

1 Introduction
Osborn, Wilkinson and Young reported the use of Rh(PPh3)3Cl3 (and other related complexes) as active hydroformylation (oxo) catalysts for the conversion of alkenes, H2, and CO into aldehyde products under mild conditions in a very short 1965 communication 〚1〛. This was followed by their seminal full paper in 1968 that identified the actual catalyst as HRh(CO)2(PPh3)2 〚2〛. While it had been known that rhodium was considerably more active towards hydroformylation relative to cobalt, the use of PPh3 as a highly effective promoter ligand to generate a catalyst system that could perform oxo catalysis at 25 °C and 1 bar of H2/CO was unprecedented. In many ways, this result was more important than that of the catalyst of Osborn and Wilkinson, RhCl(PPh3)3, and its activity for the hydrogenation of alkenes 〚3〛. Heterogeneous hydrogenation catalysts were well known and are still routinely used in most non-asymmetric hydrogenations. But for hydroformylation, homogeneous catalysts are essentially the only ones used in industry due to their higher selectivity and activity. Indeed, the exceptionally high activity of RhH(CO)2(PPh3)2, in conjunction with Pruett’s discovery 〚4〛 that excess PPh3 is required in rhodium hydroformylation in order to stabilize the catalyst and enhance the linear aldehyde product selectivity, opened the door for low pressure hydroformylation, which currently accounts for over 70% of industrial oxo production 〚5〛.
Interest in homobimetallic cooperativity in hydroformylation catalysis goes all the way back to Heck’s original 1961 mechanism for HCo(CO)4-catalyzed hydroformylation where, in addition to the now commonly accepted monometallic mechanism for both Co and Rh catalysts, he proposed an intermolecular hydride transfer from HCo(CO)4 to Co(acyl)(CO)4 leading to the reductive elimination of aldehyde and formation of Co2(CO)8 as shown in Fig. 1 〚6〛. Heck, however, did not favor the bimetallic mechanism due to the low concentrations of both species involved and subsequent spectroscopic studies have effectively confirmed this 〚7, 8〛.
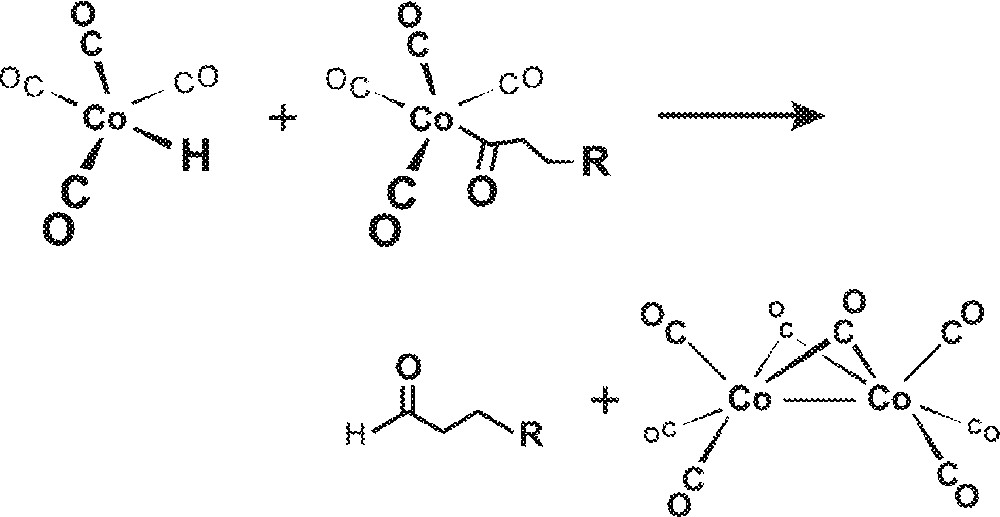
Intermolecular hydride transfer from HCo(CO)4 to Co(acyl)(CO)4 leading to the reductive elimination of aldehyde and formation of Co2(CO)8.
One strategy to enhance possible bimetallic cooperativity in the catalysis is to tether the two metal centers together using a suitable bridging ligand system. We have designed the binucleating tetraphosphine ligand (Et2CH2CH2)(Ph)PCH2P(Ph)(CH2CH2PEt2), et,ph-P4, to strongly chelate and bridge two metal centers in order to explore bimetallic cooperativity in hydroformylation catalysis 〚9〛. The two internal phosphines in the et,ph-P4 ligand are chiral, leading to the two diastereomers shown in Fig. 2.
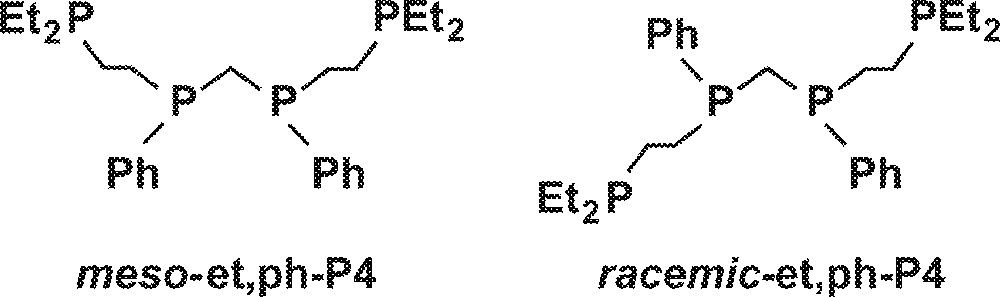
Diastereomers meso-et,ph-P4 and racemic-et,ph-P4.
