

1 Introduction
Protein-protein interactions play a key role in all biological processes, including cell growth and differentiation 〚1〛. In many cases, these interactions are mediated through a large surface area contact, as seen in protein oligomerization and antibody-antigen complexes. The design of synthetic agents that can recognize and bind to specific regions of a protein surface should offer new approaches to the development of enzyme inhibitors 〚2〛 and protein antagonists 〚3〛. We have recently introduced a new class of protein surface receptors, in which four identical peptide loops are attached to a central calixarene scaffold 〚4〛. The peptide loops are based around a cyclic tetrapeptide, which is cyclized through a 4-aminomethyl benzoic acid group. The hydrophobic spacer provides not only a rigidification of the macrocycle, but also a point of attachment through 5-amino substituents on the phenyl ring. The amine groups are then linked to the calixarene through carboxylic acid activation. The resulting design contains a concave molecular surface approximately 450–500 Å2 in area, whose recognition characteristics can be readily changed by varying the sequences of the cyclic peptides as well as attaching different peptide loops (Fig. 1).

Structure of compound 1.
For example, compound 1, containing four peptide loop with the sequence Gly–Asp–Gly–Asp, has a hydrophobic core, comprising the calixarene and aminomethylbenzoate spacer and a hydrophilic periphery, defined by the eight carboxylate groups in the peptide ring. A calculated structure for the tetra-loop derivative (Fig. 2) shows the concentric arrangement of the hydrophilic and hydrophobic domains and the large surface area (∼ 450 Å2) available for contact to a protein target. We have shown that this derivative binds tightly to the cationic surface of cytochrome c and disrupts its interaction with its native protein partner, cytochrome c peroxidase 〚5〛. The exact site on the surface of cytochrome c where 1 is fixed has not yet been definitively established. However, one strong possibility is the region close to the heme edge, surrounded by the group of positively charged residues (Lys-17, 18, 21, 77, 88), which forms the primary interaction surface with electron transfer partners such as cytochrome c oxidase and cytochrome c peroxidase. Docking a calculated structure for 1 with this region on the X-ray structure of cytochrome c 〚6〛 (Fig. 3) confirms that four peptide loops can contact four of the five lysine residues and cover a large area of the protein surface. Consistent with this structure, we have shown that compound 1 disrupts not only the cytochrome c/cytochrome c peroxidase complex 〚5〛, but also the approach of reducing agents to the heme edge 〚7〛. In phosphate buffer, Fe(III) cytochrome c (1.57 × 10–5 M) is rapidly reduced by excess ascorbate (2.0 × 10–3 M) with a pseudo first-order rate constant of 0.109 ± 0.0001 s–1. In the presence of 1 (1.91 × 10–5 M), the rate of cytochrome c reduction is diminished ten-fold (kobs = 0.010 ± 0.001 s–1) in a similar manner to that seen with the cytochrome c cytochrome c peroxidase complex 〚5〛.

The calculated structure for tetra-loop calixarene derivative 1.
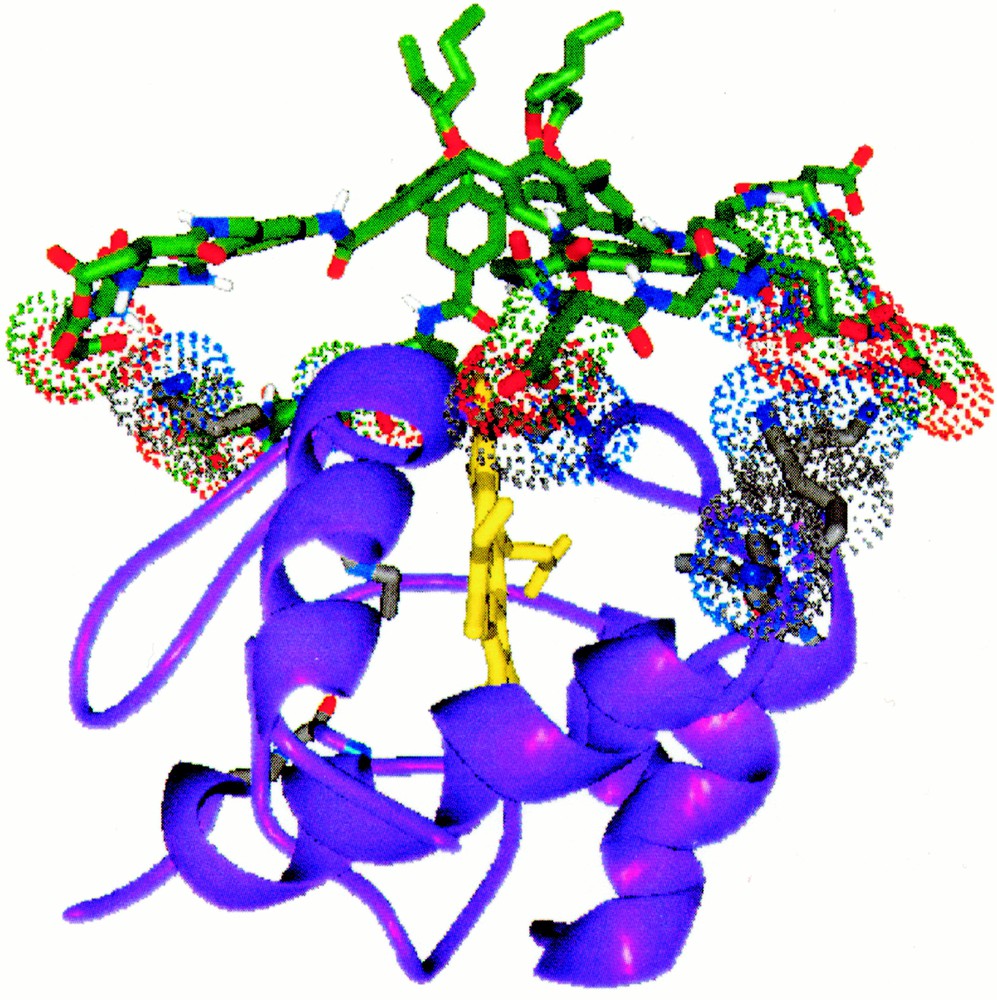
The calculated complex structure between cytochrome c and compound 1, the contacting basic residues of cytochrome c are shown in polytube model.
