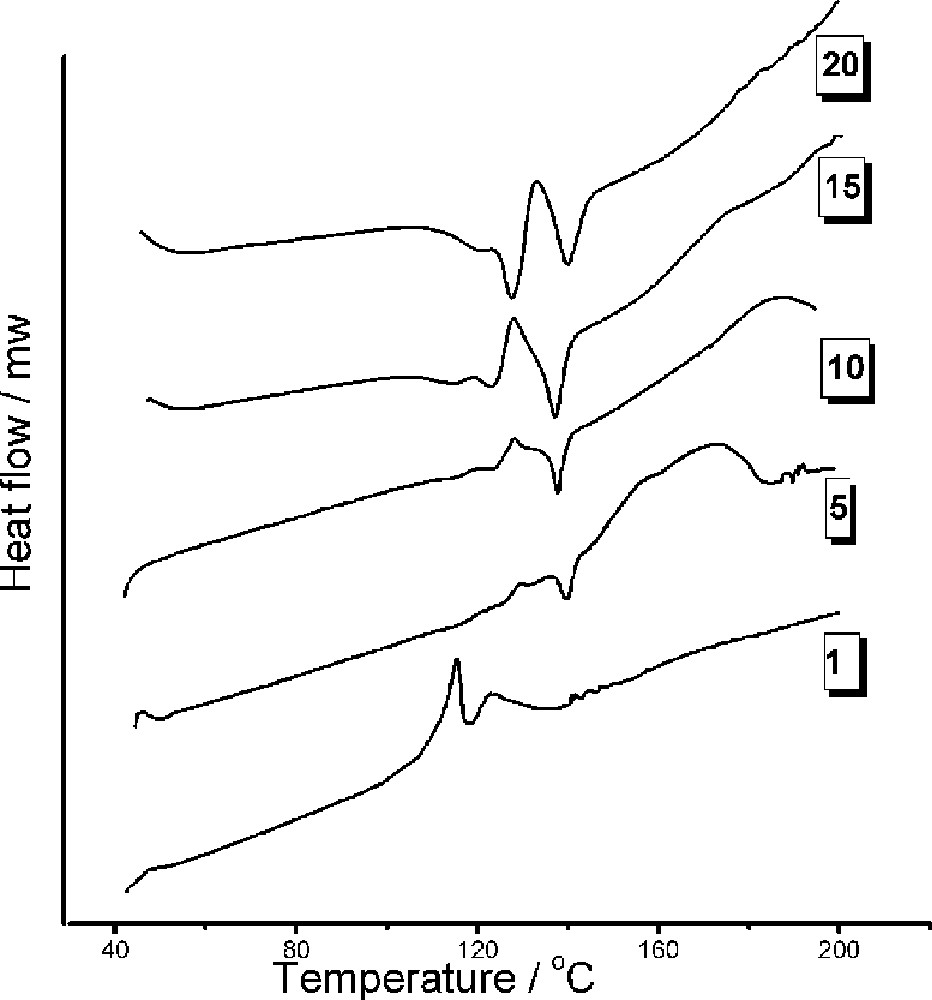
DSC profiles as a function of scanning rate (°C min–1): cis-MeCpMo(CO)2P(OiPr)3I (scanning rates indicated in the figure).
To further explore the DSC data the cis and trans isomers were heated to 70–90 °C (1 h) in an oil bath, cooled to room temperature and the DSC profiles recorded (Fig. 3). NMR spectra were recorded on the samples after the DSC reactions.
The DSC profiles obtained at different heating rates were also recorded (Figs. 4 and 5) and are discussed below.
2.2.3 Reactions monitored by powder XRD measurements
Samples (40 mg) of (η5-C5H4Me)Mo(CO)2(P(OiPr)3)I (either isomer) were packed into NMR tubes and completely immersed in an oil bath set at 90 °C. The samples were removed from the tubes after 20 min intervals. Powder diffraction data were then obtained by spreading all of the fine amorphous material, mixed with silicone grease, onto a silicon disc. After data collection the samples were removed from the sample holder, together with the grease and loaded into NMR tubes for analysis. Powder diffraction data for the cis and trans isomers as a function of time are shown in Figs. 6 and 7, whereas a comparison of the data at times t = 0 and 100 min, for both isomers, is highlighted in Fig. 8.