The reaction of 2 equiv of 〚Rh(nbd)2〛(BF4) (nbd = norbornadiene) with rac-et,ph-P4 generates the homobimetallic complex 〚rac-Rh2(nbd)2(et,ph-P4)〛(BF4)2, 1, which is the catalyst precursor to a highly selective and active hydroformylation catalyst for 1-alkenes (90 °C, 6.1 bar, 1 mM catalyst, 0.9 M 1-hexene, acetone solvent, initial turnover frequency = 640 h–1, aldehyde linear-to-branched = 27:1, 8% alkene isomerization, 3.4% alkene hydrogenation). We have reported that this represents one of the most dramatic examples of bimetallic cooperativity in homogeneous catalysis 〚10〛. Our in situ spectroscopic studies have indicated that the active catalyst is the unusual dicationic dirhodium complex 〚rac-Rh2H2(μ-CO)2(CO)2(et,ph-P4)〛2+, 2, shown in Fig. 3 〚11〛. One of the strongest and most persuasive indications that fragmentation to active monometallic catalysts is not occurring in this system was the high linear to branched (L:B) aldehyde regioselectivity of 27:1 observed for 1-hexene. Monometallic ‘half’ analogs of the bimetallic catalyst using chelating R2PCH2CH2PR2 ligands show very low activity (TOF = 1 h-1), L:B regioselectivities (3:1), and considerable alkene isomerization and hydrogenation side reactions (∼ 70%) 〚10〛.
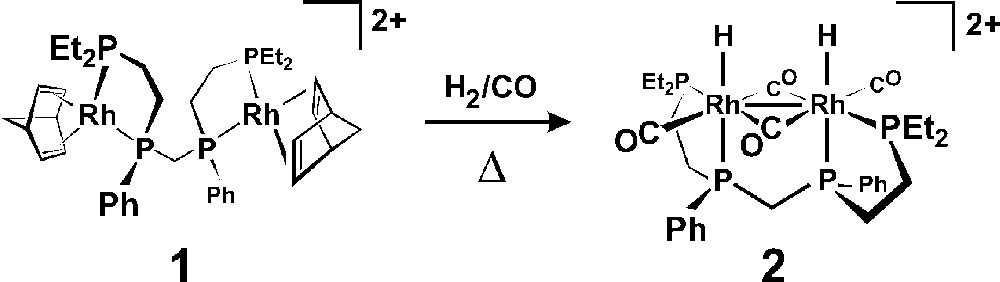
Synthesis of the dicationic dirhodium complex 〚rac-Rh2H2(μ-CO)2(CO)2(et,ph-P4)〛2+, 2, from 1.
Prior to our work, Kalck and coworkers also reported an active thiolate bridged dirhodium catalyst, Rh2(μ-SR)2(CO)4, for hydroformylation that was proposed to work via homobimetallic cooperativity 〚12, 13〛. An unusual aspect of this catalyst was that it had no hydroformylation activity until PPh3 was added, and then the activity and regioselectivity essentially mirrored that of RhH(CO)2(PPh3)2. It has been subsequently demonstrated that the thiolate-bridged binuclear complex readily fragments even under mild conditions 〚14〛 and that PPh3 simply assists in producing the active RhH(CO)2(PPh3)2 monometallic catalyst 〚15〛.
Ongoing spectroscopic studies of our bimetallic hydroformylation catalyst 〚rac-Rh2H2(μ-CO)2(CO)2(et,ph-P4)〛2+ have clearly indicated that as the initial mixture of bimetallic carbonyl and hydrido-carbonyl complexes sits under H2/CO (20 bar) that fragmentation of the catalytically active bimetallic complexes is occurring. Two of the fragmentation products that we have tentatively identified from in situ NMR studies are the Rh(III) monometallic complex 〚rac-RhH2(η4-et,ph-P4)〛+, 3, and the double ligand coordinated bimetallic complex 〚rac,rac-Rh2H2(CO)2(et,ph-P4)2〛2+, 4, shown in Fig. 4 〚16, 17〛. Neither of these is believed to be catalytically active for hydroformylation and under extended ‘soaking’ (time spent under H2/CO pressure while heating the autoclave to the reaction temperature) of the bimetallic catalyst solution under H2/CO (90 °C, 6.1 bar, > 2 h), without any alkene present, we see almost complete deactivation of the catalyst towards hydroformylation. We now report the unusual effect of added PPh3 on our bimetallic rhodium hydroformylation catalyst system that leads to loss of the high L:B aldehyde regioselectivity and eventually all catalyst activity.
