

1 Introduction
Steric effects play a central role in the chemistry of N-heterocyclic carbenes (NHCs) [1] and are largely controlled by the size and shape of the substituents of their nitrogen atoms [2–8]. Thus, bulky N-substituents provide kinetic stabilisation of free NHCs with respect to dimerisation [9,10]; when bound to a metal, they may both increase complex stability and assist specific steps in catalytic reactions [11]. Typically, bulky NHCs such as IMes, IPr, IPent or IPr∗ (Fig. 1) display a hemiovoidal umbrella shape, thereby contrasting with the conical encumbrance brought about by bulky tertiary phosphines. The most useful parameter for the measure of their steric properties is the so-called “buried volume” (%VBur), which expresses the percentage of volume occupied by the ligand within the first coordination sphere of the metal [12]. Actually, this parameter is only helpful for the rough prediction of steric effects; however, it does not enable prediction of regioselectivities of catalytic reactions. These latter essentially depend on subtle interactions in the catalytic process between specific locations of the ligand and coordinated substrates. In this regard, it is useful to remind that a number of NHCs (notably the IBiox ligands, see Fig. 1) display in their complexes the flexible steric bulk [13], a feature enabling these ligands to adapt to the steric requirements of intermediates of catalytic cycles and consequently improve the catalyst's performance. Thus, the value expressing the %VBur of a NHC may vary during a catalytic cycle. Finally, it should also be mentioned that the buried volume parameter does not integrate steric effects due to possible self-assembly phenomena.
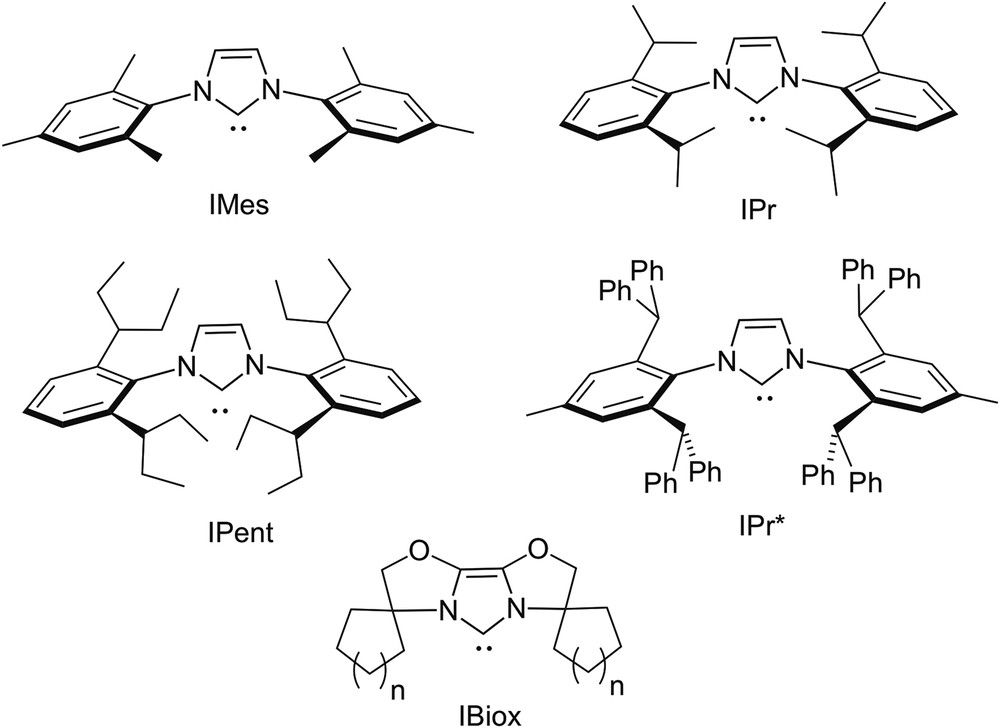
Conventional bulky NHCs.
As part of a programme aimed at the development of NHCs displaying contours that delineate a sterically embracing environment, we have prepared the NHC precursors 1–3, both characterised by the presence of cavity-shaped calixarene termini in their wingtips. We wondered whether in NHC complexes derived from them, the two bowl-shaped units could influence ligands of the first coordination sphere. It is noteworthy that to date double calixarenes [14–16] having the macrocyclic units separated by an NHC spacer have not been documented (see Fig. 2).
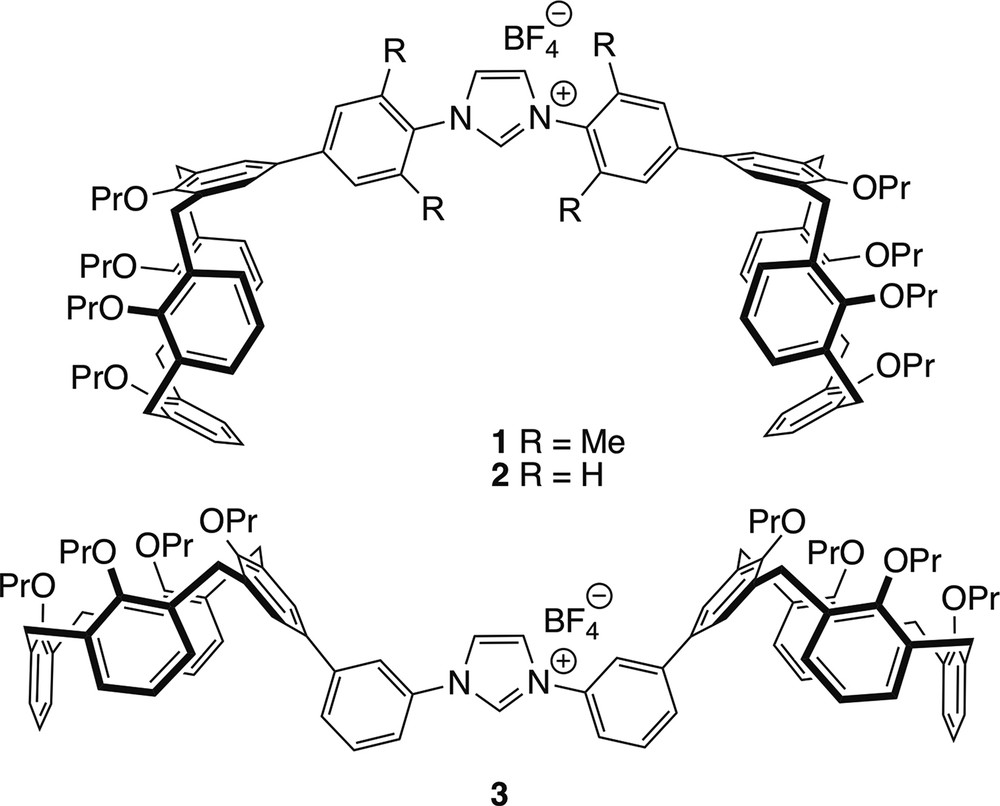
Imidazolium salts synthesised in the present study.
2 Results and discussion
The imidazolium salts 1–3 were each prepared in a multistep sequence (Schemes 1 and 2) starting from bromocalixarene 4. Only the synthesis of 1 is discussed hereafter. Full description of the preparations of 2 and 3, which are basically similar to that of 1, is given in Section 4. Thus, the preparation of 1 began with the synthesis of intermediate 5, which was obtained from 4 by Br/Li exchange, followed by reaction with B(OMe)3 and subsequent treatment with HCl (Scheme 1). The boronic acid 5, which was used in its crude form, was then reacted with tert-butyl N-(4-bromo-2,6-dimethylphenyl)carbamate under Suzuki–Miyaura coupling conditions, affording the N-protected aniline 6. Deprotection of the latter with CF3CO2H in dichloromethane gave the primary amine 7.
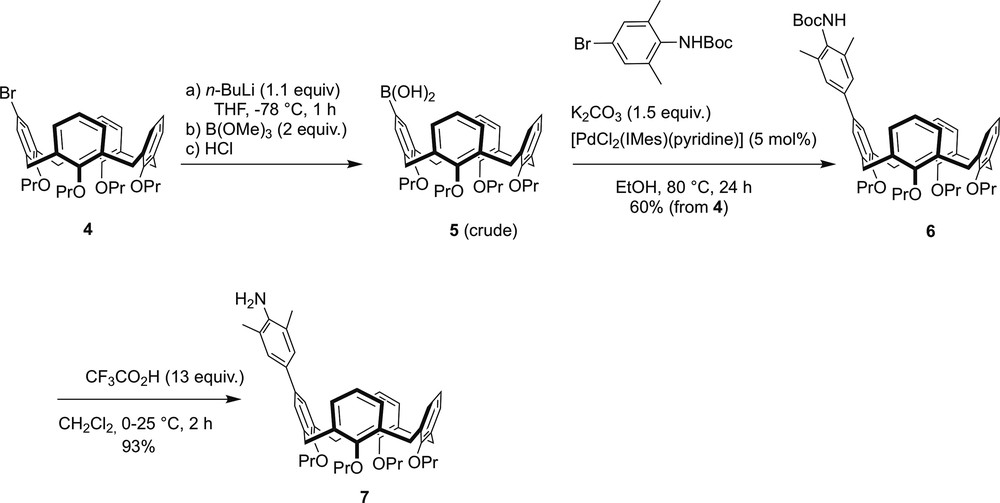
Stepwise synthesis of arylamine 7.
