

1 Introduction
Global industrial emission of sulfur(IV), the primary species responsible for acid rain, is estimated to be on the order of 1014 g annually from such sources as electricity production and vehicular fuel combustion 〚1〛. Natural sources of sulfur in the atmosphere, such as the volcanic release of SO2, can also be substantial. For example, 20 megatons of SO2 (∼1013 g) were injected into the atmosphere during the few days following the 15 June 1991 eruption of Mt. Pinatubo 〚2〛.
Hydration of sulfur dioxide leads to sulfurous acid or the corresponding sulfite salts. Sulfites are subject to further oxidation to sulfur(VI) species such as sulfates or peroxysulfates. Autoxidation of sulfite (with O2) to monoperoxysulfate is catalyzed by transition metals or light – the photochemical process accounting for about 35% of the sulfur content of remote marine clouds 〚3〛.
Manganese(III) and iron(III) compounds are the most effective transition metal catalysts of the aqueous autoxidation of sulfite 〚4〛. In general, the mechanism is thought to involve one-electron redox cycling between the M(III/II) states with formation of sulfite and monoperoxysulfate radical anions as intermediates as shown in equations (1)–(3) 〚1, 4–9〛.
This simplified version of the mechanism has been elaborated for specific metal catalysts depending upon (a) the ability of various metals to bind to sulfite, monoperoxysulfate, and the various intermediates, (b) the pH of the medium and therefore the protonation state of the various sulfur oxides, (c) other oxidation states available to the metal ions such as M(IV), and (d) the presence of other ligands such as azide or porphyrins that can influence the redox potentials of the transition metals involved. In the specific case of manganese-catalyzed sulfite autoxidation, the picture can become quite complex. For example, simple manganese salts are commonly stable in the +II oxidation state. Thus, a one-electron oxidation to Mn(III) is required to initiate the reaction. Depending upon the other species present, this might be an oxo-bridged mixed valent dimer, HO–Mn(III)–O–Mn(II) formed by catalytic oxidation by other trace metals such as Fe(III) 〚4〛. Other ways to stabilize the initial formation of Mn(III) include addition of azide ion 〚5〛, although the presence of more highly stabilizing ligands such as porphyrins can lead to a change in mechanism that includes the formation of a high-valent Mn(V)=O species 〚10–12〛.
The sulfuroxy radicals formed as intermediates in sulfite autoxidation have been proposed as key species in sulfite toxicity and carcinogenicity 〚13〛. Because simple manganese oxides may be present in aerosols and particulates generated from combustion of MMT-containing (methylcyclopentadienyl manganese tricarbonyl) fuels 〚14–16〛, we sought to determine the ability of simple manganese oxides and salts to effect oxidative DNA damage in the presence of sulfite. Previous work in collaboration with the Meunier laboratory in Toulouse had established that a manganese(III) porphyrin complex can efficiently catalyze autoxidation of sulfite generating a Mn(V) oxo intermediate capable of effecting DNA oxidation that appeared to occur at both sugar and base residues 〚10〛. Because the simple manganese salts and oxides might more likely be important in inhalation and ingestion than multidentate complexes and furthermore would be incapable of forming Mn(V) oxo species, we anticipated that their DNA reactivity would be different. The present study examines DNA damage in both plasmid DNA and a 167-bp restriction fragment induced by sulfite in the presence of various Mn(II) and Mn(III) compounds. Sulfite and manganese, present in atmospheric aerosols in urban environments, have been shown to act synergistically in redox reactions 〚17〛.
2 Results and discussion
As early as 1972, studies reported that DNA underwent oxygen-dependent strand cleavage in the presence of 10 mM sulfite and 1 mM MnCl2 〚18〛, but little was known about the mechanism. Later, a nucleoside study indicated that 2’-deoxyguanosine could be converted to 8-oxo-7,8-dihydro-2’-deoxyguanosine by the action of sulfite autoxidation in the presence chromium(VI) 〚13〛 and iron(III), but manganese(II) was much less effective 〚19〛. In related work, manganese(III)-catalyzed autoxidation of hydrazine was proposed to generate radical intermediates that could cause oxidative DNA damage 〚20〛. For the present study, two types of DNA targets were selected: pUC-19 is a circularly closed, supercoiled plasmid that is a sensitive target for assessing overall levels of oxidative damage in an agarose gel assay while polyacrylamide gel electrophoresis studies of a restriction fragment provide information on sequence and base specificity. In addition, the initial formation of sulfite radical was confirmed by an EPR trapping study.