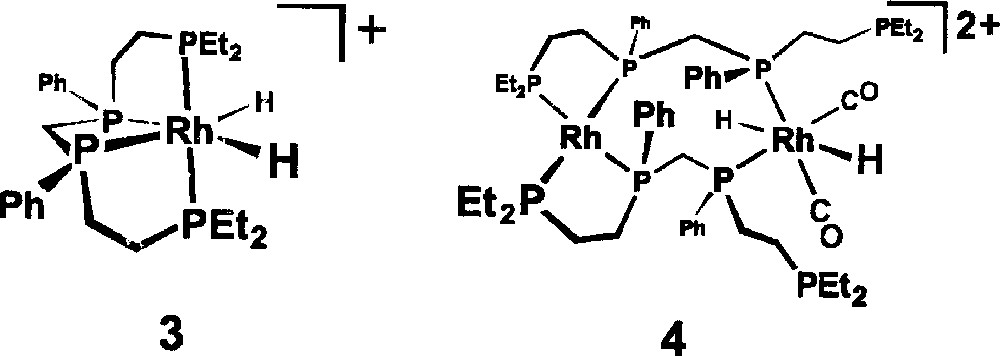
Rh(III) monometallic complex 〚rac-RhH2(η4-et,ph-P4)〛+, 3, and the double ligand coordinated bimetallic complex 〚rac,rac-Rh2H2(CO)2(et,ph-P4)2〛2+, 4.
2 Results and discussion
2.1 Addition of PPh3 to bimetallic catalyst solution
The hydroformylation reaction conditions used are very similar to that previously reported 〚10〛: 90 °C, 6.12 bar 1:1 H2/CO, acetone solvent, 1 mM 〚rac-Rh2(nbd)2(et,ph-P4)〛(BF4)2, 1, as the catalyst precursor, 1 M 1-hexene (1000 equiv), 160 ml Parr autoclave, and 1000 rpm stirring. The catalyst activity in acetone solvent is strongly influenced by the initial soaking under H2/CO prior to the addition of the 1-hexene substrate. The catalyst precursor 1 and appropriate amount of PPh3 was dissolved in 80 ml of acetone and transferred to the Parr autoclave in a glove box under inert atmosphere. The autoclave is then purged with 1:1 H2/CO, pressurized to 3 bar with H2/CO, and temperature ramped up as quickly as possible to 90 °C. This takes approximately 20 min and the pressure increases to about 5.4 bar. The 11 ml (1000 equiv) of 1-hexene is pressure added to the autoclave from a small external stainless steel reservoir to initiate the hydroformylation catalysis and raise the autoclave pressure to the operating pressure of 6.12 bar.
Allowing the bimetallic catalyst solution to sit prior to alkene addition for longer periods at 90 °C under 5.4 to 6.1 bar of H2/CO leads to steadily decreasing catalyst activity. For example, after 50 min, the dirhodium catalyst has only about 20% the hydroformylation activity of the 20-min system, and after 80 min, there is essentially no hydroformylation catalysis. This, in some ways, parallels the RhH(CO)2(PPh3)2 monometallic catalyst that also deactivates on extended soaking under H2/CO at 90 °C (8–12 h), even in the presence of excess PPh3 (400–800 equiv) 〚18, 19〛. The deactivation routes for the dirhodium and RhH(CO)(PPh3)2 monometallic catalyst, however, appear to be quite different. It has been fairly well established that RhH(CO)(PPh3)2 can attack and cleave a phenyl ring off the PPh3 ligand to ultimately form catalytically inactive phosphido-bridged rhodium dimer and cluster complexes 〚20, 21〛 or add an alkyl group to the phosphine that is a considerably poorer ligand for hydroformylation 〚22, 23〛. Another less serious deactivation route involves the formation of carbonyl-bridged dinuclear Rh(0) complexes like Rh2(μ-CO)2(CO)2(PPh3)2, which is inhibited by excess PPh3 〚2, 24〛. In marked contrast, we have not observed any Rh-induced fragmentations of our more alkylated et,ph-P4 ligand 〚11〛, but instead observed that the tetraphosphine does not appear to chelate the rhodium centers strongly enough leading to release of one Rh center and the formation of the catalytically inactive complexes shown in Fig. 4.
The catalytic results for the hydroformylation of 1-hexene starting with the bimetallic precursor 1 and monometallic precursor Rh(acac)(CO)2 (or 〚Rh(nbd)2〛+) in the presence of varying amounts of PPh3 are shown in Table 1. In all runs, we observed minimal alkene hydrogenation side reactions (less than 1%). The homobimetallic catalyst 〚rac-Rh2H2(μ-CO)2(CO)2(et,ph-P4)〛2+, 2, without any added PPh3 is quite active with an initial turnover frequency (TOF) of 1200 h–1, high L:B aldehyde regioselectivity of 25, and 2.5% alkene isomerization and 3.4% alkene hydrogenation side-reactions. This initial TOF is 88% faster than the 640 h–1 value originally reported 〚10〛 and this requires some comment.