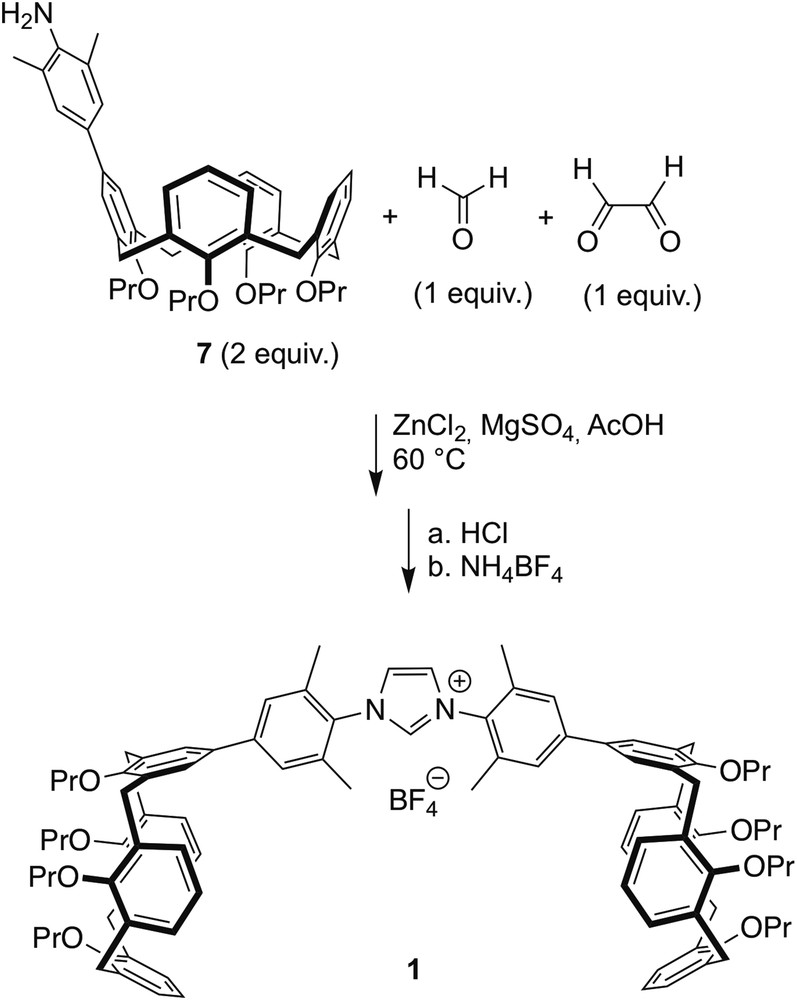
Multicomponent synthesis of imidazolium salt 1.
In the next step (Scheme 2), a solution of glyoxal, formaldehyde, acetic acid and ZnCl2 was heated at 60 °C for 5 min, and then added to a mixture heated at 60 °C of amine 7, MgSO4 and AcOH. After 0.5 h, dichloromethane was added to get a homogeneous solution. Treatment with HCl followed by addition of NH4BF4 in excess gave imidazolium salt 1.
The 1H NMR spectrum of each salt shows an imidazolium NCHN signal appearing in the usual range [17,18], and that of 1 being slightly less deshielded (δ = 8.46) than that of the other two salts (δ = 9.26 (2), δ = 9.26 (3)). For all three compounds, the observed AB patterns (with J values ranging from 13.2 to 13.5 Hz) are characterised by the separation of the methylenic signals (1.22 < Δδ < 1.34 ppm) typical of that of calix[4]arenes in the cone conformation [19,20]. As expected, the BF4 anions appear in the corresponding 19F NMR spectra as two broad peaks with relative intensities of 1:4 (separation ca. 0.05 ppm), this pattern being caused by the two boron isotopes, 10B and 11B [21].
Compounds 1–3 were converted into the Pd complexes 8–10 by their reaction with [PdCl2] in pyridine in the presence of K2CO3 (Scheme 3). All three complexes were purified by column chromatography. The reaction leading to 8 was carried out at 80 °C, affording the product in 82% yield. Those resulting in 9 and 10 required to be done at room temperature to avoid product decomposition, this presumably because of facile cyclometalation. Under these conditions, 9 and 10 were obtained in, respectively, 60% and 68% yield.
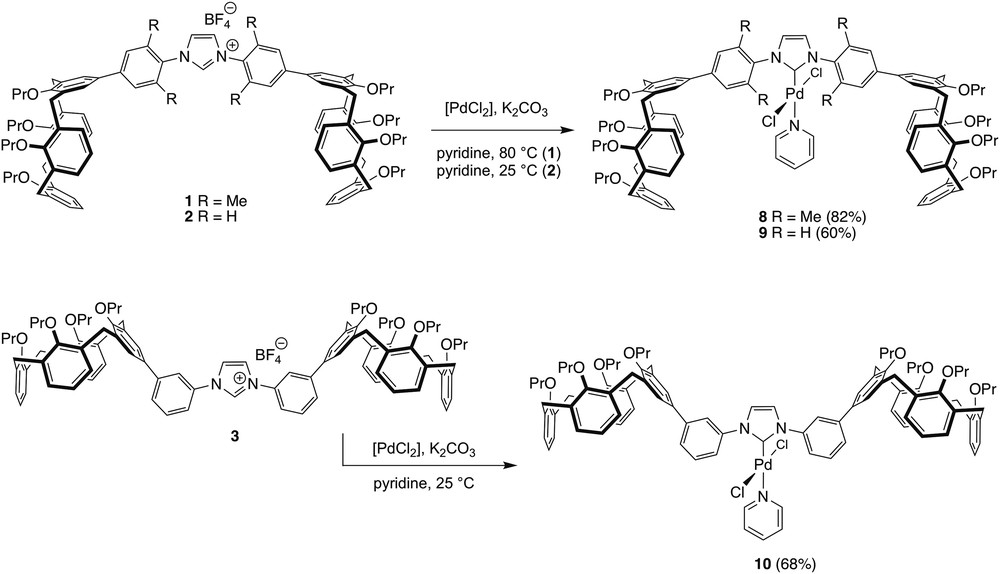
Syntheses of complexes 8–10.
The 1H and 13C{1H} NMR spectra of the three carbene complexes present no anomalies. Thus, all 1H NMR spectra show the presence of one NHC ring, one pyridine ligand and two equivalent calixarene units. The carbene carbons are observed at, respectively, 153.12 (8), 150.77 (9) and 151.53 (10) ppm in the 13C NMR spectra, these values being close to those observed for related [PdCl2(NHC)(pyridine)] complexes based on an imidazolylidene [22].
The crystal structure of complex 9 was determined by a single-crystal X-ray diffraction study (Fig. 3). Crystal and data collection are detailed in Section 4. The unit cell consists of two pairs of self-assembled molecules of 9, this considerably increases crowding about the palladium atoms. The individual molecules of each dimer (denoted as α and β) present similar structural features. Thus, in both molecules the palladium atom lies in a slightly distorted square planar ligand environment (nonstrictly linear Cl–Pd–Cl arrangements, see Table 1) with Pd–Ccarb (1.946(3) Å in α; 1.954(3) Å in β), Pd–N (2.135(3) Å in α; 2.099(3) Å in β) and Pd–Cl (2.312(1), 2.309(1) Å in α; 2.310(1), 2.312(1) Å in β) bond lengths similar to those found in related molecules.