2.1 EPR trapping study of sulfate radical
Radicals generated by 10 mM manganese(II) or manganese(III) phosphate and 0.1 M sulfite in 10 mM NaH2PO4 (pH 7.2) were trapped with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). The EPR spectra obtained from reactions catalyzed by Mn(II) vs Mn(III) are shown in Fig. 1a and b, respectively. The spectra clearly show enhanced radical formation for the use of Mn(III) phosphate. The hyperfine splitting was aN = 14.7 G, aH = 16.0 G, which compare favorably to the literature values of the DMPO/SO3•– adduct (aN = 14.7 G, aH = 15.9 G) 〚21〛. Neither the sulfate radical adduct (aN = 13.9 G, aH = 10.1 G) nor the hydroxyl radical adduct (aN = aH = 14.4 G) was observed. These results are entirely consistent with the proposed mechanism that requires oxidation of manganese(II) to manganese(III) in order to initiate sulfite oxidation. Therefore, beginning with a pre-formed manganese(III) catalyst increases the efficiency of SO3•– production.
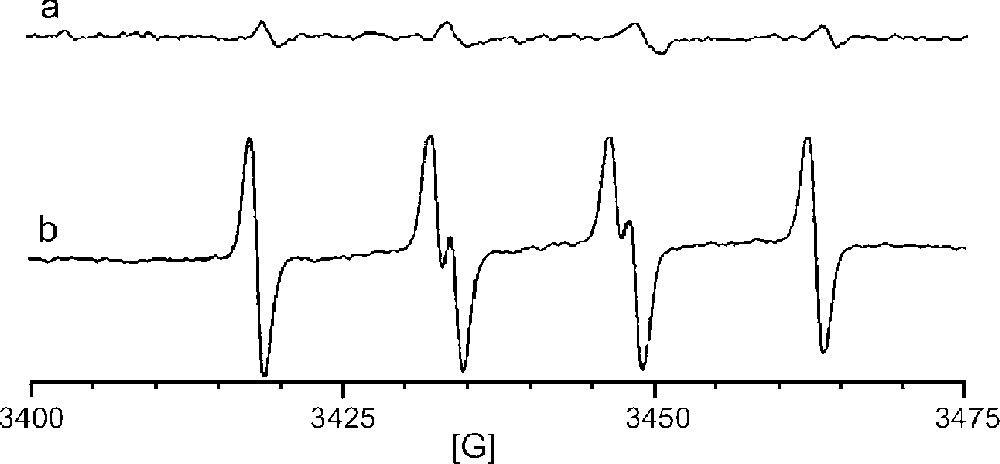
EPR spectra of radical adducts trapped by DMPO in the reaction of 0.1 M Na2SO3 in aerated aqueous NaH2PO4 buffer (pH 7.2) catalyzed by (a) 10 mM MnHPO4 (manganese(II)), or (b) by 10 mM MnPO4 (manganese(III)).
That other radicals, SO4•–, SO5•–, or HO•, were not observed is not surprising because DMPO was present in sufficient excess when the reaction was initiated to presumably trap all of the SO3•– as it was formed. In the absence of a trapping agent, however, one would expect that sulfite radical might be trapped by O2 as shown in equation (2) (〚O2〛 ∼ 250 μM; k25 = 109 M–1 s–1) faster than it encounters DNA (〚DNA nucleotides〛 < 1 μM) to generate SO5•–, another potentially damaging radical. Once SO5•– is converted to HSO5– (equation (3)), there are two additional pathways for further generation of radicals. Monoperoxysulfate can undergo reductive O–O bond cleavage in the presence of Mn(II) to generate either sulfate radical plus hydroxide ion (equation (4)) or sulfate anion plus hydroxyl radical (equation (5)).
In order to gain further information about the nature of the radicals involved in DNA damage from sulfite autoxidation, we investigated both large DNA targets (plasmid DNA) as well as DNA restriction fragments.