Hydroformylation of 1-hexene (1 M) in acetone at 90 °C and 6.12 bar 1:1 H2/CO using the bimetallic precursors: 〚rac-Rh2(nbd)2(et,ph-P4)〛(BF4)2, monometallic Rh(acac)(CO)2, or cationic monometallic 〚Rh(nbd)2〛(BF4) (all 1 mM).
| Precursor | Equivalents PPh3a | Equivalents PEt3a | Initial TOFb (h–1) | Aldehyde L:B selectivity | % Alkene isomerc |
| Bimetallic | 0 | 0 | 1200 | 25 | 2.5 |
| Bimetallic | 0.5 | 0 | 660 | 3.4 | 3.0 |
| Bimetallic | 2 | 0 | 590 | 3.1 | 5.0 |
| Bimetallic | 5 | 0 | 320 | 2.9 | 3.0 |
| Bimetallic | 10 | 0 | 220 | 3.2 | 3.3 |
| Bimetallic | 100 | 0 | 0 | — | 0 |
| Mono | 0 | 0 | 0 | — | 0 |
| Mono | 2 | 0 | 2200 | 2.6 | 6.0 |
| Mono | 5 | 0 | 5300 | 3.2 | 2.9 |
| Mono | 10 | 0 | 6800 | 3.2 | 2.0 |
| Mono | 100 | 0 | 6300 | 5.0 | < 1 |
| Mono | 400 | 0 | 780 | 9.1 | < 1 |
| Mono | 10 | 1.1 | 3100 | 3.1 | 2.0 |
| Mono | 100 | 1.1 | 780 | 4.9 | 1.5 |
| Mono | 10 | 2.2 | 2000 | 3.0 | < 1 |
| Mono | 100 | 2.2 | 450 | 3.6 | < 1 |
| Mono(+)d | 10 | 0 | 2 | 1.6 | 80 |
| Mono(+)d | 100 | 0 | 300 | 4.2 | 1.8 |
| Mono(+)d | 10 | 2.2 | 180 | 3.3 | 1.7 |
| Mono(+)d | 100 | 2.2 | 180 | 3.1 | < 1 |
The catalysis is first order in 1-hexene and a slightly higher alkene concentration was used (1.0 vs 0.91 M) in these studies, but this only accounts for a small amount of the increased rate. The main reason is tied into the importance of purifying the reagent grade 1-hexene by passing it through a fresh alumina column under inert atmosphere prior to each catalytic run. In our original work we degassed and stored the reagent grade 1-hexene under N2, but did not purify it through an alumina column, which turns out to effectively remove any peroxide impurities that can lead to partial catalyst deactivation. It is this partial deactivation that produces the higher alkene isomerization (8%) and hydrogenation (4%) side reactions originally reported 〚10〛. The difference in the L:B aldehyde regioselectivity, however, is very small (28 vs 25 here) and quite consistent within the error of our typical catalytic runs.
The addition of 2 equiv of PPh3 to the bimetallic catalyst causes a dramatic drop in the L:B aldehyde regioselectivity from 25 to 3.1 and a considerable reduction in the initial TOF to 590 h–1. Increasing amounts of PPh3 leads to steadily decreasing catalytic rates, until at 100 equiv essentially no hydroformylation is observed. This is almost opposite behavior from Kalck’s bimetallic Rh2(μ-SR)2(CO)4 system that is inactive until one adds PPh3 and then the rate and selectivity increase with increasing amounts of added PPh3 (up to a point). Van Leeuwen and Claver have demonstrated that Kalck’s bimetallic system readily fragments and reacts with PPh3 to generate the classic monometallic HRh(CO)x(PPh3)y (x = 1–2; y = 3 – x) catalyst 〚15〛.
Even the addition of only 0.5 equiv of PPh3 causes a dramatic drop in the regioselectivity and catalyst activity as shown in Table 1. The steady deactivation and essentially constant low regioselectivity of our bimetallic system with increasing amounts of PPh3 is not consistent with simple fragmentation to a monometallic Wilkinson-like catalyst system. This is illustrated by the parallel runs with the monometallic precursor Rh(acac)(CO)2 and PPh3 (Table 1). At low PPh3 ratios, one generates an extremely active, but short-lived, catalyst with relatively low selectivity. At 100 equiv of PPh3, the catalyst starts to slow down and the aldehyde L:B regioselectivity starts to increase. At 400 equiv (0.4 M) of PPh3, the catalyst initial TOF has deceased to 700 h–1, but the L:B regioselectivity has increased to 9.1. Commercial Rh/PPh3 hydroformylation processes are typically run with at least 0.4 M PPh3 and up to 50% PPh3 by solution weight when the highest L:B aldehyde regioselectivities are needed.
2.2 Mechanistic considerations
The L:B aldehyde regioselectivity data strongly indicates that the addition of even small amounts of PPh3 strongly inhibits the highly regioselective bimetallic catalyst and generates an alternate catalyst with far lower regioselectivity. The rate and regioselectivity data, however, indicates that this alternate catalyst is not HRh(CO)4, HRh(CO)2(PPh3), or HRh(CO)(PPh3)2. Even assuming that fragmentation is generating very low concentrations of a monometallic rhodium carbonyl complex that could be intercepted by PPh3, the rate and regioselectivity should increase as the PPh3 concentration increases within the range studied. Instead, the bimetallic catalytic run with 100 equiv of PPh3 is essentially inactive for hydroformylation catalysis, which is inconsistent with fragmentation to a HRh(CO)(PPh3)2-type catalyst.