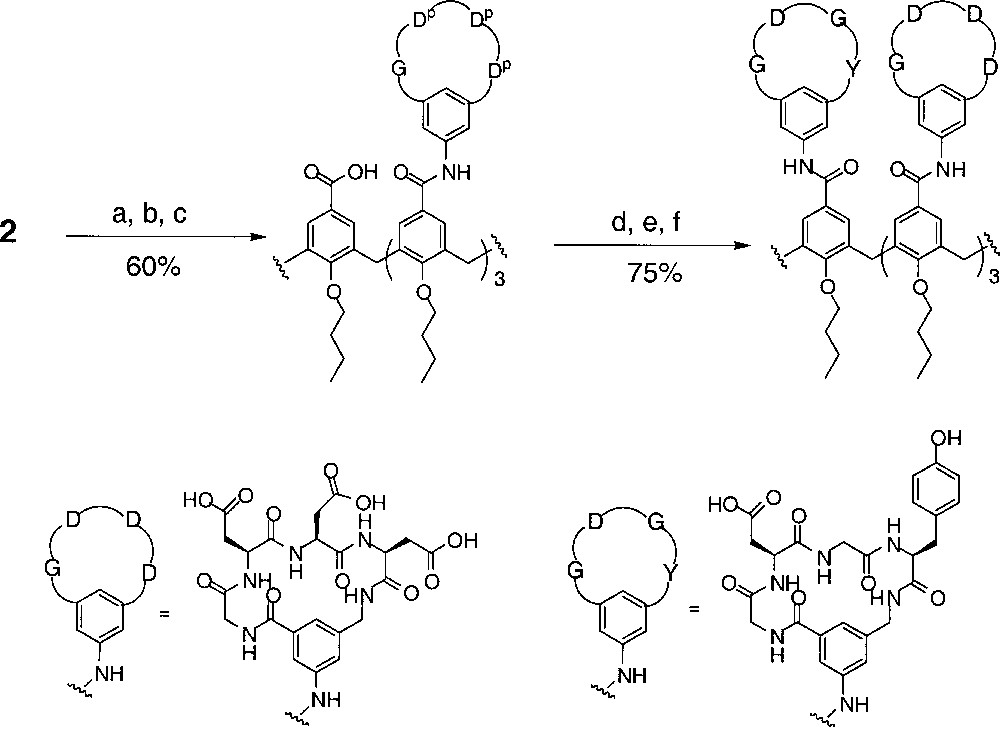
The design and simple synthesis of 1 results in a protein-binding agent with fourfold symmetry containing four identical loops on the four phenyl groups of the calix〚4〛arene core. While leading to ready access of 1 in large quantities, this feature is limiting, particularly since the distribution of charged and hydrophobic domains on protein surfaces is invariably irregular. We therefore needed a synthetic approach that would lead to a less symmetrical arrangement of the peptide loops around the core scaffold. In the present paper, we report an important step in the wider application of this strategy with the preparation of a series of unsymmetrical receptors, in which two different loops are attached to the core calixarene (Fig. 4).

Design of unsymmetrical receptors based on a calix〚4〛arene scaffold.