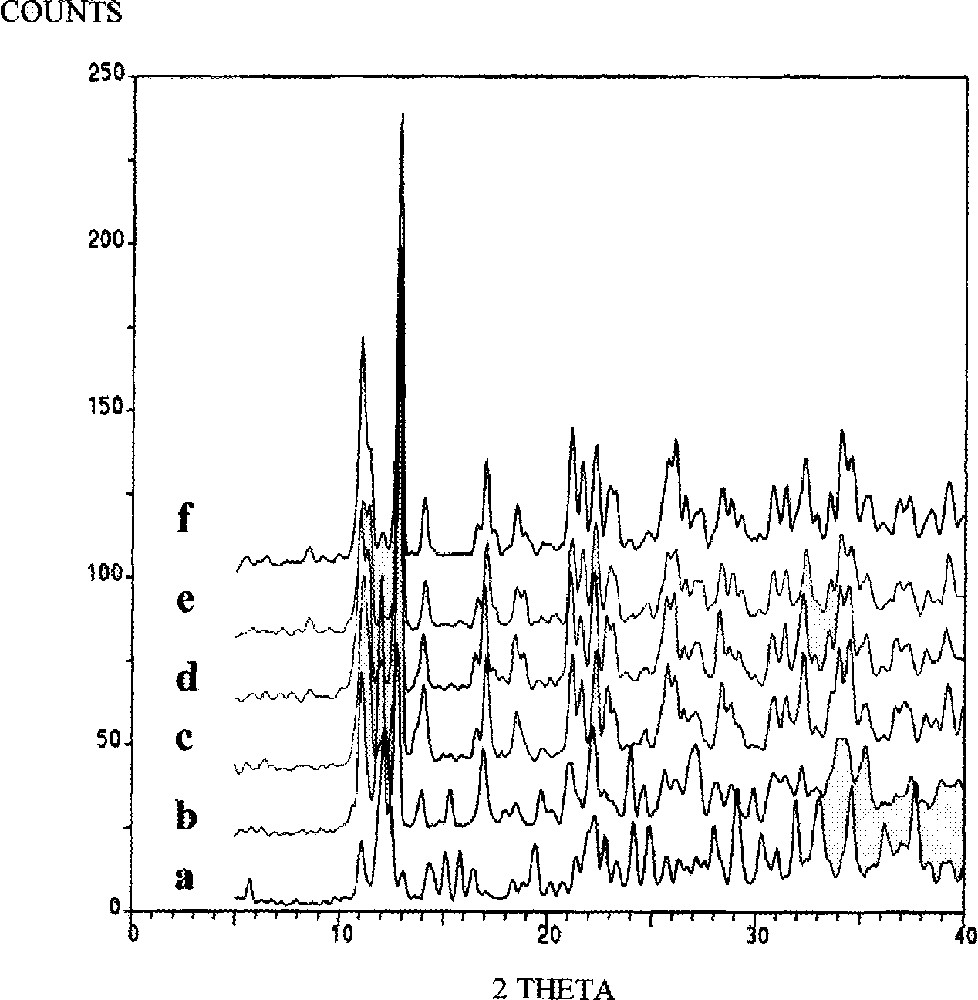
Powder XRD patterns of cis-MeCpMo(CO)2P(OiPr)3I (heated at 90 °C); (a) 0 min, (b) 20 min, (c) 40 min, (d) 60 min, (e) 80 min, (f) 100 min.