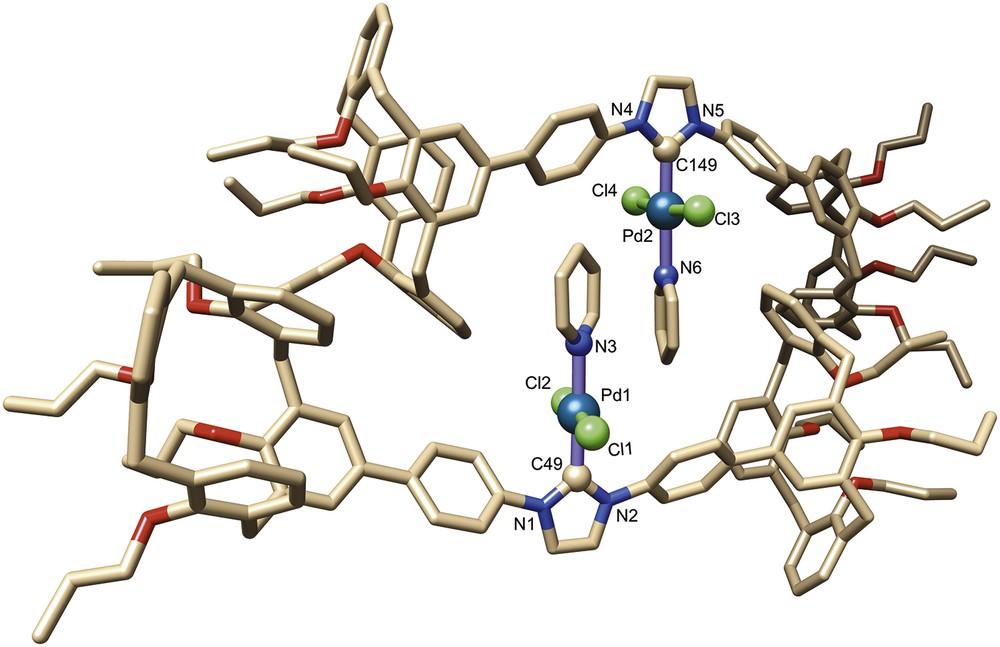
Molecular structure of complex 9. Solvent molecules have been omitted for clarity.
Selected distances (Å) and angles (°) in complex 9.
| Distances | |||
| Molecule α | |||
| C1–Pd1 | 1.946(3) | Pd1–Cl1 | 2.312(1) |
| Pd1–N3 | 2.135(3) | Pd1–Cl2 | 2.309(1) |
| Molecule β | |||
| C149–Pd2 | 1.954(3) | Pd2–Cl3 | 2.310(1) |
| Pd2–N6 | 2.099(3) | Pd2–Cl4 | 2.312(1) |
| Angles | |||
| Molecule α | |||
| C49–Pd1–N3 | 179.13(11) | N3–Pd1–Cl2 | 93.38(9) |
| C49–Pd1–Cl2 | 86.37(10) | N3–Pd1–Cl1 | 94.40(9) |
| C49–Pd1–Cl1 | 85.84(10) | Cl2–Pd1–Cl1 | 172.20(4) |
| Molecule β | |||
| C149–Pd2–N6 | 178.56(12) | N6–Pd2–Cl3 | 92.79(10) |
| C149–Pd2–Cl3 | 86.36(10) | N6–Pd2–Cl4 | 92.76(10) |
| C149–Pd2–Cl4 | 92.96(10) | Cl3–Pd2–Cl4 | 174.24(3) |
| Interplanar angles | |||
| Molecule α | |||
| Metal plane/pyridine plane | 16.3(1) | Metal plane/carbene ring | 81.7(1) |
| Carbene plane/N-aryl plane 1 | 36.2(1) | Carbene plane/N-aryl plane 2 | 36.2(1) |
| Molecule β | |||
| Metal plane/pyridine plane | 32.9(1) | Metal plane/carbene ring | 32.9(1) |
| Carbene plane/N-aryl plane 1 | 48.2(1) | Carbene plane/N-aryl plane 2 | 39.2(1) |
All calixarene units adopt a typical pinched cone conformation with interplanar angles between the facing phenoxy rings of 11.8°/79.1° and 23.4°/69.2° in α and 20.8°/72.6° and 22.0°/78.5° in β. It is interesting that the two phenoxy rings linked to the central NHC unit of each molecule are different in nature, one belonging to the pinched part of the corresponding calixarene moiety and the other to the flared part of the second calixarene end. Also noteworthy is the short separation between the o-phenylene protons and the metal centre (shortest separations 2.79(1) and 3.08(1) Å in α; 2.73(1) and 2.84(1) Å in β), suggesting that the phenylene units are prone to undergo cyclometalation (vide supra).
Examination of the solid state structure further reveals that the bulky calixarene moieties themselves are not responsible for the steric encumbrance created around the metal atoms, the observed crowding resulting in fact from the self-assembly phenomenon, which brings each pyridine ligand close to two calixarene phenoxy rings as well as to the pyridine moiety of the partner molecule. As could be inferred from the noncovalent interaction (NCI) analysis of the density functional theory (DFT)-optimised structure, the two components of the dimer are held together by a network of attractive dispersion forces (Fig. 4). For example, each pyridine ligand is wrapped in a van der Waals surface involving interactions between the pyridine ligand (π stacking), a phenylene unit and a phenoxy(calix) ring of the partner molecule. Van der Waals interactions also exist between one phenoxy ring of each calixarene end and a specific phenylene-phenoxy fragment of the other molecule. However, these steric interactions could not be identified in solution (absence of cross-peaks in the corresponding ROESY spectrum).
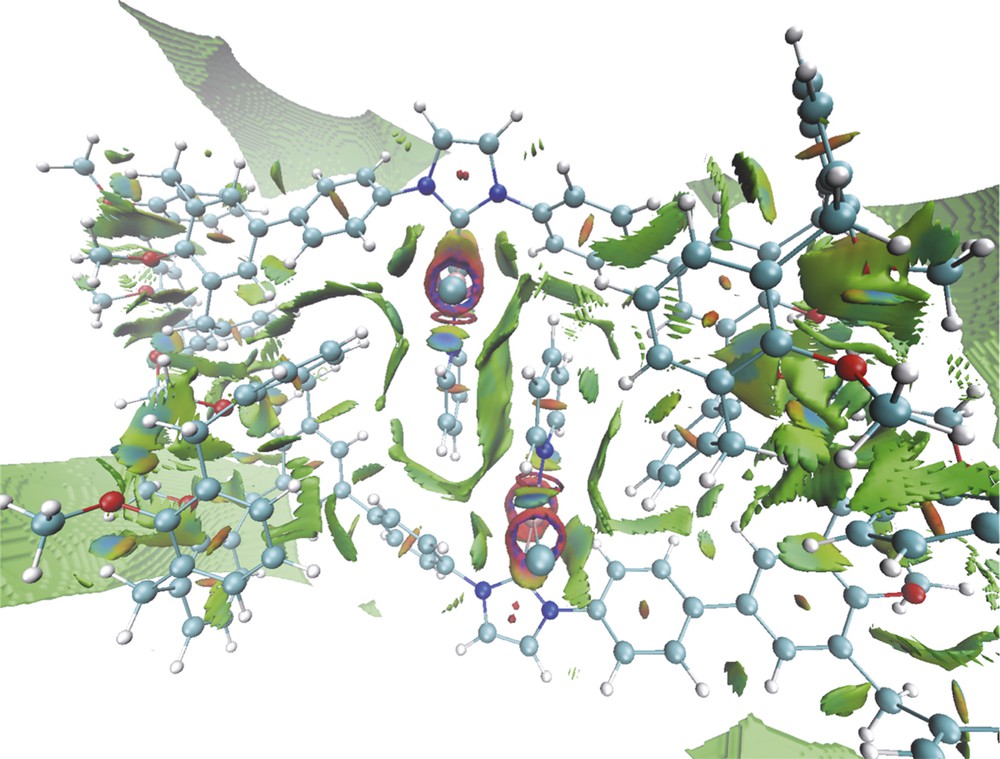
NCIs in complex 9-dimer (optimised structure), with electrostatic interactions in blue, attractive dispersion forces in green and steric repulsions in red.
Wondering whether the ability of Pd complexes of type 9 to self-assemble would influence their catalytic properties, we examined the behaviour of complex 8 in the cross-coupling of phenylboronic acid with 4-chlorotoluene at 80 °C (Scheme 4). Complexes 9 and 10 were not considered as appropriate for catalytic runs, as these readily underwent decomposition when heated. Catalysis was carried out in ethanol using 1 mol % of 8 in the presence of K2CO3 (1.5 equiv/mol of 4-chlorotoluene). After 1 h of reaction time, 4-phenyltoluene was formed in 55% yield. Under identical conditions, the related complex [PdCl2(IMes)(pyridine)] (11) [23], an analogue which is devoid of calixarene ends, resulted in 77% yield. Because carbene ligands of 8 and 11 possess quite similar electronic properties (δ(Ccarb) = 153.1 (8); 152.9 (11) ppm), one would have expected that the bulkier carbene ligand of 8 would facilitate the reductive elimination step and thus result in a higher catalytic activity. As the reverse is the case, it appears likely that at least a fraction of the catalytic species self-assemble in a manner similar to that observed for 9 in the solid state, thereby rendering the metal more difficult of access. However, one has to be careful in interpreting our findings, as other phenomena may occur in a differentiated way for the two complexes, notably the formation of nanoparticles.