2 Results and Discussion
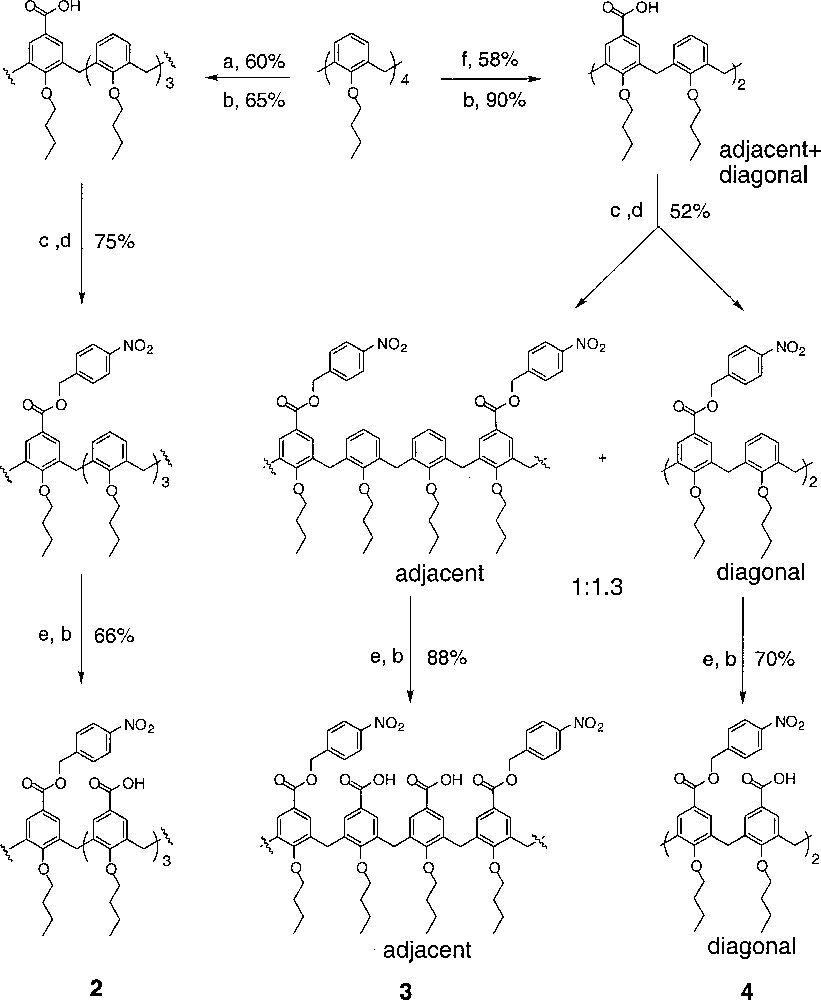
Calix〚4〛arene has been extensively used in the supramolecular chemistry, due to its well-defined shape 〚8〛. However, few examples exist in which the upper rim of calixarene has been differentially functionalized 〚9〛. In our effort to attach two different peptide loops onto the calixarene scaffold, we envisioned that three partially protected calix〚4〛arene tetracarboxylic acid derivatives, such as (2), (3) and (4), should lead to unsymmetrical receptors through sequencial coupling steps. Initial attempts to selectively protect butoxycalix〚4〛arene tetracarboxylic acid were unsuccessful, resulting in inseparable mixtures. However, stepwise functionalization of the upper rim of calix〚4〛arene could be carried out efficiently and gave all three partially protected derivatives in good yields (Fig. 5) 〚10〛. Treating butoxycalix〚4〛arene with 1 equiv Cl2CHOCH3 in the presence of 1 equiv TiCl4 gave primarily mono-formylated product in 60% yield. Subsequent oxidation and protection afforded a mono-calix〚4〛arene carboxylester. p-Nitro benzyl ester was chosen due to its easy removal by hydrogenation after coupling to the peptide loops (Fig. 6). Further formylation with excess TiCl4 and oxidation with NaClO2 furnished the final product as a mono-protected butoxycalix〚4〛arene tetracarboxylic acid (2). The other scaffolds were prepared in an analogous manner. Formylation of butylcalix〚4〛arene with SnCl4 gave a mixture of bis-formylated derivatives, which could not be separated by flash chromatography. The mixture was carried on to the bis-ester stage, after which the two isomers could be easily separated by column in a 1:1.3 cis/trans ratio. The bis-acids were protected as para-nitrobenzyl esters respectively. Further formylation and oxidation afforded the final products, cis-calix〚4〛arene bisacid bisester (3) and trans-calix〚4〛arene bisacid bisester (4), which set up all three required scaffolds for our receptor constructions.
(a) 1 equiv Cl2CHOCH3, 1 equiv TiCl4, CH2Cl2, –10 °C; (b) NaClO2, H2NSO3H, CH2Cl2, acetone, H2O; (c) (COCl)2, cat. DMF, CH2Cl2; (d) p-nitrobenzyl alcohol, N(iPr)2Et, CH2Cl2; (e) Cl2CHOCH3, TiCl4 (excess), CH2Cl2, –10 °C; (f) Cl2CHOCH3, SnCl4, CH2Cl2, –10 °C.
(a) (COCl)2, cat. DMF, CH2Cl2; (b) cyclo-(Gly–Asp(OtBu)–Asp(OtBu)–Asp(OtBu)–Spc)–NH2, N(iPr)2Et, CH2Cl2; (c) 10% Pd/C, H2, MeOH; (d) 2,4,6-trichlorobenzoyl chloride, NEt3, THF; (e) cyclo-(Gly–Asp(OtBu)–Gly–Tyr(OtBu)–Spc)–NH2, DMAP, benzene/CH2Cl2; (f) 25% TFA/CH2Cl2.
The use of the scaffold 2 for the preparation of the unsymmetrical receptors is illustrated in Fig. 6. The first coupling proceeded using a straightforward acid chloride method 〚11〛. After removal of the para-nitrobenzyl protecting group under hydrogenation conditions, the acid was converted to a Yamaguchi type anhydride 〚12〛, and subsequently reacted with the second peptide loop to give the fully protected calix〚4〛arene tetra-cyclic peptide receptor. Removal of the protecting groups by TFA treatment gave the final product. A series of A3B receptors were prepared in good yields by this route (Table 1).
Unsymmetrical receptors.
| Sequence | Yield | Molecular weight | |
| expected (M+H+) | determined (ES–MS) | ||
| X-3(GDDD)-GDGY | 45% | 2945.82 | 2945.50 ± 0.55 |
| X-3(GDGY)-GDDG | 45% | 2867.88 | 2867.37 ± 0.46 |
| X-3(GDGY)-GDDY | 52% | 2974.00 | 2973.75 ± 0.87 |
| X-3(GDGY)-GDDD | 51% | 2925.91 | 2926.01 ± 0.31 |
| X-3(GDGD)-GDGY | 43% | 2771.71 | 2771.22 ± 1.30 |
| X-3(GDGD)-GDDD | 57% | 2781.65 | ? |
| X-3(GDGD)-GDDY | 54% | 2829.75 | 2828.74 ± 0.45 |
| trans-X-2(GDDG)-2(GDGY) | 26% | 2819.80 | 2818.79 ± 0.72 |
| cis-X-2(GDDG)-2(GDDY) | 19% | 2935.88 | 2935.62 ± 0.07 |
Similar routes were successfully performed for the preparations of cis-A2B2 and trans-A2B2 types of receptors (Fig. 7).