2.2 Plasmid nicking by manganese-catalyzed sulfite autoxidation
Plasmid DNA provides a very sensitive assay of DNA damage because an oxidative event occurring anywhere in the few thousand base pairs of the circularly closed duplex leads to formation of the same new band on a gel. No information is gained about site specificity, but plasmid nicking is nevertheless a convenient means of comparing the relatively reactivity of various metal complexes and oxidation conditions. In the present study, the pUC-19 plasmid was used in conjunction with a variety of manganese oxides and salts. Manganese oxides, including MnO, MnO2, and Mn3O4, are the principal components of combustion of MMT-containing fuel. Unfortunately, MnO and MnO2 were insufficiently soluble to give reproducible results under the buffer conditions employed, although the mixed valence compound Mn3O4 was well behaved. Other compounds selected for study included the simple manganese(II) salts MnCl2 and MnSO4. Manganese(III) salts of phosphate and pyrophosphate are able to be prepared, and so these were compared to the corresponding Mn(II) phosphate and pyrophosphate compounds. Because radical formation is dependent upon the presence of both Mn(II) and Mn(III) oxidation states at various points in the mechanism, studies were also conducted using a 1:1 mixture of Mn(II) and Mn(III) compounds for the phosphate and pyrophosphate anions.
Experiments were conducted with 50 μM manganese and 5 mM sulfite at pH 7.2 in phosphate buffer (10 mM) at 37 °C for 40 min. Oxidative damage to the plasmid was assayed as the amount of conversion of circularly closed, double-stranded DNA (form I) compared to nicked DNA (form II), as observed on an agarose gel. Background cleavage by sulfite in the absence of metal ions was subtracted from the raw data in order to obtain the values shown in Fig. 2. Consistent with the EPR data and the proposed mechanism, manganese(III) compounds are found to be more effective in generating oxidative damage to DNA. The use of mixtures of Mn(II) and Mn(III) compounds had essentially no effect compared to the use of Mn(III) alone. Interestingly, the results indicate that the fuel additive product Mn3O4 was reasonably active as a sulfite autoxidation catalyst according to this assay.
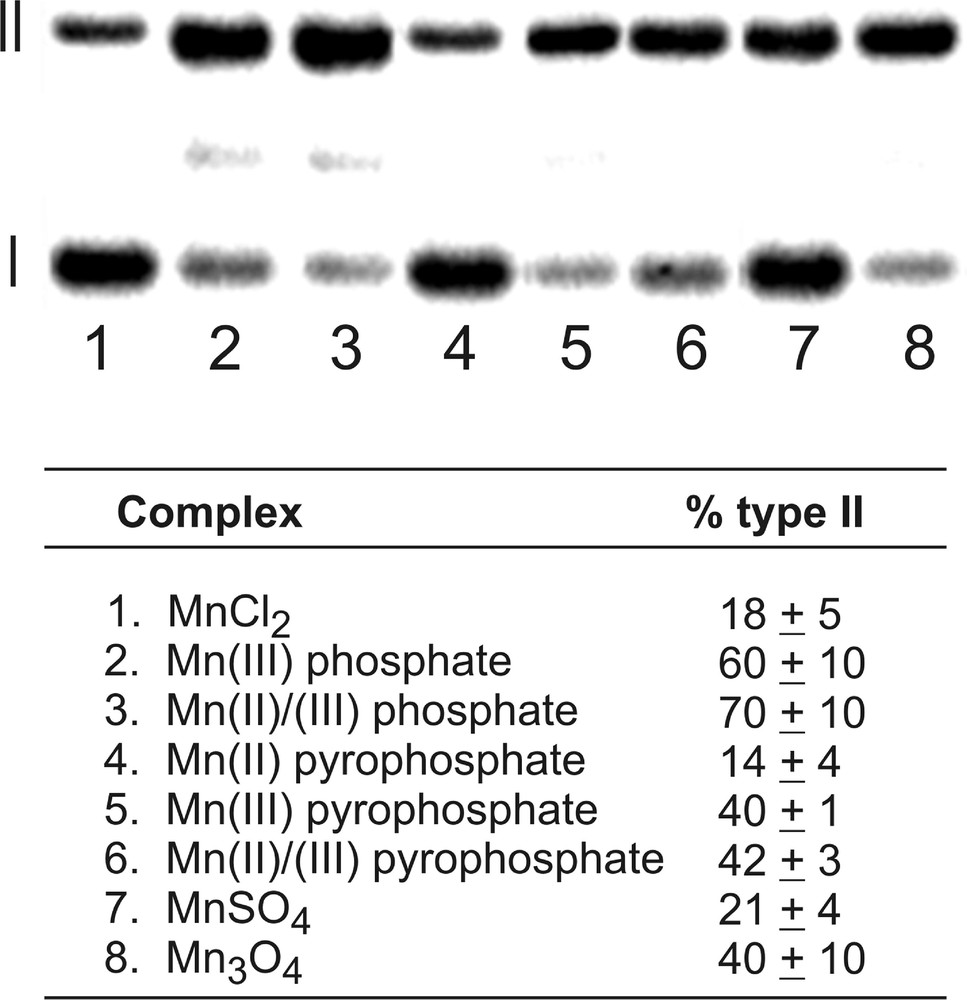
Manganese-catalyzed sulfite autoxidation mediates conversion of form I (closed, circular duplex DNA) to form II (nicked DNA). Reactions were carried out with 50 μM Mn and 5 mM Na2SO3 in 10 mM NaH2PO4 (pH 7.2) with 100 mM NaCl at 37 °C for 1 h. Percentages listed are the averages of three trials and are normalized with respect to a background reaction in the absence of manganese.