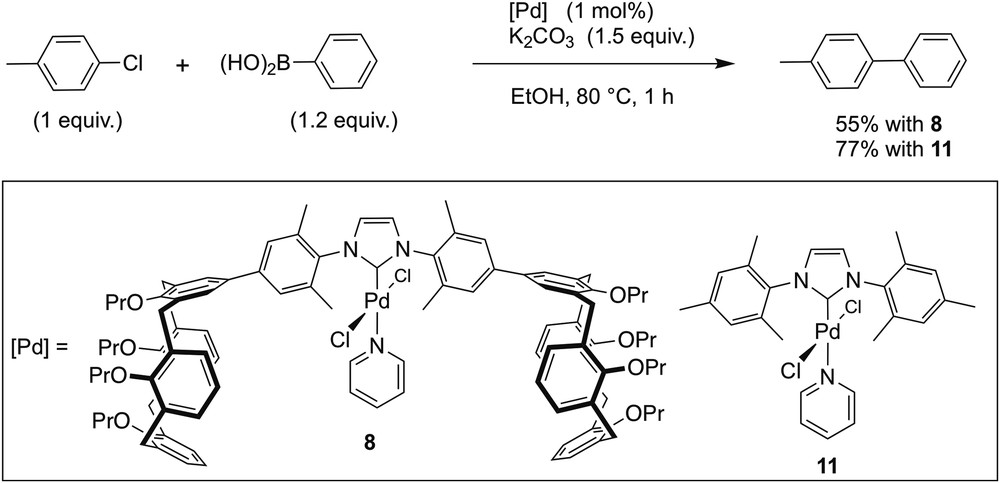
Suzuki–Miyaura cross-coupling reaction with 8 and 11.
3 Conclusions
We have synthesised the first double calixarenes having their macrocyclic units linked through a symmetrically substituted NHC spacer. The structural study of the [PdCl2(NHC)(pyridine)] complex 9 unexpectedly revealed how the presence of the calixarene termini may assist dimer formation in the solid state, thereby considerably increasing the steric crowding about the metal centres. Consistent with a possible competitive aggregation phenomenon of this type in solution, the performance of 8 in a Suzuki cross-coupling reaction was lower than that observed for a related, cavity-devoid analogue. Overall, the present study provides a new example of the difficulty in predicting steric effects in NHC complexes.
4 Experimental section
4.1 General procedures
The syntheses were performed in Schlenk-type flasks under dry nitrogen. Solvents were dried by conventional methods and distilled immediately before use. Routine 1H and 13C{1H} NMR spectra were recorded using FT Bruker Avance 500, 400 or 300 instruments at 25 °C. 1H NMR spectral data were referenced to residual protonated solvent (CHCl3, δ 7.26); 13C chemical shifts are reported relative to deuterated solvent (CDCl3, δ 77.16). The 19F NMR spectra of 1–3 were recorded using a FT Bruker Avance 300 apparatus (external reference CFCl3). In the NMR data given hereafter, Cq denotes a quaternary carbon atom. The bridging CH2 groups of the calixarene backbone are designated by the abbreviation ArCH2Ar. For column chromatography, Geduran SI (E. Merck, 0.040–0.063 mm) silica was used. Routine thin-layer chromatography analyses were carried out using plates coated with Merck Kieselgel 60 GF254. Elemental analyses were performed by the Service de Microanalyse, Institut de Chimie (Unistra-CNRS), Strasbourg. X-ray diffraction analysis of complex 9 was performed by the Service de Radiocrystallographie, Institut de Chimie de Strasbourg. tert-Butyl N-(4-bromo-2,6-dimethylphenyl)carbamate, tert-butyl N-(4-iodophenyl)carbamate and tert-butyl N-(3-bromophenyl)carbamate were obtained according to the procedure reported for a related compound [24] (vide infra). The procedure for the preparation of the symmetrical imidazolium salts 1–3 was inspired by that applied by Tarrieu et al. [25] for the synthesis of unsymmetrically substituted imidazolium salts. The structural formula of the N-protected compounds 6′ and 6″, both related to calixarene 6 and the amines 7′ and 7″, analogues of 7, are given in the Supplementary data. 5-Bromo-25,26,27,28-tetrapropyloxycalix[4]arene 4 [26] and [PdCl2(IMes)(pyridine)] (11) [23] were synthesised according to the published methods. In the following “Py” stands for pyridine.
4.1.1 tert-Butyl N-(4-bromo-2,6-dimethylphenyl)carbamate
Boc2O (4.40 g, 20.0 mmol) was added to a solution of 4-bromo-2,6-dimethylaniline (4.00 g, 20.0 mmol) in THF (20 mL). The resulting mixture was stirred for 18 h at 65 °C. The solvent was removed in vacuo and the resulting residue was recrystallised from CH2Cl2/petroleum ether affording the aryl carbamate as a white solid (4.12 g, 69%). The product did not require further purification. 1H NMR (400 MHz, CDCl3), δ 7.19 (2H, s, ArH), 5.83 (1H, s, NH), 2.22 (6H, s, 2CH3), 1.48 (9H, s, C(CH3)3). NMR spectroscopic data are consistent with those of the literature [27].
4.1.2 tert-Butyl N-(4-iodophenyl)carbamate
Boc2O (5.00 g, 22.9 mmol) was added to a solution of 4-iodoaniline (5.00 g, 22.8 mmol) in THF (20 mL). The resulting mixture was stirred for 18 h at 65 °C. Then the solvent was removed in vacuo and the resulting residue was recrystallised from CH2Cl2/petroleum ether affording the aryl carbamate as a white solid (5.11 g, 70%). The product did not require further purification. 1H NMR (500 MHz, CDCl3), δ 7.55 (2H, d, 3J = 8.5 Hz, ArH), 7.14 (2H, d, 3J = 8.5 Hz, ArH), 6.62 (1H, s, NH), 1.49 (9H, s, CH3). NMR spectroscopic data are consistent with those of the literature [28].
4.1.3 tert-Butyl N-(3-bromophenyl)carbamate
Boc2O (10.00 g, 58.1 mmol) was added to a solution of 3-bromoaniline (12.70 g, 58.1 mmol) in THF (40 mL). The resulting mixture was stirred for 18 h at 65 °C. Then the solvent was removed in vacuo and the resulting residue was recrystallised from CH2Cl2/petroleum ether affording the aryl carbamate as a white solid (10.30 g, 65%). The product did not require further purification. 1H NMR (300 MHz, CDCl3), δ 7.66 (1H, s, ArH), 7.23–7.08 (3H, m, ArH), 6.47 (1H, s, NH), 1.51 (9H, s, CH3). NMR spectroscopic data are consistent with those of the literature [29].
4.1.4 5-(3,5-Dimethyl-4-tert-butyloxycarbonylaminophenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (6)
A 1.5 M solution of n-BuLi in hexanes (3.1 mL, 4.90 mmol) was added dropwise to a solution of 5-bromo-25,26,27,28-tetrapropyloxycalix[4]arene (4) (3.00 g, 4.46 mmol) in THF (50 mL) at −78 °C. After 40 min, B(OMe)3 (1.00 mL, 0.92 g, 8.95 mmol) was added dropwise to the solution. After stirring for 1 h at room temperature, the solution was quenched with aq HCl (1 M, 100 mL) and the product extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over MgSO4. The solvent was removed in vacuum yielding the crude boronic acid 5 as a white powder, which was used without further purification. tert-Butyl (4-bromo-2,6-dimethylphenyl)carbamate (1.34 g, 4.46 mmol), boronic acid 5, K2CO3 (0.92 g, 6.69 mmol) and [PdCl2(IMes)(pyridine)] (0.125 g, 0.222 mmol) were introduced in a Schlenk flask containing absolute EtOH (50 mL). The mixture was stirred at 80 °C for 24 h. After cooling to room temperature the mixture was passed through Celite and the filtrate evaporated to dryness. The crude product was purified by flash chromatography (SiO2; AcOEt/petroleum ether, 7:93; Rf = 0.28) to afford 6 as a white solid (2.18 g, 60%). 1H NMR (CDCl3, 400 MHz), δ 6.98 (2H, s, ArH), 6.85–6.63 (8H, m, ArH), 6.56 (2H, d, 3J = 7.3 Hz, ArH), 6.40 (1H, t, 3J = 7.3 Hz, ArH), 5.92 (1H, br s, NH), 4.55 and 3.26 (4H, d, AB spin system, 2JAB = 13.4 Hz, ArCH2Ar), 4.53 and 3.21 (4H, d, AB spin system, 2JAB = 13.4 Hz, ArCH2Ar), 4.01–3.83 (8H, m, OCH2), 2.32 (6H, s, o-CH3), 2.07–1.92 (8H, m, CH2CH3), 1.56 (9H, br s, C(CH3)3), 1.13–1.01 (12H, m, CH2CH3) ppm. Because of the dynamic conformation of the carbamate group, some 1H NMR peaks were very broad. 13C{1H} NMR (CDCl3, 125 MHz), δ 156.83 (2 × arom. Cq–O), 156.36 (arom. Cq–O), 156.06 (arom. Cq–O), 153.83 (NHCO), 140.42 (arom. Cq–N), 135.50 (arom. Cq), 135.35 (arom. Cq), 135.34 (arom. Cq), 134.95 (arom. Cq), 134.82 (arom. Cq), 132.33 (arom. Cq), 128.41 (2 × arom. CH), 127.99 (arom. CH), 127.09 (arom. CH), 126.97 (arom. CH), 122.04 (2 × arom. CH), 79.84 (C(CH3)3), 76.88 (OCH2), 76.77 (3 × OCH2), 31.18 (ArCH2Ar), 31.07 (ArCH2Ar), 28.42 (CH3)3, 23.43 (CH2CH3), 23.40 (CH2CH3), 23.31 (2 ×CH2CH3), 18.61 (o-CH3) 10.53 (CH2CH3), 10.37 (CH2CH3) ppm. Found C, 77.89; H, 8.05; N, 1.82. Calcd for C53H65NO6 (Mr = 811.48): C, 78.38; H, 8.07; N, 1.73%. Despite several recrystallisations, elemental C-analyses remained unsatisfactory.