(a) (COCl)2, cat. DMF, CH2Cl2; (b) cyclo-(Gly–Asp(OtBu)–Asp(OtBu)–Gly–Spc)–NH2, N(iPr)2Et, CH2Cl2; (c) 10% Pd/C, H2, MeOH; (d) 2,4,6-trichlorobenzoyl chloride, NEt3, THF; (e) cyclo-(Gly–Asp(OtBu)–Asp(OtBu)–Tyr(OtBu)–Spc)–NH2, DMAP, benzene/CH2Cl2; (f) 25% TFA/CH2Cl2; (g) cyclo-(Gly–Asp(OtBu)–Gly–Tyr(OtBu)–Spc)–NH2, DMAP, CH2Cl2.
In summary, we have developed an efficient route for stepwise functionalization of the upper rim of calix〚4〛arene. It should be noted that functional groups other than carboxylate can also be incorporated into the synthetic scheme. A series of unsymmetrical receptors based on partially protected calix〚4〛arene tetra-carboxylic acid was thus constructed. The biological testing of the unsymmetrical receptors for various protein targets is underway and will be reported in due course.
3 Experimental Details
3.1 Butoxycalix〚4〛arene mono carboxylic acid
A 50-ml round flask was charged Cl2CHOCH3 (98 μl, 1.08 mmol), butoxycalix〚4〛arene (0.636 g, 0.98 mmol) and dry CH2Cl2 (20 ml), and the solution was cooled to –10 °C. TiCl4 (0.13 ml, 1.19 mmol) was added and the mixture was stirred at –10 °C → –5 °C for 1 h. The reaction was quenched with 50 ml H2O, and the organic layer was separated and dried over Na2SO4. After evaporation solvents, the residue was purified by flash chromatography (SiO2, 5% EtOAc/hexane) to give the monoaldehyde as an oil (0.393 g, 60%): 1H NMR (CDCl3, 500 MHz) δ 9.60 (s, 1H), 7.03 (s, 2H), 6.77 (m, 4H), 6.71 (m, 2H), 6.44 (m, 3H), 4.52 (d, J = 13.6 Hz, 2H), 4.47 (d, J = 13.4 Hz, 2H), 3.97 (m, 4H), 3.94 (m, 6H), 3.92 (m, 2H), 3.26 (d, J = 13.6 Hz, 2H), 3.19 (d, J = 13.5 Hz, 2H), 1.91 (m, 8H), 1.53 (m, 4H), 1.44 (m, 4H), 1.05 (m, 12H); 13C NMR (CDCl3, 125 MHz) δ 191.7, 162.0, 156.7, 156.2, 136.0, 135.8, 134.7, 134.6, 130.9, 130.0, 128.8, 128.2, 127.9, 122.2, 121.8, 75.0, 74.8(2), 32.4, 32.3(2), 32.2, 30.9, 30.7, 19.6, 19.4, 19.3, 19.2, 14.1, 14.0, 13.9. To the solution of the aldehyde (0.39 g, 0.58 mmol) in 10 ml CH2Cl2 and 30 ml acetone was added H2NSO3H (0.60 g, 6.2 mmol) in 5 ml H2O and NaClO2 (0.64 g, 5.8 mmol) in 5 ml H2O, and the mixture was stirred at room temperature overnight. The organic solvents were then removed under reduced pressure and the aqueous solution was extracted with CH2Cl2. The organic layer was separated and dried over Na2SO4. After evaporation solvents, the residue was purified by flash chromatography (SiO2, 3% MeOH/CH2Cl2) to give the final product as a white powder (0.260 g, 65%): mp 187-188 °C; 1H NMR (CDCl3, 300 MHz) δ 11.13 (s, b, 1H), 7.36 (s, 2H), 6.67 (m, 6H), 6.53 (m, 3H), 4.49 (d, J = 13.5 Hz, 2H), 4.46 (d, J = 13.2 Hz, 2H), 4.00 (t, J = 7.2 Hz, 2H), 3.90 (m, 6H), 3.24 (d, J = 13.5 Hz, 2H), 3.18 (d, J = 13.5 Hz, 2H), 1.95 (m, 8H), 1.48 (m, 8H), 1.03 (m, 12H); 13C NMR (CDCl3, 75 MHz) δ 172.3, 161.5, 156.3, 135.4, 135.2, 134.8, 134.2, 130.3, 128.3, 128.0, 127.9, 122.5, 122.0, 121.7, 74.7, 74.6, 32.1, 30.7, 19.1, 13.9(2); HR FAB-MS m/e calculated for C45H56O6 692.4077 〚M〛+, found 692.4079.