Hydroxyl radical is sufficiently reactive to be quenched by the addition of virtually any alcohol; both ethanol and tert-butyl alcohol react with HO• with second-order rate constants that are nearly diffusion controlled, 2 × 109 and 6 × 108 M–1s–1, respectively 〚22〛. On the other hand, sulfate radical is considerably less reactive, and is quenched more rapidly by primary alcohols such as ethanol compared to tert-butyl alcohol (2 × 107 vs 4 × 105 M–1s–1, respectively) 〚23〛. Rate constants for ethanol quenching of SO3•– and SO5•– are much lower, ∼103 M–1s–1 or less 〚23〛, and quenching by tert-butyl alcohol is presumably slower. We have previously employed alcohol quenching studies to help elucidate the radical species responsible for DNA oxidation 〚24, 25〛. Thus, the reactivity of plasmid DNA toward manganese-catalyzed sulfite autoxidation in the presence of alcohols could provide additional insight. Accordingly, experiments were conducted under the same conditions as those for the data obtained in Fig. 2 with the addition of either 25 mM ethanol or 25 mM tert-butyl alcohol. In all cases, the amount of plasmid nicking was reduced to essentially background levels. The interpretation most consistent with this data would be that hydroxyl radical is generated and is responsible for DNA damage. It therefore would be effectively quenched by the addition of either ethanol or tert-butyl alcohol. In order to obtain further evidence for the involvement of hydroxyl rather than sulfate radical, we turned to higher resolution DNA experiments to examine base specificity.
2.3 Oxidation of a DNA restriction fragment by manganese-catalyzed sulfite autoxidation
Rate constants are known for the oxidation of various nucleosides by sulfate radical and show an order of reactivity of dG > dA > dC ∼ dT >> ribose 〚23, 26〛. Because sulfate radical shows pronounced base selectivity and because purine oxidation typically requires post-treatment with a base such as piperidine to reveal DNA strand scission 〚27〛, we would predict that DNA damage by sulfate vs hydroxyl radical would be further distinguished by base specificity. Experiments were therefore conducted with a 167-bp duplex DNA restriction fragment obtained from enzymatic cleavage of pBR322. Sulfite autoxidation was performed using Mn(III) phosphate (50 μM manganese and 5 mM sulfite at pH 7.2 in phosphate buffer (10 mM) at 37 °C for 20 min, and the DNA samples were analyzed by polyacrylamide gel electrophoresis both before and after hot piperidine treatment.