4.1.5 5-(4-tert-Butyloxycarbonylaminophenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (6′)
A 1.5 M solution of n-BuLi in hexanes (6.2 mL, 9.80 mmol) was added dropwise to a solution of 5-bromo-25,26,27,28-tetrapropyloxycalix[4]arene (4) (6.00 g, 8.93 mmol) in THF (120 mL) at −78 °C. After 40 min, B(OMe)3 (2.00 mL, 1.84 g, 17.9 mmol) was added dropwise to the solution. After stirring for 1 h at room temperature, the solution was quenched with aq HCl (1 M, 240 mL) and the product extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over MgSO4. The solvent was removed in vacuum yielding the crude boronic acid 5 as a white powder, which was used without further purification. tert-Butyl (4-iodophenyl)carbamate (2.85 g, 8.93 mmol), crude boronic acid 5, K2CO3 (1.85 g, 13.39 mmol) and [PdCl2(IMes)(pyridine)] (0.250 g, 0.445 mmol) were introduced in a Schlenk flask containing absolute EtOH (50 mL). The mixture was then stirred at 80 °C for 24 h. After cooling to room temperature the solution was passed through Celite and the filtrate evaporated under reduced pressure. The residue was purified by flash chromatography (SiO2; CH2Cl2/petroleum ether, 40:60; Rf = 0.33) to afford 6′ as a white solid (4.58 g, 65%). 1H NMR (CDCl3, 500 MHz), δ 7.39 (2H, d, 3J = 8.5 Hz, ArH), 7.25 (2H, d, 3J = 8.5 Hz, ArH), 6.93–6.83 (4H, m, ArH), 6.78 (2H, dd, 3J = 3J’ =7.4 Hz, ArH), 6.73 (2H, s, ArH), 6.64 (1H, s, NH), 6.50 (2H, d, 3J = 7.4 Hz, ArH), 6.33 (1H, t, 3J = 7.4 Hz, ArH), 4.59 and 3.29 (4H, d, AB spin system, 2JAB = 13.5 Hz, ArCH2Ar), 4.57 and 3.25 (4H, d, AB spin system, 2JAB = 13.5 Hz, ArCH2Ar), 4.07–3.98 (4H, m, OCH2), 3.97–3.86 (4H, m, OCH2), 2.13–1.95 (8H, m, CH2CH3), 1.62 (9H, s, C(CH3)3), 1.19–1.04 (12H, m, CH2CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 157.07 (2 × arom. Cq–O), 156.17 (arom. Cq–O), 155.79 (arom. Cq–O), 152.91 (NHCO), 136.70 (arom. Cq–N), 136.45 (arom. Cq), 135.79 (arom. Cq), 135.60 (arom. Cq), 134.86 (arom. Cq), 134.56 (arom. Cq), 134.23 (arom. Cq), 128.51 (arom. CH), 128.49 (arom. CH), 127.83 (arom. CH), 127.34 (arom. CH), 126.42 (arom. CH), 122.10 (arom. CH), 121.99 (arom. CH), 118.57 (arom. CH), 80.46 (C(CH3)3), 76.86 (OCH2), 76.73 (OCH2), 76.71 (2 × OCH2), 31.19 (ArCH2Ar), 31.07 (ArCH2Ar), 28.42 (C(CH3)3), 23.43 (CH2CH3), 23.26 (CH2CH3), 10.58 (CH2CH3), 10.28 (CH2CH3) ppm. Found C, 77.97; H, 7.99; N, 1.66. Calcd for C51H61NO6 (Mr = 784.05): C, 78.13; H, 7.84; N, 1.79%.
4.1.6 5-(3-tert-Butyloxycarbonylaminophenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (6″)
A 1.5 M solution of n-BuLi in hexanes (6.2 mL, 9.80 mmol) was added dropwise to a solution of 5-bromo-25,26,27,28-tetrapropyloxycalix[4]arene (4) (6.00 g, 8.93 mmol) in THF (120 mL) at −78 °C. After 40 min, B(OMe)3 (2.00 mL, 1.84 g, 17.9 mmol) was added dropwise to the solution. After stirring for 1 h at room temperature, the solution was quenched with aq HCl (1 M, 240 mL) and the product extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over MgSO4. The solvent was removed in vacuum yielding the crude boronic acid 5 as a white powder, which was used without further purification. tert-Butyl (3-bromophenyl)carbamate (2.43 g, 8.93 mmol), boronic acid 5, K2CO3 (1.850 g, 13.39 mmol) and [PdCl2(IMes)(pyridine)] (0.250 g, 0.445 mmol) were introduced in a Schlenk flask containing absolute EtOH (50 mL). The mixture was stirred at 80 °C for 24 h. After cooling to room temperature the mixture was passed through Celite and the filtrate evaporated to dryness. The crude product was purified by flash chromatography (SiO2; CH2Cl2/petroleum ether, 40:60; Rf = 0.27) to afford 6″ as a white solid (4.55 g, 65%). 1H NMR (CDCl3, 500 MHz), δ 7.38–7.30 (1H, m, ArH), 7.27 (1H, dd, 3J = 3J’ = 7.6 Hz, ArH), 7.23 (1H, s, ArH), 6.94 (2H, d, 3J = 7.6 Hz, ArH), 6.77 (2H, s, ArH), 6.74–6.67 (4H, m, ArH), 6.64 (2H, dd, 3J = 3J’ = 7.5 Hz, ArH), 6.53 (2H, d, 3J = 7.5 Hz, ArH), 6.50 (1H, s, NH), 6.37 (1H, t, 3J = 7.5 Hz, ArH), 4.51 and 3.22 (4H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 4.48 and 3.17 (4H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 3.95–3.81 (8H, m, OCH2), 2.03–1.88 (8H, m, CH2CH3), 1.55 (9H, s, C(CH3)3), 1.08–0.97 (12H, m, CH2CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 156.82 (2 × arom. Cq–O), 156.49 (arom. Cq–O), 156.45 (arom. Cq–O), 152.83 (NHCO), 142.55 (arom. Cq–N), 138.42 (arom. Cq), 135.49 (arom. Cq), 135.26 (arom. Cq), 135.23 (arom. Cq), 135.01 (arom. Cq), 134.48 (arom. Cq), 129.01 (arom. CH), 128.43 (arom. CH), 128.39 (arom. CH), 128.07 (arom. CH), 126.94 (arom. CH), 122.08 (2 × arom. CH), 121.92 (arom. CH), 117.21 (arom. CH), 116.58 (arom. CH), 80.53 (C(CH3)3), 76.89 (OCH2), 76.80 (3 × OCH2), 31.28 (ArCH2Ar), 31.10 (ArCH2Ar), 28.49 ((CH3)3), 23.44 (CH2CH3), 23.41 (CH2CH3), 23.35 (2 × CH2CH3), 10.54 (CH2CH3), 10.42 (CH2CH3) ppm. Found C, 77.92; H, 7.82; N, 1.98. Calcd for C51H61NO6 (Mr = 784.05): C, 78.13; H, 7.84; N, 1.79%.