3.2 Butoxycalix〚4〛arene mono carboxylic acid 4-nitrobenzyl ester
To a solution of butoxycalix〚4〛arene mono carboxylic acid (0.255 g, 0.37 mmol) in 20 ml dry CH2Cl2 was added oxalyl chloride (0.40 ml, 4.4 mmol) and a catalytic amount of DMF (0.4 μl), and the mixture was stirred at room temperature overnight. The solvent and excess reagent were removed in vacuo and the residue was redissolved in 10 ml dry CH2Cl2; 4-nitrobenzyl alcohol (0.58 g, 3.8 mmol) and DIEA (0.2 ml, 1.1 mmol) were added to the above solution, and the mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was purified by flash chromatography to give the title compound as a white powder (0.227 g, 75%): mp 57–58 °C; 1H NMR (CDCl3, 300 MHz) δ 8.27 (d, J = 8.7 Hz, 2H), 7.50 (d, J = 8.7 Hz, 2H), 7.22 (s, 2H), 6.76 (m, 6H), 6.40 (d, J = 7.5 Hz, 2H), 6.22 (t, J = 7.5 Hz, 1H), 5.34 (s, 2H), 4.48 (d, J = 12.9 Hz, 2H), 4.44 (d, J = 12.3 Hz, 2H), 3.98–3.82 (m, 8H), 3.22 (d, J = 13.5 Hz, 2H), 3.16 (d, J = 13.5 Hz, 2H), 1.89 (m, 8H), 1.46 (m, 8H), 1.01 (m, 12H); 13C NMR (CDCl3, 75 MHz) δ 165.9, 161.0, 156.8, 156.3, 147.5, 143.9, 135.8, 135.4, 134.9, 134.7, 129.8, 128.6, 128.3, 127.8, 123.7, 122.8, 122.0, 121.5, 74.9, 74.8, 64.3, 32.3, 32.2, 30.9, 19.4, 19.3, 19.2, 14.1, 14.0(2); HR FAB-MS m/e calculated for C52H62NO6 828.4475 〚M+H〛+, found 828.4476.
3.3 Butoxycalix〚4〛arene mono-4-nitrobenzyl carboxylate tris-carboxylic acid (2)
The solution of butoxycalix〚4〛arene mono carboxylic acid 4-nitrobenzyl ester (0.226 g, 0.27 mmol), dichloromethyl methyl ether (0.30 ml, 3.3 mmol) in 20 ml dry CH2Cl2 was cooled to –10 °C, and TiCl4 (0.40 ml, 3.65 mmol) was added. The mixture was stirred overnight while warming up to room temperature. The reaction was then quenched with 20 ml 1.0 N HCl and the organic layer was separated and dried over Na2SO4. After evaporation of solvents, the residue was purified by flash chromatography (SiO2, 30% EtOAc/hexane) to give the trisaldehyde as a white foam (0.193 g, 78%): 1H NMR (CDCl3, 300 MHz) δ 9.62 (s, 2H), 9.55 (s, 1H), 8.24 (d, J = 8.7 Hz, 2H), 7.53 (d, J = 8.7 Hz, 2H), 7.30 (s, 2H), 7.22 (s, 4H), 7.12 (s, 2H), 5.32 (s, 2H), 4.52 (d, J = 7.8 Hz, 2H), 4.47 (d, J = 7.8 Hz, 2H), 3.97 (m, 8H), 3.37 (d, J = 9.6 Hz, 2H), 3.32 (d, J = 9.6 Hz, 2H), 1.88 (m, 8H), 1.45 (m, 8H), 1.01 (t, J = 7.2 Hz, 12H); To the above aldehyde in 6 ml CH2Cl2 and 18 ml acetone was added H2NSO3H (0.30 g, 3.1 mmol) in 2 ml H2O and NaClO2 (0.32 g, 2.9 mmol) in 2 ml H2O, and the mixture was stirred at room temperature overnight. The organic solvents were then removed under reduced pressure and the precipitate was filtered off and dried in vacuo to give the title compound as an off-white powder (0.173 g, 85%): mp > 300 °C (dec.); 1H NMR (DMSO-d6, 500 MHz) δ 8.22 (d, J = 7.9 Hz, 2H), 7.73 (s, 2H), 7.71 (s, 2H), 7.47 (d, J = 8.2 Hz, 2H), 6.89 (s, 2H), 6.88 (s, 2H), 5.20 (s, 2H), 4.36 (d, J = 6.0 Hz, 2H), 4.34 (d, J = 5.7 Hz, 2H), 4.06 (m, 4H), 3.79 (t, J = 6.5 Hz, 2H), 3.76 (t, J = 6.7 Hz, 2H), 3.42 (d, J = 12.6 Hz, 2H), 3.40 (d, J = 12.8 Hz, 2H), 1.86 (m, 8H), 1.57 (m, 4H), 1.33 (m, 4H), 0.97 (m, 12H); LR FAB-MS m/e calculated for C55H61NO14 959.4 〚M〛+, found 959.5.