Our proposed bimetallic hydroformylation catalytic cycle is shown in Fig. 5 with the Et and Ph groups on the et,ph-P4 ligand omitted for clarity. The in situ NMR studies indicate that the resting state of the catalyst is the simple open-mode dicationic carbonyl complex 〚rac-Rh2(CO)5(et,ph-P4)〛2+, 5, which we have crystallographically characterized 〚16〛. There is a very facile CO-based on-off equilibrium between the tetracarbonyl, pentacarbonyl 5, and the transient hexacarbonyl bimetallic complexes. The localized cationic charge on each rhodium atom compensates for the strongly electron-donating nature of the et,ph-P4 ligand reducing the CO π-backbonding and enabling facile CO dissociation. Oxidative addition of H2 to one of the Rh centers generates the dihydride complex A. This can readily rotate to form the bridged complex 2*, which can rearrange to form the symmetric Rh(II) terminal dihydride 2.
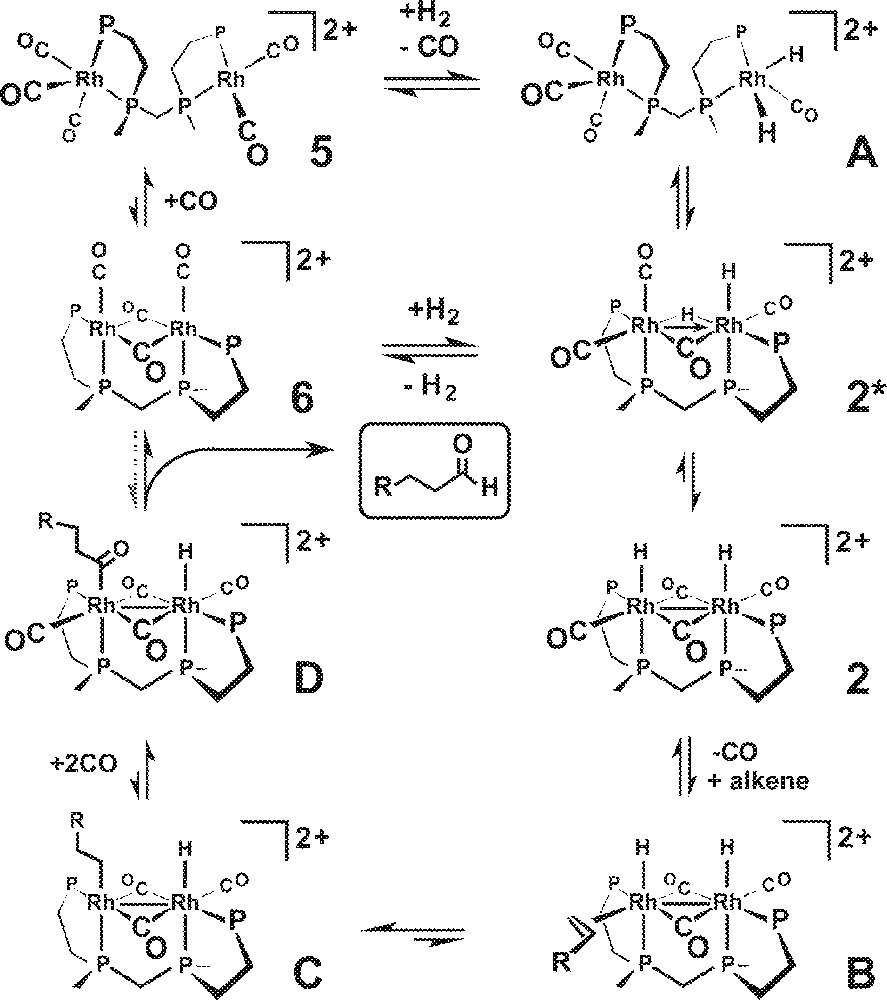
Bimetallic hydroformylation catalytic cycle proposed in this article.
The proposed edge-sharing bioctahedral structure for 2 is quite different from the far more common D4h symmetry dirhodium tetracarboxylate-like systems that have been extensively characterized and studied 〚25〛. Rh2(O2CR)4 and related compounds almost always have weakly coordinated ligands in the two axial coordination sites trans to the Rh–Rh bond. When one transforms this D4h-like structure into an edge-sharing bioctahedral structure seen for 2, there are now two equivalent coordination sites on each metal that are roughly trans to the Rh–Rh bond and should have enhanced lability relative to the other coordination sites (Fig. 6). We will refer to these two sites, coordinated with the terminal PEt2-group of the et,ph-P4 ligand and a CO, also as axial sites.
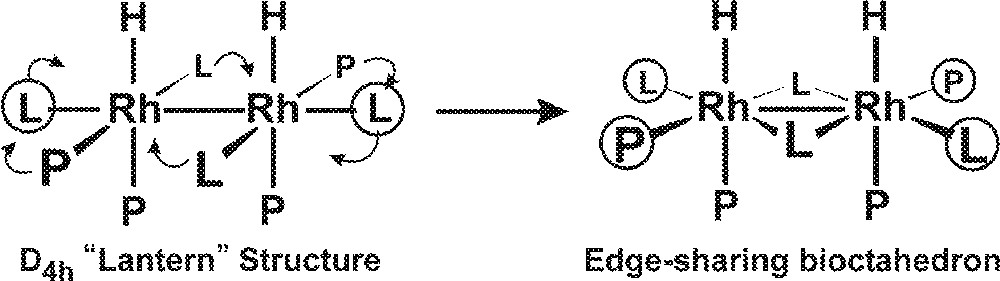
Transformation of a D4h-like structure into an edge-sharing bioctahedral structure.