4.1.7 5-(4-Amino-3,5-dimethylphenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (7)
Trifluoroacetic acid (1.82 g, 15.9 mmol) was added dropwise to a stirred solution of calix[4]arene 6 (1.00 g, 1.23 mmol) in CH2Cl2 (12 mL) cooled at 0 °C. After 15 min, the reaction mixture was allowed to reach room temperature and stirred for a further 2 h. The mixture was washed with an aqueous solution of K2CO3 (20%, 150 mL) and the two phases were separated. The aqueous phase was treated with dichloromethane (2 × 100 mL) and the organic phases were then combined. The solvent was removed under reduced pressure to afford 7 as a beige solid (0.816 g, 93%), which did not need further purification. 1H NMR (CDCl3, 500 MHz), δ 6.98 (2H, s, ArH), 6.84 (2H, s, ArH), 6.67 (4H, d, 3J = 7.4 Hz, ArH), 6.63–6.55 (4H, m, ArH), 6.53 (1H, t, 3J = 7.4 Hz, ArH), 4.53 and 3.24 (4H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 4.51 and 3.20 (4H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 3.98–3.85 (8H, m, OCH2), 3.50 (2H, br s, NH2), 2.26 (6H, s, o-CH3), 2.06–1.92 (8H, m, CH2CH3), 1.11–0.99 (12H, m, CH2CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 156.76 (arom. Cq–O), 156.54 (2 × arom. Cq–O), 155.69 (arom. Cq–O), 141.41 (arom. Cq–N), 135.38 (arom. Cq), 135.27 (arom. Cq), 135.03 (3 × arom. Cq), 131.89 (arom. Cq), 128.30 (arom. CH), 128.21 (arom. CH), 128.13 (arom. CH), 127.12 (arom. CH), 126.62 (arom. CH), 122.00 (2 × arom. CH), 121.76 (arom. Cq), 76.81 (3 × OCH2), 76.72 (OCH2), 31.22 (ArCH2Ar), 31.10 (ArCH2Ar), 23.39 (3 ×CH2CH3), 23.35 (CH2CH3), 17.92 (o-CH3), 10.51 (2 × CH2CH3), 10.44 (CH2CH3), 10.43 (CH2CH3) ppm. Found C, 80.69; H, 8.11; N, 2.09. Calcd for C48H57NO4 (Mr = 711.99): C, 80.97; H, 8.07; N, 1.97%.
4.1.8 5-(4-Aminophenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (7′)
Trifluoroacetic acid (4.20 g, 36.8 mmol) was added dropwise to a stirred solution of calix[4]arene 6′ (2.17 g, 2.77 mmol) in CH2Cl2 (20 mL) cooled at 0 °C. After 15 min, the reaction mixture was allowed to reach room temperature and stirred for a further 2 h. The mixture was washed with an aqueous solution of K2CO3 (20%, 100 mL) and the two phases were separated. The aqueous phase was treated with dichloromethane (2 × 100 mL) and the organic phases were then combined. The solvent was removed under reduced pressure to afford 7′ as a white solid (1.66 g, 88%), which did not need further purification. 1H NMR (CDCl3, 500 MHz), δ 7.16 (2H, d, 3J = 8.3 Hz, ArH), 6.80 (2H, d, 3J = 7.5 Hz, ArH), 6.76 (2H, s, ArH), 6.75 (2H, d, 3J = 8.3 Hz, ArH), 6.72–6.66 (4H, m, ArH), 6.55 (2H, d, 3J = 7.5 Hz, ArH), 6.41 (1H, t, 3J = 7.5 Hz, ArH), 4.55 and 3.26 (4H, d, AB spin system, 2JAB = 13.4 Hz, ArCH2Ar), 4.54 and 3.22 (4H, d, AB spin system, 2JAB = 13.4 Hz, ArCH2Ar), 3.96 (4H, t, 3J = 7.6 Hz, OCH2), 3.93–3.87 (4H, m, OCH2), 3.63 (2H, br s, NH2), 2.06–1.94 (8H, m, CH2CH3), 1.14–1.01 (12H, m, CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 156.87 (2 × arom. Cq–O), 156.41 (arom. Cq–O), 155.51 (arom. Cq–O), 144.94 (arom. Cq–N), 135.50 (arom. Cq), 135.40 (arom. Cq), 134.98 (arom. Cq), 134.92 (arom. Cq), 134.78 (arom. Cq), 132.20 (arom. Cq), 128.40 (arom. CH), 128.35 (arom. CH), 127.99 (arom. CH), 127.81 (arom. CH), 126.20 (arom. CH), 122.06 (arom. CH), 121.97 (arom. CH), 115.17 (arom. CH), 76.86 (OCH2), 76.75 (3 × OCH2), 31.21 (ArCH2Ar), 31.08 (ArCH2Ar), 23.43 (CH2CH3), 23.39 (CH2CH3), 23.32 (2 ×CH2CH3), 10.53 (CH3), 10.38 (CH3) ppm. Found C, 80.67; H, 7.82; N, 2.06. Calcd for C46H53NO4 (Mr = 683.93): C, 80.78; H, 7.81; N, 2.05%.
4.1.9 5-(3-Aminophenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (7″)
Trifluoroacetic acid (7.56 g, 66.3 mmol) was added dropwise to a stirred solution of calix[4]arene 6″ (4.00 g, 5.10 mmol) in CH2Cl2 (20 mL) cooled at 0 °C. After 15 min, the reaction mixture was allowed to reach room temperature and stirred for a further 2 h. The mixture was washed with an aqueous solution of K2CO3 (20%, 150 mL) and the two phases were separated. The aqueous phase was treated with dichloromethane (2 × 100 mL) and the organic phases were then combined. The solvent was removed under reduced pressure to afford 7″ as a white solid (3.43 g, 97%), which did not need further purification. 1H NMR (CDCl3, 500 MHz), δ 7.14 (1H, dd, 3J = 3J’ = 7.7 Hz, ArH), 6.77–6.68 (7H, m, ArH), 6.66–6.60 (3H, m, ArH), 6.59 (1H, dd, 3J = 7.7 Hz, 4J = 1.9 Hz, ArH), 6.51 (2H, d, 3J = 7.4 Hz, ArH), 6.38 (1H, t, 3J = 7.4 Hz, ArH), 4.51 and 3.21 (4H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 4.48 and 3.17 (4H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 3.94–3.81 (8H, m, OCH2), 3.65 (2H, br s, NH2), 2.02–1.89 (8H, m, CH2CH3), 1.09–0.96 (12H, m, CH3), ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 156.88 (2 × arom. Cq–O), 156.44 (arom. Cq–O), 156.24 (arom. Cq–O), 146.26 (arom. Cq–N), 142.87 (arom. Cq), 135.56 (arom. Cq), 135.34 (arom. Cq), 135.06 (arom. Cq), 134.95 (arom. Cq), 134.93 (arom. Cq), 129.28 (arom. CH), 128.42 (2 × arom. CH), 128.03 (arom. CH), 126.87 (arom. CH), 122.08 (arom. CH), 122.05 (arom. CH), 117.78 (arom. CH), 114.03 (arom. CH), 113.30 (arom. CH), 76.92 (OCH2), 76.79 (3 × OCH2), 31.23 (ArCH2Ar), 31.11 (ArCH2Ar), 23.46 (CH2CH3), 23.42 (CH2CH3), 23.34 (2 ×CH2CH3), 10.55 (CH3), 10.40 (CH3) ppm. Found C, 80.57; H, 7.76; N, 2.09. Calcd for C46H53NO4 (Mr = 683.93): C, 80.78; H, 7.81; N, 2.05%.
4.1.10 1,3-Bis[2,6-dimethyl-4-(25,26,27,28-tetrapropyloxycalix[4]aren-5-yl)phenyl]imidazolium tetrafluoroborate (1)
A mixture of 5-(4-amino-3,5-dimethylphenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (7) (1.71 g, 2.41 mmol) and acetic acid (0.310 mL, 0.325 g, 5.42 mmol) was heated at 60 °C for 5 min and MgSO4 (0.290 g, 2.41 mmol) was added (mixture A). In another flask, a mixture of glyoxal (0.137 mL, 0.174 g, 1.20 mmol, 40 wt % in water), formaldehyde (0.090 mL, 0.097 g, 1.20 mmol, 37 wt % in water) and acetic acid (0.310 mL, 0.325 g, 5.42 mmol) was heated at 60 °C for 5 min and ZnCl2 (0.196 g, 1.44 mmol) was added (mixture B). At 60 °C mixture B was added to mixture A, the resulting mixture was stirred at 60 °C for 25 min and then cooled to room temperature. CH2Cl2 (30 mL) and a 3 M aqueous solution of HCl (50 mL) were added and the resulting mixture was stirred for 1 h at room temperature. The organic layer was separated. Water (20 mL) and ammonium tetrafluoroborate (0.313 g, 2.99 mmol) were added, and the mixture was stirred at room temperature for 1 h. The organic layer was separated, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2; EtOAc/petroleum ether, 50:50; Rf = 0–0.47) to afford 1 as a tan solid (1.33 g, 71%). 1H NMR (CDCl3, 500 MHz), δ 8.45 (1H, s, NCHN), 7.67 (2H, s, NCH), 7.14 (4H, s, ArH), 6.81 (4H, s, ArH), 6.73 (4H, d, 3J = 7.4 Hz, ArH), 6.69–6.61 (8H, m, ArH), 6.57 (4H, d, 3J = 7.4 Hz, ArH), 6.40 (2H, t, 3J = 7.4 Hz, ArH), 4.54 and 3.23 (8H, d, AB spin system, 2JAB = 13.5 Hz, ArCH2Ar), 4.50 and 3.16 (8H, d, AB spin system, 2JAB = 13.4 Hz, ArCH2Ar), 3.99–3.82 (16H, m, OCH2), 2.18 (12H, s, o-CH3), 2.05–1.91 (16H, m, CH2CH3), 1.13–0.97 (24H, m, CH2CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 157.12 (arom. Cq–O), 156.65 (2 × arom. Cq–O), 156.53 (arom. Cq–O), 144.63 (arom. Cq–N), 136.54 (NCHN), 135.67 (arom. Cq), 135.33 (arom. Cq), 135.05 (arom. Cq), 134.91 (arom. Cq), 134.23 (arom. Cq), 132.98 (arom. Cq), 131.10 (arom. Cq), 128.43 (arom. CH), 128.36 (arom. CH), 127.99 (arom. CH), 127.79 (arom. CH), 127.12 (arom. CH), 125.52 (arom. CH), 122.15 (arom. CH), 121.85 (arom. CH), 76.91 (OCH2), 76.80 (2 × OCH2), 76.77 (OCH2), 31.12 (ArCH2Ar), 31.00 (ArCH2Ar), 23.39 (CH2CH3), 23.34 (CH2CH3), 23.30 (2 ×CH2CH3), 17.57 (o-CH3), 10.45 (CH2CH3), 10.37 (CH2CH3) ppm. 19F NMR (CDCl3, 282 MHz), −152.32 (br s, 19F-10B), −152.37 to −152.39 (m, 19F-11B) ppm. Found C, 76.55; H, 7.40; N, 1.75. Calcd for C99H113BF4N2O8 (Mr = 1545.80): C, 76.92; H, 7.37; N, 1.81%.