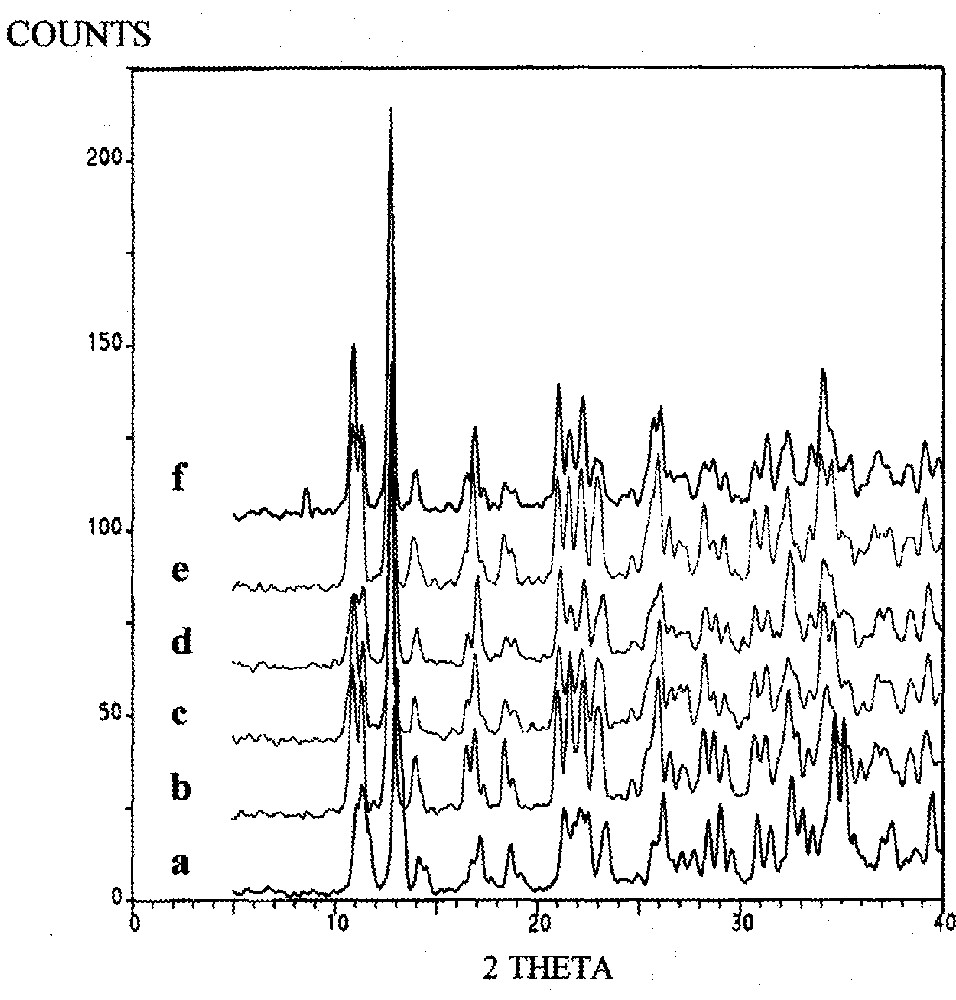
Powder XRD patterns of trans-MeCpMo(CO)2P(OiPr)3I (heated at 90 °C); (a) 0 min, (b) 20 min, (c) 40 min, (d) 60 min, (e) 80 min, (f) 100 min.