CO dissociation from 2 is followed by alkene coordination to form B, leading to a migratory insertion that generates the alkyl complex C. CO coordination is followed by a migratory insertion with the alkyl to produce the acyl complex D. This can then do a bimetallic reductive elimination of the aldehyde to form the bridging carbonyl Rh(I) complex 6, that can break open to form 5 or directly react with H2 to make the hydride complexes 2* and/or 2. Hoffmann has calculated that a direct bimetallic reductive elimination is formally symmetry forbidden and can have a high activation barrier 〚26〛. But he also found that π-backbonding ligands like CO, along with lower symmetries, could substantially reduce the barrier for this process. We are currently performing high-level density functional theory (DFT) calculations on this dirhodium catalyst and will be studying the possibility of direct bimetallic reductive elimination.
One obvious effect of added PPh3 on this catalytic cycle would be to simply block the empty axial coordination site formed when CO dissociates and prevent the alkene from coordinating. This should simply slow down the catalyst and not lower the intrinsic regioselectivity. Aside from blocking the alkene-binding site, PPh3 wouldn’t seem to have much impact on most of the other proposed catalytic steps. The high regioselectivity of the dirhodium catalyst is believed to arise from the Rh–Rh bond, bridging carbonyls, and Rh(II) oxidation state. These work together to create an extremely well-defined binding site that does not electronically reorganize or distort upon coordination of the alkene to the empty axial coordination site maximizing the steric directing effects of the relatively small ethyl and phenyl groups.
This is quite unlike square-planar Rh(I) monometallic catalysts that electronically distort upon coordination of the alkene to form a 5-coordinate geometry, as illustrated in Fig. 7. The electronic distortion of a square-planar complex to a 5-coordinate structure causes the sterically directing R groups on the phosphine ligands to be moved further away from the alkene. This minimizes the phosphine ligand steric orienting effect for the subsequent hydride–alkene migratory insertion that is the regioselectivity determining step.
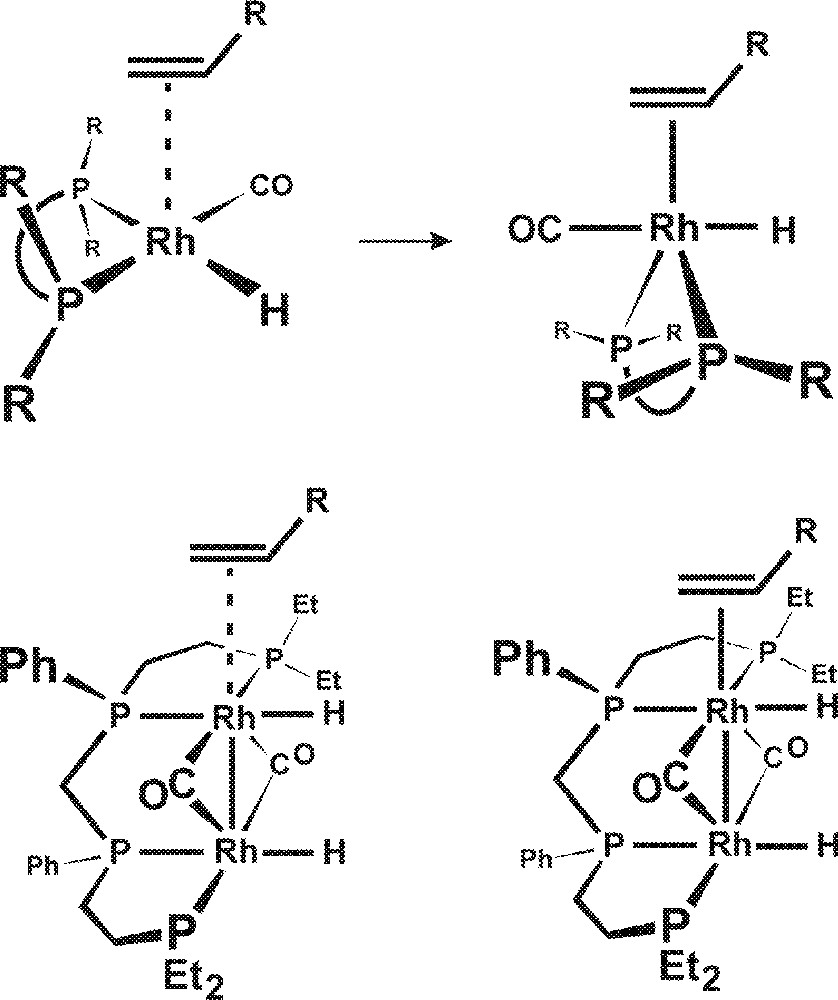
Electronic distortion of a square-planar complex to a 5-coordinate structure.