4.1.11 1,3-Bis[4-(25,26,27,28-tetrapropyloxycalix[4]aren-5-yl)phenyl]imidazolium tetrafluoroborate (2)
A mixture of 5-(4-aminophenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (7′) (6.65 g, 9.72 mmol) and acetic acid (1.25 mL, 1.31 g, 21.9 mmol) was heated at 60 °C for 5 min and MgSO4 (1.17 g, 9.72 mmol) was added (mixture A). In another flask, a mixture of glyoxal (0.555 mL, 0.705 g, 4.86 mmol, 40 wt % in water), formaldehyde (0.361 mL, 0.394 g, 4.86 mmol, 37 wt % in water) and acetic acid (1.25 mL, 1.31 g, 21.9 mmol) was heated at 60 °C for 5 min and ZnCl2 (0.794 g, 5.83 mmol) was added (mixture B). At 60 °C mixture B was added to mixture A, the resulting mixture was stirred at 60 °C for 25 min and then cooled to room temperature. CH2Cl2 (70 mL) and a 3 M aqueous solution of HCl (100 mL) were added and the resulting mixture was stirred for 1 h at room temperature. The organic layer was separated. Water (40 mL) and ammonium tetrafluoroborate (1.02 g, 9.72 mmol) were added, and the mixture was stirred at room temperature for 1 h. The organic layer was separated, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2; EtOAc/petroleum ether, 50:50, Rf = 0–0.51) to afford 2 as a tan solid (4.40 g, 61%). 1H NMR (CDCl3, 500 MHz), δ 9.26 (1H, s, NCHN), 7.73 (2H, s, NCH), 7.64 (4H, d, 3J = 7.7 Hz, ArH), 7.39 (4H, d, 3J = 7.7 Hz, ArH), 6.80 (4H, d, 3J = 7.4 Hz, ArH), 6.77 (4H, d, 3J = 7.4 Hz, ArH), 6.70 (4H, dd, 3J = 3J’ = 7.4 Hz), 6.65 (4H, s, ArH), 6.36 (4H, d, 3J = 7.4 Hz, ArH), 6.16 (2H, t, 3J = 7.4 Hz, ArH), 4.50 and 3.19 (8H, d, AB spin system, 2JAB = 13.4 Hz, ArCH2Ar), 4.45 and 3.12 (8H, d, AB spin system, 2JAB = 13.4 Hz, ArCH2Ar), 3.97–3.88 (8H, m, OCH2), 3.85 (4H, t, 3J = 7.2 Hz, OCH2), 3.77 (4H, t, 3J = 7.2 Hz, OCH2), 2.01–1.85 (16H, m, CH2CH3), 1.07 (6H, t, 3J = 7.4 Hz, CH3), 1.04 (6H, t, 3J = 7.4 Hz, CH3), 0.98 (12H, t, 3J = 7.4 Hz, CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 157.03 (2 × arom. Cq–O), 157.00 (arom. Cq–O), 156.20 (arom. Cq–O), 144.02 (arom. Cq–N), 135.89 (arom. Cq), 135.48 (arom. Cq), 135.37 (arom. Cq), 134.60 (arom. Cq), 132.35 (arom. Cq), 131.95 (NCHN), 128.73 (arom. CH), 128.71 (arom. CH), 128.51 (arom. CH), 127.79 (arom. CH), 126.76 (arom. CH), 122.39 (arom. CH), 122.24 (arom. CH), 122.16 (arom. CH), 121.89 (arom. CH), 77.00 (OCH2), 76.85 (OCH2), 76.75 (2 × OCH2), 31.17 (ArCH2Ar), 31.04 (ArCH2Ar), 23.50 (CH2CH3), 23.45 (CH2CH3), 23.26 (2 ×CH2CH3), 10.63 (CH3), 10.27 (CH3) ppm. 19F NMR (CDCl3, 282 MHz), −151.00 (br s, 19F-10B), −151.05 (br s, 19F-11B) ppm. Found C, 76.23; H, 7.22; N, 2.10. Calcd for C95H105BF4N2O8 (Mr = 1489.69): C, 76.60; H, 7.10; N, 1.88%.
4.1.12 1,3-Bis[3-(25,26,27,28-tetrapropyloxycalix[4]aren-5-yl)phenyl]imidazolium tetrafluoroborate (3)
A mixture of 5-(3-aminophenyl)-25,26,27,28-tetrapropyloxycalix[4]arene (7″) (2.04 g, 2.99 mmol) and acetic acid (0.384 mL, 0.404 g, 6.73 mmol) was heated at 60 °C for 5 min and MgSO4 (0.359 g, 2.99 mmol) was added (mixture A). In another flask, a mixture of glyoxal (0.170 mL, 0.216 g, 1.49 mmol, 40 wt % in water), formaldehyde (0.111 mL, 0.121 g, 1.49 mmol, 37 wt % in water) and acetic acid (0.384 mL, 0.404 g, 6.73 mmol) was heated at 60 °C for 5 min and ZnCl2 (0.244 g, 1.79 mmol) was added (mixture B). At 60 °C mixture B was added to mixture A, the resulting mixture was stirred at 60 °C for 25 min and then cooled to room temperature. CH2Cl2 (30 mL) and a 3 M aqueous solution of HCl (50 mL) were added and the resulting mixture was stirred for 1 h at room temperature. The organic layer was separated. Water (20 mL) and ammonium tetrafluoroborate (0.313 g, 2.99 mmol) were added, and the mixture was stirred at room temperature for 1 h. The organic layer was separated, dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2; EtOAc/petroleum ether, 50:50; Rf = 0–0.5) to afford 3 as a tan solid (0.508 g, 23%). 1H NMR (CDCl3, 500 MHz), δ 9.26 (1H, s, NCHN), 7.74 (2H, s, NCH), 7.63–7.42 (4H, m, ArH), 7.40–7.20 (4H, m, ArH), 6.93–6.75 (8H, m, ArH), 6.71 (4H, d, 3J = 7.3 Hz, ArH), 6.66 (4H, dd, 3J = 3J’ = 7.3 Hz), 6.49 (4H, d, 3J = 7.3 Hz, ArH), 6.26 (2H, t, 3J = 7.3 Hz, ArH), 4.51 and 3.29 (8H, d, AB spin system, 2JAB = 13.2 Hz, ArCH2Ar), 4.48 and 3.16 (8H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 3.97–3.85 (12H, m, OCH2), 3.83 (4H, t, 3J = 7.4 Hz, OCH2), 2.03–1.88 (16H, m, CH2CH3), 1.10–0.96 (24H, m, CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 157.25 (arom. Cq–O), 156.85 (2 × arom. Cq–O), 156.45 (arom. Cq–O), 144.06 (arom. Cq–N), 135.80 (arom. Cq), 135.53 (arom. Cq), 135.25 (arom. Cq), 134.96 (arom. Cq), 134.44 (arom. Cq), 132.37 (NCHN), 132.19 (arom. Cq), 130.76 (arom. CH), 128.86 (arom. CH), 128.56 (arom. CH), 128.47 (arom. CH), 127.88 (arom. CH), 126.98 (arom. CH), 122.72 (arom. CH), 122.14 (arom. CH), 121.77 (arom. CH), 120.27 (arom. CH), 120.10 (arom. CH), 76.96 (OCH2), 76.85 (OCH2), 76.77 (2 × OCH2), 31.10 (ArCH2Ar), 31.05 (ArCH2Ar), 23.43 (CH2CH3), 23.39 (CH2CH3), 23.29 (2 ×CH2CH3), 10.52 (CH3), 10.34 (CH3) ppm. 19F NMR (CDCl3, 282 MHz), −150.31 (br s, 19F-10B), −150.36 (br s, 19F-11B) ppm. Found C, 72.97; H, 6.92; N, 1.73. Calcd for C95H105BF4N2O8·CH2Cl2 (Mr = 1489.69 + 84.93): C, 73.23; H, 6.85; N, 1.78%.