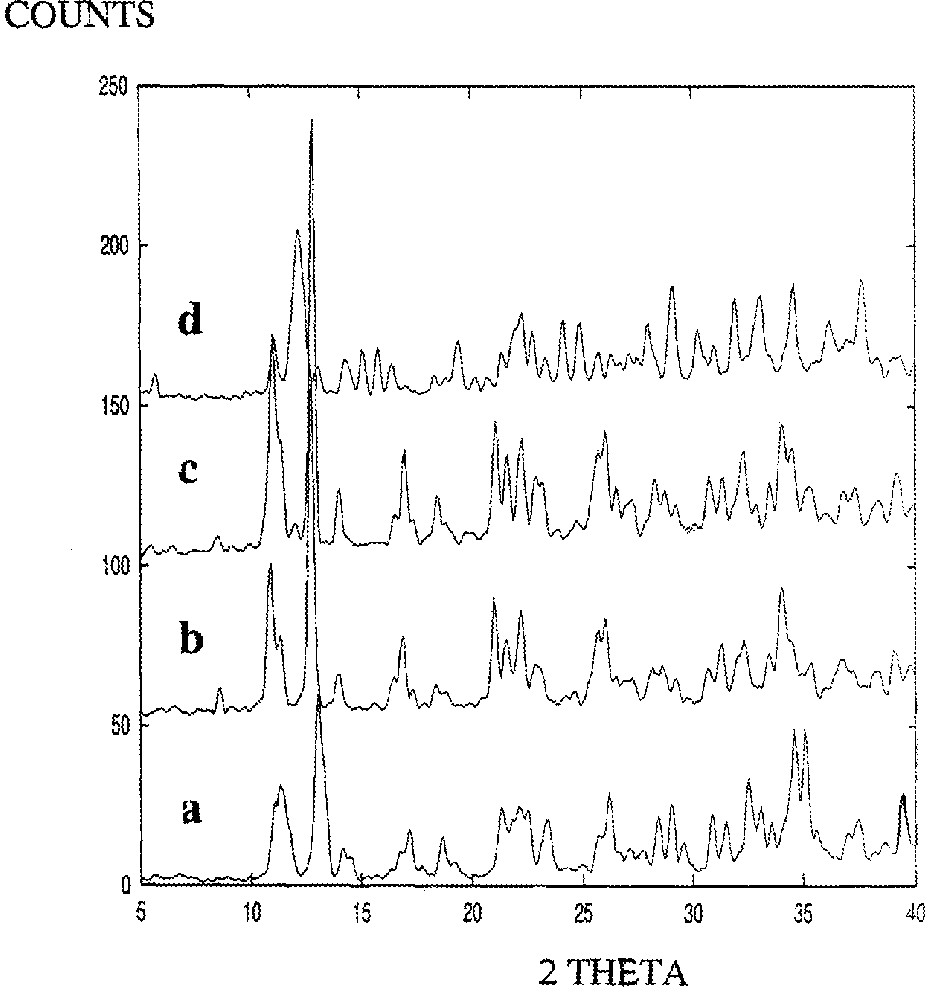
Powder XRD patterns of MeCpMo(CO)2P(OiPr)3I at 90 °C. Spectra of the trans isomer at (a) t = 0 min, (b) t = 100 min. Spectra of the cis isomer at (c) t = 100 min, (d) t = 0 min.
3 Results and discussion
3.1 Preparation of cis- and trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I
Cis- and trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I were prepared in good yield (70%) by reaction of (η5-C5H4Me)Mo(CO)3I with a five-fold excess of P(OiPr)3 and trimethylamine N-oxide in dichloromethane at room temperature 〚11,13〛. The reaction proceeded slowly to completion if only a stoichiometric amount of ligand or trimethylamine N-oxide was used. The separation of the cis and trans isomers was achieved by silica gel column chromatography. The isomers were stable at room temperature in the solid state and had good solubility in organic solvents, including hexane.
The assignments of the cis and trans isomers of (η5-C5H4Me)Mo(CO)2(P(OiPr)3)I (Table 2) were based on well-established infrared and proton NMR criteria 〚15,16,20〛. The NMR spectrum of the cis isomer shows four complex non-equivalent resonances between 5–5.5 ppm corresponding to the four non-equivalent protons on the cyclopentadienyl ring. The trans isomer shows the expected two resonances for the four ring protons 〚18〛. At equilibrium (90 °C, 100 min) the ratio of the two isomers can readily be determined (30:70, cis/trans) from the intensity of the resonances associated with the ring Me groups situated at about 2.3 ppm. This data has been re-plotted in Fig. 2 and it can be seen that the same equilibrium isomer ratio is obtained starting from either isomer. Data for the reaction performed at 80 °C are also shown in Fig. 1.