3.4 trans-Butoxycalix〚4〛arene bis(4-nitrobenzylcarboxylate) (A); cis-butoxycalix〚4〛arene bis(4-nitrobenzylcarboxylate) (B)
To a 100-ml flask was charged butoxycalix〚4〛arene (1.35 g, 2.1 mmol) Cl2CHOCH3 (0.76 ml, 8.4 mmol) and 50 ml dry CH2Cl2, and the solution was cooled to –10 °C. SnCl4 (1.0 M in CH2Cl2, 8.4 ml, 8.4 mmol) was added and the mixture was stirred at –10 °C for 30 min. The reaction was then quenched with 30 ml H2O, and the organic layer was separated and dried over Na2SO4. After evaporation of solvents, the residue was purified by flash chromatography (SiO2, 10% EtOAc/hexane) to give the bis-aldehyde as an oil (0.76 g, 52%). To the solution of the aldehyde (0.76 g, 1.1 mmol) in 20 ml CH2Cl2 and 60 ml acetone was added H2NSO3H (0.90 g, 9.3 mmol, dissolved in 5 ml H2O and NaClO2 (0.96 g, 8.7 mmol, dissolved in 5 ml H2O), and the mixture was stirred at room temperature overnight. The organic solvents were then removed under reduced pressure and the precipitate was filtered off and dried in vacuo to give the mixed cis- and trans-biscarboxylic acid product as a white powder (0.78 g, 98%): mp > 290 °C (dec.); LR FAB-MS m/e calculated for C46H56O8 736.4 〚M〛+, found 736.3. To a solution of butoxycalix〚4〛arene bis-carboxylic acid (0.780 g, 1.06 mmol) in 30 ml dry CH2Cl2 was added oxalyl chloride (0.80 ml, 9.2 mmol) and catalytic amount of DMF (0.2 μl), and the mixture was stirred at room temperature overnight. The solvent and excess reagent were removed in vacuo and the residue was redissolved in 30 ml dry CH2Cl2. 4-Nitrobenzyl alcohol (0.86 g, 5.6 mmol) and DIEA (0.6 ml, 3.3 mmol) were added to above solution, and the mixture was stirred at room temperature overnight. The solvent was evaporated and the residue was applied to a flash chromatography column to give the trans-(A) and cis-(B) title compounds: A (0.244 g, 23%); 1H NMR (CDCl3, 500 MHz) δ 8.27 (d, J = 8.6 Hz, 4H), 7.54 (d, J = 8.5 Hz, 4H), 7.35 (s, 4H), 6.69 (m, 6H), 5.32 (s, 4H), 4.48 (d, J = 13.5 Hz, 4H), 3.95 (m, 8H), 3.24 (d, J = 13.6 Hz, 4H), 1.90 (m, 8H), 1.47 (m, 8H), 1.02 (m, 12H); 13C NMR (CDCl3, 75 MHz) δ 165.9, 161.3, 156.6, 147.6, 144.0, 135.6, 134.8, 130.0, 128.6, 128.3, 123.8, 122.9, 122.3, 75.2, 75.1, 64.8, 32.4, 32.3, 31.0, 19.4, 14.2, 14.1; B (0.312 g, 29%): 1H NMR (CDCl3, 500 MHz) δ 8.24 (d, J = 8.4 Hz, 4H), 7.51 (d, J = 8.4 Hz, 4H), 7.37 (s, 2H), 7.36 (s, 2H), 6.55 (d, J = 7.4 Hz, 2H), 6.54 (d, J = 7.3 Hz, 2H), 6.39 (t, J = 7.4 Hz, 2H), 5.37 (s, 4H), 4.50 (d, J = 13.8 Hz, 1H), 4.46 (d, J= 13.7 Hz, 2H), 4.42 (d, J = 13.6 Hz, 1H), 4.01–3.83 (m, 8H), 3.29 (d, J = 13.8 Hz, 1H), 3.22 (d, J = 13.7 Hz, 2H), 3.15 (d, J = 13.6 Hz, 1H), 1.87 (m, 8H), 1.45 (m, 8H), 1.00 (m, 12H); 13C NMR (CDCl3, 125 MHz) δ 165.9, 161.4, 156.6, 147.6, 143.8, 136.0, 135.3, 135.1, 134.4, 130.3, 129.9, 128.5, 128.1, 127.9, 123.8, 123.0, 121.8, 75.0, 74.9, 64.6, 32.3, 31.0 (2), 19.3 (2), 14.0 (2).
3.5 cis-Butoxycalix〚4〛arene bis(4-nitrobenzylcarboxylate) biscarboxylic acid (3)
To a 250-ml flask was added cis-butoxycalix〚4〛 arene bis(4-nitrobenzylcarboxylate) (0.422 g, 0.42 mmol), dichloromethyl methyl ether (0.33 mL, 3.7 mmol) and 30 ml dry CH2Cl2, and the solution was cooled to –10 °C. TiCl4 (0.40 ml, 3.7 mmol) was added and the mixture was stirred at –10 °C → room temperature overnight. The reaction was quenched with 10 ml 1.0 N HCl, and the organic layer was separated and dried over Na2SO4. The solvent was evaporated and the residue was purified by flash chromatography (SiO2, 30% EtOAc/hexane) to give the aldehyde as an oil (0.270 g, 61%); The aldehyde was dissolved in 15 ml CH2Cl2 and 45 ml acetone. H2NSO3H (0.45 g, 4.7 mmol, dissolved in 3 ml H2O) and NaClO2 (0.48 g, 4.4 mmol, dissolved in 3 ml H2O) were then added to the solution and the mixture was stirred at room temperature overnight. The organic solvents were removed under reduced pressure and the precipitate was filtered off and dried in vacuo to give the title compound as a yellow powder (0.245 g, 88%): mp 168-170 °C; 1H NMR (DMSO-d6, 500 MHz) δ 8.19 (d, J = 7.2 Hz, 4H), 7.57 (d, J = 7.6 Hz, 4H), 7.37 (s, 2H), 7.35 (s, 2H), 7.30 (s, 4H), 5.34 (s, 4H), 4.35 (m, 4H), 3.92 (m, 8H), 3.43 (m, 4H), 1.85 (m, 8H), 1.44 (m, 8H), 0.97 (m, 12H); LR FAB-MS m/e calculated for C62H66N2O16 1095.2 〚M〛+, found 1094.5.