While the added PPh3 ligand probably inhibits the active bimetallic catalyst, it also appears to short-circuit the catalyst into another structure that has low regioselectivity and moderate activity, but one that is eventually completely inhibited by increasing amounts of PPh3. The most likely entry point for disrupting the formation of the Rh–Rh bonded complex 2 involves the starting carbonyl complex 5. PPh3 coordination could easily disrupt the rotation to 2* after H2 oxidative addition. Molecular modeling studies indicate that this open- to closed-mode rotation is strongly influenced by steric effects. For example, the simple tetracarbonyl 〚rac-Rh2(CO)4(et,ph-P4)〛2+ (or pentacarbonyl 5) shows no sign of rotation into a bridged-carbonyl structure like 6 (Fig. 5) in the presence of CO. Only when H2 is added do we observe rhodium complexes with bridging carbonyls. Indeed, the activity of the catalyst appears to directly track with the presence and intensity of the bridging carbonyl bands in the IR spectrum. The modeling studies clearly demonstrate that it is much easier for the bimetallic complex to rotate into a closed-mode structure when there are two small hydride ligands present on the one rhodium center instead of carbonyls. If a PPh3 ligand was present on one or both of the Rh atoms that could easily sterically block the ability of the complex to form a closed-mode Rh–Rh bonded structure, which we believe is necessary for the high catalyst regioselectivity (and activity).
The coordination of PPh3 is also proposed to open up the bimetallic catalyst chelate structure to form, in essence, a monometallic catalyst center with one alkylated phosphine (PEt3) and a PPh3. This is shown in Fig. 8 for one half of 5, although it could, of course, happen to both sides of the bimetallic complex. One needs to have loss of a proton in order to generate the neutral Rh-H side of the complex. Cationic monometallic precursors like 〚Rh(nbd)2〛+ (Table 1, bottom four experiments) typically generate terrible hydroformylation catalysts unless one can deprotonate the resulting saturated cationic Rh(III) dihydrides (e.g., 〚RhH2(CO)2(PPh3)2〛+) produced from the oxidative addition of H2 〚21〛. PPh3 is marginally basic enough for this, with 10 equiv giving a catalyst that is barely active for hydroformylation, but quite good at alkene isomerization. The addition of 100 equiv of PPh3, however, does shift the deprotonation equilibrium enough to give modest hydroformylation activity (initial TOF = 300 h–1) and dramatically reduced alkene isomerization.
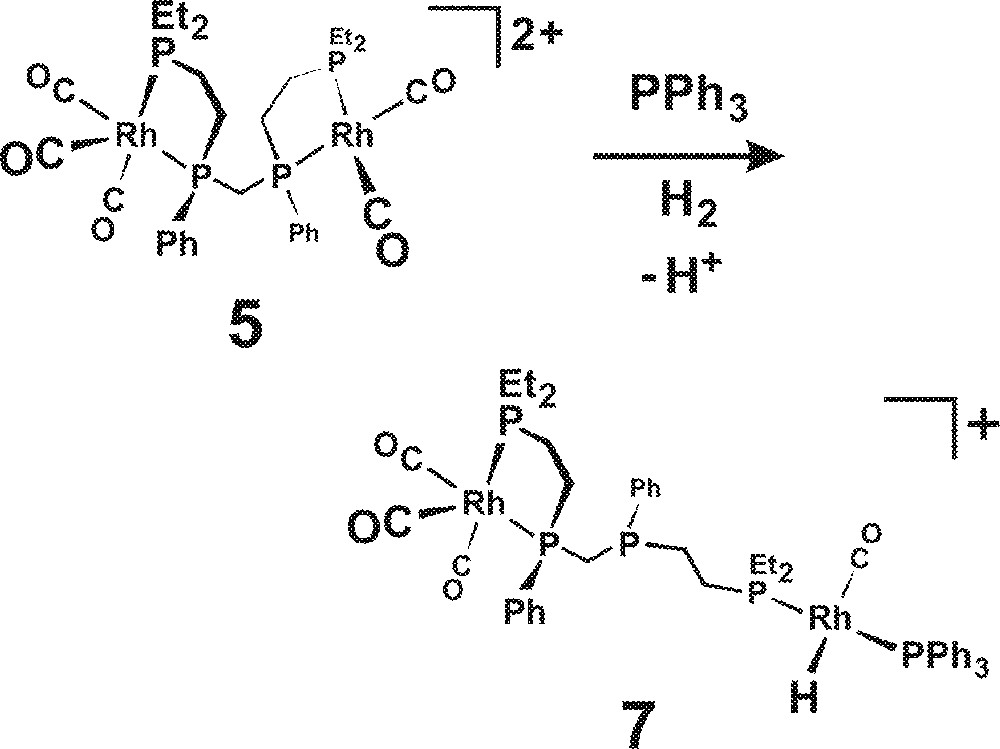
Opening up of the bimetallic catalyst chelate structure (5) to form a monometallic catalyst center with one alkylated phosphine (PEt3) and a PPh3 (7).