Table 1 shows the relative reactivity of the four bases as well as the total extents of direct strand scission and piperidine-induced strand scission for manganese-catalyzed sulfite autoxidation compared to a system known to generate HO• (FeEDTA with H2O2 〚28〛) and a system known to generate SO4•– (sulfite autoxidation catalyzed by a nickel peptide 〚24〛). The studies on base selectivity were conducted after piperidine treatment for all three systems and are reported as relative reactivities with guanosine arbitrarily set to 1. Since sulfate radical reacts almost exclusively at DNA bases, principally guanine, no strand scission is observed unless treated with piperidine. Hydroxyl radical reacts at both sugar and base residues, and the total cleavage of DNA is enhanced nearly threefold by piperidine treatment, although substantial direct strand scission was also observed. An additional characteristic of hydroxyl radical is its base neutrality; essentially all nucleosides are equally reactive. The data obtained for manganese(III)-catalyzed sulfite autoxidation are most consistent with generation of hydroxyl radical because direct strand scission and diminished base selectivity are observed. However, the slight preference for dG oxidation and the nearly five-fold enhancement upon piperidine treatment suggest that the mechanism may be complicated by the additional formation of sulfate radical to some extent.
Relative base specificity of reactive intermediates generated in three different systemsa.
| Target | MnIIIPO4/ SO32– | FeEDTA/H2O2 | NiIIKGH/SO32– |
| (?) | (HO•) | (SO4•–) | |
| G | 1.0 | 1.0 | 1.0 |
| A | 0.7 | 0.8 | 0.1 |
| C | 0.7 | 1.1 | 0.2 |
| T | 0.7 | 0.7 | 0.1 |
| Total direct scission | 4 | 28 | not detected |
| Total piperidine-induced scission | 19 | 64 | 35 |
2.4 Mechanistic conclusions
As proposed by previous investigations 〚1, 4–6〛, it is evident from these studies that manganese(III) is much more active to initiate sulfite autoxidation compared to manganese(II). In a weak ligand field, manganese(II) is the preferred oxidation state; however, anions such as phosphate and pyrophosphate make the +III oxidation state more accessible. The higher reactivity of manganese(III) phosphate and pyrophosphate compounds in this study suggest that binding of Mn to the phosphate backbone of DNA or to other nucleotides in the cell could direct the sulfite autoxidation catalyst to its eventual target for oxidative damage. This differs from the chemistry of sulfite autoxidation catalyzed by nickel(II) complexes in which guanine-specific oxidative damage is observed 〚24〛.
That SO3•– is initially formed in the Mn-catalyzed oxidation of sulfite is evident from the EPR studies in which DMPO, present in excess, generated a characteristic spectrum of the adduct. However, this radical is unlikely to be the actual species that reacts with DNA because of its low redox potential (∼0.6 V/NHE at pH 7 〚23〛) and its high reactivity with O2. The only kinetic data available for reaction of sulfite radical with a DNA base is for adenine in which the rate constant is <106 〚23〛, while the O2 reaction has a rate constant is essentially diffusion controlled. Little is known about the DNA reactivity of the subsequently formed SO5•–, but its redox potential (∼1.1 V/NHE at pH 7 〚23〛) is still low enough compared to DNA bases (1.29 V. vs NHE for G 〚29〛) that it should react in a G-specific manner with DNA, if it reacts at all. It is well known that guanosine has the lowest redox potential of the four nucleosides in DNA, and reactions that proceed by a process of one-electron abstraction do so with high G selectivity. This is the case for the nickel(II) peptide-catalyzed sulfite autoxidation system, for example 〚24〛.
In the redox cycling of manganese-catalyzed sulfite autoxidation, monoperoxysulfate anion (HSO5–) is formed by re-oxidation of manganese(II). This anion has very low reactivity with DNA by itself, but in the presence of transition metal ions can generate either SO4•– and/or HO•. The fact that both ethanol and tert-butyl alcohol are effective quenchers of the species involved in DNA oxidation as well as the observation of direct strand scission and low base selectivity point to the involvement of hydroxyl radical as the key species producing DNA damage. The results are not as clear as in Fenton chemistry (FeEDTA + H2O2), and it is likely that some formation of sulfate (or another) radical competes. Overall, the results suggest that equation (5) is more important than equation (4) in generating DNA damaging agents.