Decomposition was observed after 150 min reaction. It is pertinent to note that at 70 °C little isomerization was noted (60 min, < 5%) and also that the complexes decomposed rapidly at temperatures above 100 °C, as confirmed by both NMR spectra and TGA analyses.
3.2 Solution state isomerization of cis- and trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I
The cis- and trans-(η5-C5H4Me)Mo(CO)2(P(OiPr)3)I isomers studied are stable in solution (e.g. CHCl3, benzene) at room temperature for a period of hours. However, when either isomer is refluxed in benzene they quickly isomerized (< 2 h) to a thermodynamic equilibrium of isomers (〚cis〛/〚trans〛 ratio, 70:30). This result shows that the solution state isomerization is bidirectional. Similar equilibrium studies on (η5-C5H5)Mo(CO)2(L)X (L = PPh3, PBu3; X = Br, I) 〚15〛 and (η5-C5H4R)M(CO)2(L)I (R = Me, tBu, SiMe3; L = isonitrile, phosphine, phosphite) 〚12〛 in refluxing benzene have also been reported.
3.3 Solid state isomerization of cis- and trans-(η5-C5H4Me)Mo(CO)2((OiPr)3)I
Both cis and trans isomers were tested for solid-state isomerization behavior (Table 1). It was found that the thermal solid-state isomerization reaction is also bidirectional – with an equilibrium ratio established from either isomer (equilibrium ratio: 〚cis〛/〚trans〛 = 30:70). This ratio differs from the solution (benzene) data, thus revealing phase-dependent isomerization isomer ratios.
The k values for the reaction were calculated assuming first order kinetics and the Arrhenius plot yielded an activation energy for the reaction = 68 ± 10 kJ mol–1. This is to be compared to the value of 100 ± 9 kJ mol–1, found for the (η5-C5H4Me)Re(CO)(P(OPh)3Br2 complex 〚23〛.
XRD data for the conversion of the cis to the trans isomer of (η5-C5H4Me)Mo(CO)2(P(OiPr)3)I (90 °C) were recorded and reveal dramatic changes in the peak positions/intensities as a function of time (Fig. 6). Particular peaks associated with the cis isomer are reduced in intensity, while new peaks grow (e.g. at 11° and 13°, see §2.2). The XRD data for the trans isomer were also monitored as a function of time and spectra showing the changes in peak intensity/position are shown in Fig. 7. Two key observations from the figures are (i) the XRD patterns after 100 min reaction are identical in the two spectra (Figs. 6 and 7) and (ii) the XRD pattern of the original cis and trans isomers are no longer present. This is more clearly seen in Fig. 8.
NMR spectra of all the above samples were recorded after the XRD experiment by solution NMR spectroscopy and the data indicated that both the cis and the trans isomers were present in the samples (similar NMR results were also obtained from the solid state reaction performed in NMR tubes, see above). The implication from the XRD and NMR data is that the cis isomer has either converted to a new cis (and trans) polymorph 〚24〛 or to a new crystal form in the solid-state reaction. Heating of the trans isomer also yields similar changes (see Figs. 7 and 8). This contrasts with an earlier study on (η5-C5H4Me)Re(CO)(L)Br2 (L = CO, CNC6H3Me2, P(OR)3, PPh3) complexes 〚18–20〛, in which the isomerization reaction was unidirectional and no polymorphs or new crystal forms could be detected.
A DSC study was carried out to obtain further information on the reaction and data are shown in Figs. 3–5.
Trans isomer (Figs. 3 and 5). An initial interpretation of the DSC profile recorded at 10 °C min–1 (Fig. 3b) would suggest that the pure trans isomer melts at 138 °C. However, the NMR spectrum of the material recorded after DSC experiments were terminated (between 120 and 140 °C), reveals that in all cases the material comprises of a 30:70 cis/trans mixture (some decomposition was also noted in the NMR spectrum when the reaction was performed at the higher temperatures). The NMR data imply that isomerization has taken place in the solid state even though no exotherm is to be seen in the DSC profile! Samples were also preheated to 80 °C for 1 h in an NMR tube and the DSC profile (Fig. 3d) and NMR spectra then recorded. The NMR spectra revealed a 30:70 cis/trans isomer ratio and the DSC profile revealed that the endotherm had moved to a lower temperature suggestive of the presence of a mixture.