Our homobimetallic catalyst normally avoids this cationic dihydride ‘trap’ by performing an intramolecular hydride transfer from complex A in Fig. 5 to ultimately form the symmetrical bimetallic dihydride 2. This is one of the key bimetallic cooperativity steps operating in our catalyst. While PPh3 can deprotonate 〚RhH2(CO)2(PPh3)2〛+ when present in high enough concentrations, it probably would not be effective for the less acidic cationic dihydride precursor that leads to 7 due to the more strongly donating et,ph-P4 ligand. The considerably more basic et,ph-P4 ligand, however, may well be able to act as a suitable base for this deprotonation. Indeed, the internal free phosphine in 7 is well situated to act as an intramolecular deprotonating agent, although it was not drawn as such in Fig. 8.
These mechanistic proposals have been partially tested by adding 1.1 and 2.2 equiv of PEt3 to the neutral and cationic monometallic precursor catalytic reactions. In the case of Rh(CO)2(acac), the addition of PEt3 causes a dramatic drop in activity and a considerably smaller (sometimes negligible) drop in regioselectivity (Table 1). For example, the initial TOF with 100 equiv of PPh3 drops from 6300 h–1 down to 450 h–1 when 2.2 equiv of PEt3 are added, while the L:B aldehyde regioselectivity is reduced from 5.0 to 3.6. The deprotonating ability of 2.2 equiv of PEt3 (one to deprotonate, one to coordinate to the Rh) was tested on the cationic 〚Rh(nbd)2〛+ precursor system and it increased the initial rate with 10 equiv of PPh3 from 2 h–1 up to 180 h–1. Comparing this to the 10 equiv of PPh3 and 1.1 equiv of PEt3 neutral Rh(CO)2(acac) experiment that has an initial rate of 3100 h–1, clearly demonstrates that the PEt3 is only partially deprotonating the cationic rhodium complex and that there is an equilibrium between the cationic and neutral catalyst species. Interestingly, with 100 equiv of PPh3 and 2.2 equiv of PEt3, the cationic catalyst precursor has the same initial TOF as with 10 equiv of PPh3 and 2.2 equiv of PEt3, but actually shows a slight decrease in the regioselectivity (3.3 down to 3.1), along with an expected drop in the alkene isomerization.
These simple model studies offer reasonable support for the proposed formation of an inefficient monometallic-like catalyst similar to 7. The essentially complete catalytic inhibition upon addition of 100 equiv of PPh3 is consistent with the coordination of a second PPh3 to generate a sterically hindered HRh(η1-et,ph-P4)(PPh3)2 complex, which should be essentially inactive for hydroformylation catalysis. Note that the tris-PPh3 analog of this complex only forms under considerably higher PPh3 concentrations and has rather facile PPh3 dissociation in order to relieve the steric strain of having three bulky PPh3 coordinated in a square planar environment. The smaller ‘PEt2’ arm of our ligand, however, should make the catalytically inactive HRh(η1-et,ph-P4)(PPh3)2 complex considerably more stable at lower PPh3 concentrations. The low CO pressure being used will also help stabilize this inactive tris-phosphine complex. The inability of the simple PEt3 model system to mimic the zero activity with 100 equiv of PPh3 is reasonable given that PEt3 is certainly not the same as an η1-et,ph-P4 ligand that has a cationic Rh(CO)3 moiety coordinated to the other side, as proposed for 7. The dramatic regioselectivity lowering effect of even a 0.5 equiv of added PPh3 must point to the extremely efficient inhibition of the active and selective homobimetallic catalyst, which is probably only present in relatively small amounts compared to the total amount of catalyst precursor being used.
In summary, even small amounts of PPh3 disrupt the active and regioselective bimetallic catalyst 2 through a combination of blocking effects that includes coordination to the bimetallic binding site preventing alkene coordination along with coordination to 5 that inhibits the formation of the closed-mode catalyst 2. In concert with this, PPh3 is also proposed to crack open 5 (with deprotonation) to form 7, which contains the active neutral mono-hydride HRh(CO)(η1-et,ph-P4)(PPh3) species which is moderately active for hydroformylation, but not regioselective. Continued addition of PPh3 rapidly leads to catalyst deactivation by ‘saturating’ the neutral half of 7 to form HRh(CO)(η1-et,ph-P4)(PPh3)2. In situ NMR and FT–IR studies will be performed to further probe the validity of these proposals. We are also designing and preparing more strongly chelating analogs of our ligand to minimize these types of side reactions.
3 In memory of John Osborn
The corresponding author (GGS) had the great privilege of working with John Osborn (Fig. 9) as a NATO and CNRS postdoctoral fellow between 1979 and 1981. Those were two of the best years of my life. John and Karen Osborn (along with their sons Michael and Nicholas) basically adopted me into their family and I was routinely invited to their apartment for dinner and to join them on weekend family trips to the surrounding area. He taught me that the quality of one’s science and life is far more important than quantity. John also loved catalysis and infected me forever with that same love. Considering that John’s original research is the foundation of modern rhodium hydroformylation, I’m quite pleased that my own career has ended up centered on an unusual permutation of this (homobimetallic cooperativity). John certainly lived life to the fullest and even though his life ended far too soon and on a tragic note, he certainly packed more into it than anyone I know. I will always remember him as bigger than life, with a smile on his face (although rarely for photographs), and one of the best catalytic chemists I have known.

John A. Osborn in 1980.
Acknowledgements
We would like to thank the National Science Foundation for financial support (CHE-97-00526 and CHE-01-11117).