The coordination of manganese(III) to stronger-field, multidentate ligands such as porphyrins opens different avenues in the mechanism of sulfite autoxidation and DNA damage, notably the possibility to bind in specific ways to duplex DNA and to form a Mn(V)=O species, the chemistry of which has been richly explored 〚10, 30〛. The studies reported herein demonstrate that even simple manganese compounds relevant to environmental pathology may act in conjunction with sulfite leading to DNA oxidation. Oxidative DNA damage has been identified as a contributor to aging, neurological disease and cancer.
3 Experimental section
3.1 General
Sodium pyrophosphate was obtained from Acros, MnCl2·4 HCl (99.99%), MnO, MnO2, Mn3O4, Mn5(PO4)〚PO3(OH)〛2 (manganese(II) phosphate) from Aldrich, and diethylenetriaminepentaacetic acid (DTPA) from Sigma. Luria Broth, ampicillin, pBR322 DNA plasmid and maximum efficiency DH5α E. coli cells were purchased from Gibco BRL. All buffer solutions and the anion-exchange resin used in the preparation of pUC-19 plasmid DNA were furnished in the Qiagen Plasmid Maxi Kit. A 167-bp restriction fragment from the pBR322 plasmid DNA was prepared as previously reported 〚24〛. Manganese(III) pyrophosphate and manganese(III) phosphate were prepared by previously described methods 〚31〛.
All solutions and materials for use with plasmids were sterilized in an autoclave prior to use. All buffer solutions and water (Millipore 18 MΩ cm–1) used in reactions were passed through a Chelex-100 (Bio-Rad) column and contained 1 μM DTPA.
Restriction fragment DNA and plasmid DNA were visualized by phosphorimagery (Molecular Dynamics Storm 840) using ImagequaNT software. UV–vis spectra were obtained with a Beckman DU650. EPR spectra were obtained with a Bruker EMX Spectrometer at X-band (9.6 GHz). The spectrometer was equipped with a TM110 cylindrical cavity. Spectra were analyzed with WinEPR software.
3.2 Trapping of sulfite radical with DMPO
A solution of Na2SO3 (0.1 M, 13 mg) in 10 mM NaH2PO4 (pH 7.2) was aerated with O2 for 1 min; a final concentration of 0.1 M DMPO (5,5-dimethyl-1-pyrroline-N-oxide) was added followed by 1 mM of metal complex. Reactions of 1-ml volume were carried out at room temperature in a 1.5 ml microcentrifuge tube and then transferred to a flat cell immediately prior to attaining a spectrum.
3.3 Cleavage of pUC-19 DNA plasmid by Mn(II)/(III) and complexes
The plasmid (150 pmole/reaction) was diluted in 10 mM NaH2PO4 buffer at pH 7.2. The reactions were incubated at 37 °C for 40 min in a total volume of 50 μl. The reactions were quenched with 2 mM HEPES/10 mM EDTA and precipitated with iso-propanol. Loading dye (10% glycerol, 0.05% bromophenol blue, 0.05% xylene cyanol) was added, and the samples run on a 1% agarose gel. The gels were dyed for 2 h with SyberGreen, and visualized by luminescence.
3.4 Cleavage of 167-bp fragment of pBR322 plasmid DNA by Mn(II)/(III) and complexes
Reactions were carried out in 10 mM NaH2PO4 (pH 7.2), 100 mM NaCl, 5’-〚32P〛 end-labeled restriction fragment (20 000 cpm/reaction) and 100 nM calf thymus DNA in 50 μl total volume. They were incubated at 37 °C for 40 min, quenched with 2 mM HEPES + 10 mM EDTA, and dialyzed with 3 × 3.5 l H2O. The dialyzed samples were lyophilized to dryness, treated with 1 M piperidine for 30 min at 90 °C, lyophilized to dryness again and washed with 2 × 20 μl H2O. Fragments were separated on an 8% polyacrylamide/7 M urea denaturing gel and visualized by phosphorimagery.
Acknowledgements
This research was supported by a grant from the US National Science Foundation (to CJB) and funds (to RAJ) from a National Institutes of Health training grant (GM08537) and an EPA-STAR graduate fellowship from the US Environmental Protection Agency.