The above data (together with the XRD data) are entirely consistent with an isomerization occurring between 80 and 100 °C, but the event is not reflected in the DSC profiles.
DSC profiles were also measured as a function of heating rate (Fig. 4). At low heating rates of 1 °C (and 5 °C min –1), more complex behavior is detected with the endotherm (melting point) situated on top of an exotherm. A TGA profile recorded at 1 °C min–1 (not shown) indicates 50% mass loss that commences at about 100 °C, indicating that the endotherm is due to a decomposition reaction. At heating rates of 10°C min–1 and above, only the melting point of the mixed isomer mixture and a decomposition endotherm (T > 140 °C) is to be seen.
Cis isomer (Figs. 3 and 5). The DSC profile of the cis isomer (at a heating rate of 10 °C min–1) shows complex behavior. Of note is the endotherm at 135 °C which is similar to that observed for the trans isomer. Also of note are the exotherms to be seen at 113 and 124 °C.
Samples of the cis isomer were heated at 80 °C for 1 h and analyzed: (i) solution NMR spectra of a sample indicated that it was comprised of a mixture of cis/trans isomers (ratio: 30:70) and (ii) a DSC profile revealed that most of the peaks below 130 °C had been eliminated (compare Fig. 3a and Fig. 3c) and only a single endotherm similar to that observed for the trans isomer (Fig. 3d) was to be seen.
DSC data were recorded on the cis isomer as a function of DSC heating rate. At 1 °C min–1 the profile is similar to that recorded for the trans isomer (exotherm superimposed on an endotherm), again suggesting that the isomerization is bi-directional and not detected in the DSC profiles. Interestingly, the DSC profiles at 15 and 20 °C min–1 show a complex behavior that arises from 70% of the cis isomer having to isomerize, in a cis matrix, to the trans isomer.
At a heating rate of 20 °C min–1, two exotherms are to be seen. It can be speculated that at this heating rate that the first melting point (endotherm) relates to the cis (or cis-rich) isomer.
In conclusion, all the data are consistent with a bi-directional cis–trans isomerization occurring in the solid state between 70 and 100 °C, followed by product decomposition.
Mechanism. We have reported on the solid-state behavior of over 20 rhenium piano stool complexes 〚18–20〛. The data suggests, in contrast to the solution phase studies, that an understanding of the isomerization mechanism must entail an understanding of the intramolecular forces at play in the solid state. The intermolecular forces that override the intramolecular forces, which dominate in the solution phase, could be both steric (phase rebuilding 〚25〛, topotatic 〚26〛) and electronic, in nature. Thus, an understanding of the reaction will only be possible from the knowledge of solid-state packing information 〚27〛, and crystal structure studies are presently underway to explore this reaction in more detail.
We recently proposed that the isomerization of (η5-C5H4R)Re(CO)(L)X2 complexes involved only one polymorph/crystal form in the reaction 〚20〛. The XRD data recorded on the Mo complex suggests that a new crystal form/polymorph has been formed in the reaction, prior to product decomposition. These possibilities are shown in Fig. 9. In this figure, we have indicated that while a separate mixture of cis and trans isomers is possible, the isomers would have to be polymorphs.