3.6 trans-Butoxycalix〚4〛arene bis(4-nitrobenzylcarboxylate) biscarboxylic acid (4)
The solution of trans-butoxycalix〚4〛arene bis(4-nitrobenzylcarboxylate) (0.231 g, 0.23 mmol), dichloromethyl methyl ether (0.18 ml, 2.0 mmol) in 20 ml dry CH2Cl2 was cooled to –10 °C. TiCl4 (0.22 ml, 2.0 mmol) was added and the mixture was stirred at –10 °C → room temperature overnight. The reaction was quenched with 10 ml 1.0 N HCl, and the organic layer was separated and dried over Na2SO4. After evaporation of solvents, the residue was purified by flash chromatography (SiO2, 25% EtOAc/hexane, then 30% EtOAc/hexane) to give the aldehyde (0.170 g, 70%); 1H NMR (CDCl3, 300 MHz) δ 9.38 (s, 2H), 8.27 (d, J = 8.1 Hz, 4H), 7.65 (s, 4H), 7.61 (d, J = 8.7 Hz, 4H), 6.89 (s, 4H), 5.42 (s, 4H), 4.49 (d, J = 13.5 Hz, 4H), 4.05 (t, J = 7.5 Hz, 4H), 3.88 (t, J = 6.9 Hz, 4H), 3.33 (d, J = 13.8 Hz, 4H), 1.87 (m, 8H), 1.51 (m, 4H), 1.36 (m, 4H), 1.02 (t, J = 6.8 Hz, 6H), 0.99 (t, J = 6.9 Hz, 6H); To the solution of the aldehyde in 12 ml CH2Cl2 and 36 ml acetone was added H2NSO3H (0.45 g, 4.7 mmol, dissolved in 3 ml H2O) and NaClO2 (0.48 g, 4.4 mmol, dissolved in 3 ml H2O), and the mixture was stirred at room temperature overnight. The organic solvents were removed under reduced pressure and the precipitate was filtered off and dried in vacuo to give the title compound as a white powder (0.123 g, 70%): mp 300–305 °C; 1H NMR (DMSO-d6, 300 MHz) δ 8.18 (d, J = 8.7 Hz, 4H), 7.85 (s, 4H), 7.41 (d, J = 8.7 Hz, 4H), 6.85 (s, 4H), 5.12 (s, 4H), 4.36 (d, J = 13.5 Hz, 4H), 4.10 (t, J = 8.0 Hz, 4H), 3.76 (t, J = 6.2 Hz, 4H), 3.45 (d, J = 13.8 Hz, 4H), 1.85 (m, 8H), 1.59 (m, 4H), 1.30 (m, 4H), 0.99 (t, J = 7.5 Hz, 6H), 0.96 (t, J = 7.5 Hz, 6H); LR FAB-MS m/e calculated for C62H66N2O16 1095.2 〚M〛+, found 1094.4
3.7 General procedure for preparation of A3B type receptors
3.7.1 5,11,17-Tris(cyclo-GDDDSp)-23-cyclo-GDGYSp-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
A solution of 2 (6.4 mg, 0.0067 mmol), (COCl)2 (20 μl, 0.23 mmol), DMF (0.1 μl) in 2.0 ml dry CH2Cl2 was stirred at room temperature overnight. The solution was evaporated to dryness and the residue was then redissolved in 2.0 ml dry CH2Cl2. To the solution was added cyclo-GDPDPDPSp-NH2 (13.8 mg, 0.02 mmol), DIEA (8.0 μl, 0.046 mmol), and the mixture was stirred at room temperature for 20 h. The solvent was then removed by evaporation and the residue was purified by gel filtration (Sephadex LH-20, CH2Cl2 as eluent) to give a white solid (20 mg, 100%). The solid was then dissolved in 5 ml MeOH, and the solution was added 10% Pd/C (15.0 mg, 0.014 mmol) and stirred under H2 atmosphere for 12 h. After TLC showed the disappearance of starting material, the catalyst was removed by filtration through celite and the solution was collected and evaporated. The residue was purified by gel filtration (Sephadex LH-20, CH2Cl2 as eluent) to afford tris-loop mono-acid product (11.3 mg, 60% for three steps). The monoacid was dissolved in 1 ml dry THF, and the solution was added 2,4,6-trichlorobenzyl chloride (1.0 μl, 0.006 mmol), TEA (1.0 μl) and the mixture was stirred at room temperature for 2 h. The solvent and reagents were removed in vacuo and the residue was dissolved in 2.0 ml dry CH2Cl2. To the solution was added cyclo-GDPGYPSp-NH2 (7.5 mg, 0.012 mmol), DMAP (2.0 mg, 0.016 mmol), and the mixture was stirred at room temperature for 4 h. The solvent was then removed by evaporation and the residue was purified by gel filtration (Sephadex LH-20, CH2Cl2 as eluent) to give the fully protected product as a white solid (10.8 mg). Further treatment of the product with 25% TFA/CH2Cl2 for 1.5 h afforded the title compound as a white solid (8.8 mg, 75% for three steps): analysical HPLC showed a single peak; ES-MS m/e calculated for C139H154N24O49 〚M〛– 2944.82, found 2945.50 ± 0.55.
3.7.2 5,11,17-Tris(cyclo-GDGYSp)-23-carboxylic acid-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
ES-MS m/e calculated for C123H134N18O33 〚M〛– 2392.45, found 2393.6.
3.7.3 5,11,17-Tris(cyclo-GDGYSp)-23-cyclo-GDDGSp-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
ES-MS m/e calculated for C143H156N24O41 〚M〛– 2866.88, found 2867.37 ± 0.46.
3.7.4 5,11,17-Tris(cyclo-GDGYSp)-23-cyclo-GDDYSp-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
ES-MS m/e calculated for C150H162N24O42 〚M〛– 2973.00, found 2973.75 ± 0.87.