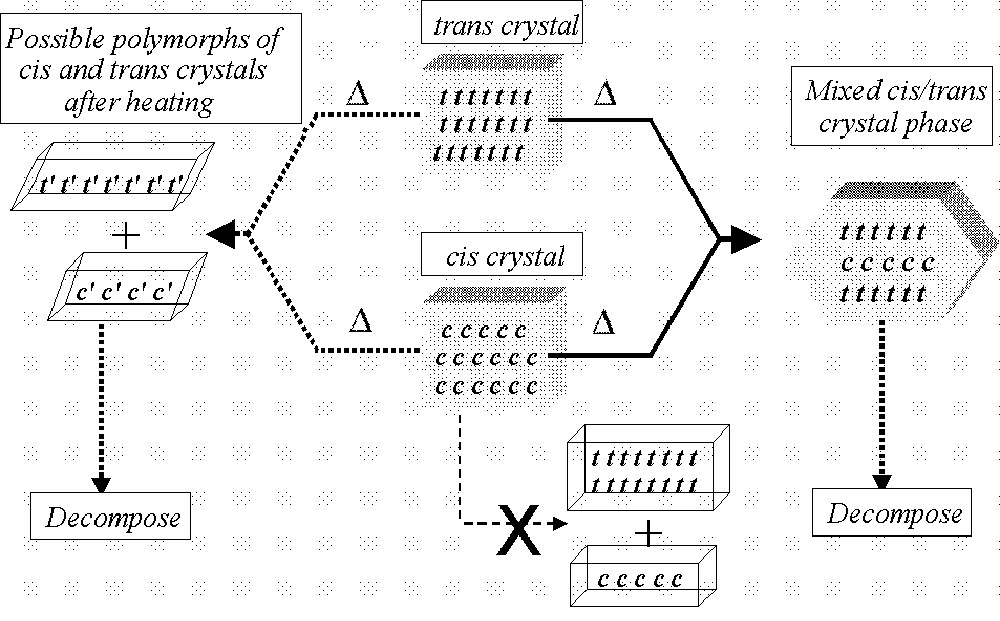
Cartoon showing possible routes for the isomerization reaction in the solid state.
If indeed polymorphs are generated, these could relate to the different orientations of the phosphite ligand. In previous studies we have shown that phosphite groups can show different arrangements of the OR groups in the solid state and that these arrangements for P(OMe)3 and P(OEt)3 can be influenced by intermolecular interactions 〚28,29〛. Presently there are insufficient structures containing the P(OiPr)3 ligand available to propose the specific arrangements of the OiPr groups that could give rise to the different polymorphs that may have been formed in this study.
Whereas the melting point data for the Re complexes allowed for prediction of the direction of the isomerization reaction, the use of this measurement is not as obvious for the Mo complexes. Thus, the melting point measured on a Kofler melting point apparatus (10 °C min–1) indicates that the trans isomer has a marginally higher melting point than the cis isomer (Table 1). The DSC ‘melting point’ data reveal a more complex issue. This arises since the Mo complex isomerizes from both directions on heating and the endotherms measured (Table 1) refer to the melting points of the equilibrium isomer mixtures. Our data suggests that many melting point measurements that have previously been described for this class of organometallic complexes (and other complexes in general?) may in actual fact relate to the melting points of an isomer mixture.
Neither isomer is more stable than the other in the solid-state environment. The stable environment is the environment in which an approx 30:70 ratio of cis/trans isomers is formed in the solid state for this complex. The solution (refluxing benzene) isomer ratio involving a dynamic equilibrium is quite different to that found in the solid-state. It is not clear as to whether the solid-state data refer to a dynamic equilibrium at the high temperature, or whether this is a static equilibrium determined by the most stable arrangement of isomers in the solid mixture.
4 Conclusions
Unlike the rhenium complexes (η5-C5H4R)Re(CO)(L)X2, the molybdenum complex (η5-C5H4Me)Mo(CO)2(P(OiPr)3)I undergoes a bidirectional thermal solid-state isomerization reaction. Both the DSC and XRD powder data are consistent with this view. Two key findings from this study are:
- • DSC data alone do not allow for identification of cis/trans isomer reactions;
- • melting point data recorded on complexes that can undergo isomerization reactions in the solid state may relate to isomer mixtures.
Our results further suggest that thermal solid-state cis–trans isomerization reactions should be a common reaction type for cyclopentadienyl four-legged piano stool transition metal complexes and for many other complexes with geometries that permit for cis/trans isomers.
Acknowledgements
We wish to thank the NRF, THRIP and the University for financial support and Prof. FRL Schöning for helpful discussions.