3.7.5 5,11,17-Tris(cyclo-GDGYSp)-23-cyclo-GDDDSp-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
ES-MS m/e calculated for C145H158N24O43 〚M〛– 2924.91, found 2926.01 ± 0.31.
3.7.6 5,11,17-Tris(cyclo-GDGDSp)-23-cyclo-GDDYSp-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
ES-MS m/e calculated for C135H150N24O45 〚M〛– 2828.75, found 2828.74 ± 0.45.
3.7.7 5,11,17-Tris(cyclo-GDGDSp)-23-cyclo-GDGYSp-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
ES-MS m/e calculated for C133H148N24O43 〚M〛– 2770.71, found 2771.22 ± 1.30.
3.8 General procedure for preparation of A2B2 type receptors
3.8.1 5,11,-Bis(cyclo-GDDGSp)-17,23-bis(cyclo-GDDYSp)-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
A solution of 3 (11.2 mg, 0.011 mmol), (COCl)2 (30 μl, 0.35 mmol), DMF (0.1 μl) in dry CH2Cl2 (2.0 ml) was stirred at room temperature overnight. The solution was evaporated and dried. The residue was redissolved in dry CH2Cl2 (2.0 ml), and cyclo-GDPDPGSp-NH2 (16.0 mg, 0.026 mmol), DIEA (10 μl, 0.058 mmol) wad added and stirred at room temperature for 20 h. The mixture was then applied to a Sephadex LH-20 column with CH2Cl2 eluent, and the product was collected and applied to a flash chromatography column (SiO2, 10% MeOH/CH2Cl2) to give a white solid (12.7 mg). The solid was redissolved in MeOH (5 ml), was added 10% Pd/C (15.0 mg, 0.014 mmol), and stirred under H2 atmosphere for 8 h. After TLC showed the disappearance of starting material, the catalyst was removed by filtration through celite and the solution was collected and evaporated to dryness. The residue was applied to Sephadex LH-20 column to give the bis-acid intermediate (7.0 mg, 33% for three steps). The bis-acid was dissolved in 2 ml dry THF, and the solution was added 2,4,6-trichlorobenzyl chloride (4.0 μl, 0.024 mmol), DIEA (5.0 μl) and the mixture was stirred at room temperature for 2 h. The solution was then evaporated and dried in vacuo. The residue was dissolved in dry CH2Cl2 (2.0 ml) and to the solution was added cyclo-GDPDPYPSp-NH2 (20.0 mg, 0.026 mmol), DMAP (5.0 mg, 0.04 mmol). The mixture was stirred at room temperature overnight. The solution was then applied to a Sephadex LH-20 column with CH2Cl2 as the eluent. The appropriate portion was collected to give the fully protected product as a white solid (8.0 mg). Further treatment with 25% TFA/CH2Cl2 (4 ml) for 2 h afforded the title compound as a white solid (6.0 mg, 58% for three steps): analysical HPLC showed a single peak; ES-MS m/e calculated for C142H156N24O46 〚M〛– 2934.87, found 2935.78.
3.8.2 5,17,-Bis(cyclo-GDGYSp)-11,23-bis(cyclo-GDDGSp)-25,26,27,28-tetrakis(butoxy)calix〚4〛arene
A solution of 4 (8.0 mg, 0.017 mmol), (COCl)2 (20 μl, 0.23 mmol), DMF (0.1 μl) in dry CH2Cl2 (2.0 ml) was stirred at room temperature overnight. The solution was evaporated to dryness and the residue was then redissolved in 2.0 ml dry CH2Cl2. To the solution was added cyclo-GDPDPGSp-NH2 (25.0 mg, 0.041 mmol), DIEA (8.0 μl, 0.046 mmol) and stirred at room temperature for 20 h. The mixture was applied first to a gel filtration column (Sephadex LH-20, CH2Cl2), then a silica gel column (10% MeOH/CH2Cl2) to give a white solid (13.5 mg). The solid was then dissolved in 5 ml MeOH, and the solution was treated with 10% Pd/C (15.0 mg, 0.014 mmol) and stirred under H2 atmosphere for 8 h. After TLC showed the disappearance of starting material, the catalyst was removed by filtration through celite and the solution was collected and evaporated. The residue was purified by gel filtration (Sephadex LH-20, CH2Cl2 as eluent) to give the bis-acid product (18.4 mg, 61% for three steps). The product was dissolved in dry THF (2 ml), and the solution was added 2,4,6-trichlorobenzyl chloride (4.0 μl, 0.024 mmol), DIEA (5.0 μl) and the mixture was stirred at room temperature for 2.5 h. The solution was then evaporated and dried in vacuo. The residue was dissolved in 2.0 ml dry CH2Cl2 and the solution was treated with cyclo-GDPGYPSp-NH2 (20.0 mg, 0.031 mmol), DMAP (6.0 mg, 0.048 mmol). The mixture was stirred at room temperature overnight. The solution was then applied to a Sephadex LH-20 column with CH2Cl2 as the eluent. The appropriate portion was collected to give the fully protected product as a white solid (10.8 mg). Further treatment of the solid with 25% TFA/CH2Cl2 (4 ml) for 2 h afforded the title compound as a white solid (12.4 mg, 42% for three steps): analysis by HPLC showed a single peak; ES-MS m/e calculated for C138H152N24O42 〚M〛– 2818.80, found 2818.79 ± 0.72.
Acknowledgements
We thank the National Institutes of Health (GM35208) and the US Army (DAMD17-99-1-9458) for financial support of this work.