4.1.13 trans-[1,3-Bis-{2,6-dimethyl-4-(25,26,27,28-tetrapropyloxycalix[4]aren-5-yl)phenyl}imidazol-2-ylidene](pyridine)palladium(II) dichloride (8)
A suspension of imidazolium salt 1 (0.200 g, 0.133 mmol), finely crushed K2CO3 (0.092 g, 0.665 mmol) and PdCl2 (0.028 g, 0.159 mmol) in pyridine (4 mL) was heated at 80 °C for 18 h under vigorous stirring. The mixture was cooled to room temperature, filtered through Celite and the collected solid washed with CH2Cl2 (ca. 20 mL). The filtrate was evaporated to dryness and the residue purified by flash chromatography (SiO2; CH2Cl2/petroleum ether, 60:40; Rf = 0.68) to afford 8 as a yellow solid (0.187 g, 82%). 1H NMR (CDCl3, 500 MHz), δ 8.51–8.47 (2H, m, o-NC5H5), 7.54 (1H, tt, 3J = 7.6 Hz, 4J = 1.7 Hz, p-NC5H5), 7.19 (4H, s, ArH), 7.13 (2H, s, NCH), 7.10–7.05 (2H, m, m-NC5H5), 6.93 (4H, s, ArH), 6.73–6.66 (4H, m, ArH), 6.64–6.57 (12H, m, ArH), 6.48 (2H, t, 3J = 7.5 Hz, ArH), 4.53 and 3.25 (8H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 4.48 and 3.17 (8H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 3.93 (4H, t, 3J = 7.4 Hz, OCH2), 3.90–3.84 (12H, m, OCH2), 2.43 (12H, s, o-CH3), 2.02–1.88 (16H, m, CH2CH3), 1.06–0.98 (24H, m, CH2CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 156.81 (arom. Cq–O), 156.72 (arom. Cq–O), 156.65 (2 × arom. Cq–O), 153.12 (NCN), 151.67 (arom. CH o-Py), 142.39 (arom. Cq–N), 137.52 (arom. CH p-Py), 136.67 (arom. Cq), 136.01 (arom. Cq), 135.62 (arom. Cq), 135.33 (arom. Cq), 135.23 (arom. Cq), 135.00 (arom. Cq), 134.05 (arom. Cq), 128.47 (arom. CH), 128.33 (arom. CH), 128.17 (arom. CH), 127.20 (arom. CH), 127.18 (arom. CH), 124.34 (arom. CH), 124.05 (arom. CH), 122.22 (arom. CH), 122.13 (arom. CH), 76.91 (OCH2), 76.87 (2 × OCH2), 76.79 (OCH2), 31.29 (ArCH2Ar), 31.16 (ArCH2Ar), 23.46 (CH2CH3), 23.41 (3 ×CH2CH3), 19.52 (o-CH3), 10.50 (CH2CH3), 10.48 (CH2CH3) ppm. Found C, 72.64; H, 6.97; N, 2.53. Calcd for C104H117Cl2N3O8Pd (Mr = 1714.41): C, 72.86; H, 6.88; N, 2.45%.
4.1.14 trans-[1,3-Bis{4-(25,26,27,28-tetrapropyloxycalix[4]aren-5-yl)phenyl}imidazol-2-ylidene](pyridine)palladium(II) dichloride (9)
A suspension of imidazolium salt 2 (0.429 g, 0.298 mmol), finely crushed K2CO3 (0.206 g, 1.49 mmol) and PdCl2 (0.063 g, 0.357 mmol) in pyridine (6 mL) was stirred at room temperature for 18 h. The mixture was filtered through Celite and the filtered solid washed with CH2Cl2 (ca. 20 mL). The filtrate was evaporated to dryness and the residue purified by flash chromatography (SiO2; CH2Cl2/petroleum ether, 50:50; Rf = 0.59) leading to 9 as a yellow solid (0.296 g, 60%). 1H NMR (CDCl3, 500 MHz), δ 8.69 (2H, d, 3J = 5.2 Hz, o-NC5H5), 8.10 (4H, d, 3J = 8.2 Hz, ArH), 7.66 (1H, t, 3J = 7.7 Hz, p-NC5H5), 7.47 (4H, d, 3J = 8.2 Hz, ArH), 7.33 (2H, s, NCH), 7.22 (2H, dd, 3J = 7.7 Hz, 3J’ = 5.2 Hz, m-NC5H5), 6.90 (4H, d, 3J = 7.4 Hz, ArH), 6.85 (4H, d, 3J = 7.4 Hz, ArH), 6.82–6.74 (7H, m, ArH), 6.45 (4H, d, 3J = 7.4 Hz, ArH), 6.30 (2H, t, 3J = 7.4 Hz, ArH), 4.58 and 3.29 (8H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 4.52 and 3.21 (8H, d, AB spin system, 2JAB = 13.3 Hz, ArCH2Ar), 3.99 (8H, dd, 3J = 3J’ =7.1 Hz, OCH2), 3.91 (4H, t, 3J = 7.1 Hz, OCH2), 3.85 (8H, t, 3J = 7.1 Hz, OCH2), 2.12–1.91 (16H, m, CH2CH3), 1.12 (6H, t, 3J = 7.3 Hz, CH3), 1.09 (6H, t, 3J = 7.3 Hz, CH3), 1.03 (12H, t, 3J = 7.3 Hz, CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 157.07 (2 × arom. Cq–O), 156.43 (arom. Cq–O), 156.14 (arom. Cq–O), 151.25 (arom. CH o-Py), 150.77 (NCN), 142.05 (arom. Cq–N), 137.77 (arom. CH p-Py), 137.64 (arom. Cq), 135.89 (arom. Cq), 135.51 (arom. Cq), 135.11 (arom. Cq), 134.53 (arom. Cq), 133.47 (arom. Cq), 128.67 (arom. CH), 128.52 (arom. CH), 127.85 (arom. CH), 127.61 (arom. CH), 126.81 (arom. CH), 125.94 (arom. CH), 124.36 (arom. CH), 123.56 (arom. CH), 122.18 (arom. CH), 122.04 (arom. CH), 76.94 (OCH2), 76.78 (OCH2), 76.72 (2 × OCH2), 31.21 (ArCH2Ar), 31.07 (ArCH2Ar), 23.47 (CH2CH3), 23.43 (CH2CH3), 23.25 (2 × CH2CH3), 10.61 (CH3), 10.26 (CH3) ppm. Found C, 72.22; H, 6.71; N, 2.45. Calcd for C100H109Cl2N3O8Pd (Mr = 1658.31): C, 72.43; H, 6.63; N, 2.53%.
4.1.15 trans-[1,3-Bis-{3-(25,26,27,28-tetrapropyloxycalix[4]aren-5-yl)phenyl}imidazol-2-ylidene](pyridine)palladium(II) dichloride (10)
A suspension of imidazolium salt 3 (0.200 g, 0.139 mmol), finely crushed K2CO3 (0.096 g, 0.695 mmol) and PdCl2 (0.029 g, 0.166 mmol) in pyridine (4 mL) was stirred at room temperature for 18 h. The mixture was filtered through Celite and the collected solid washed with CH2Cl2 (ca. 20 mL). The filtrate was evaporated to dryness and the residue purified by flash chromatography (SiO2; CH2Cl2/petroleum ether, 60:40; Rf = 0.56) to afford 10 as a yellow solid (0.157 g, 68%). 1H NMR (CDCl3, 500 MHz), δ 8.68 (2H, d, 3J = 5.2 Hz, o-NC5H5), 8.32 (2H, s, ArH), 8.14 (2H, d, 3J = 7.5 Hz, ArH), 7.65–7.55 (4H, m, ArH), 7.53 (1H, t, 3J = 7.3 Hz, p-NC5H5), 7.42 (2H, s, NCH), 7.18 (4H, s, ArH), 7.06 (2H, dd, 3J = 7.3 Hz, 3J’ = 5.2 Hz, m-NC5H5), 6.76 (4H, d, 3J = 7.5 Hz, ArH), 6.60 (2H, t, 3J = 7.4 Hz, ArH), 6.57–6.53 (4H, m, ArH), 6.41–6.43 (8H, m, ArH), 4.51 and 3.21 (8H, d, AB spin system, 2JAB = 13.2 Hz, ArCH2Ar), 4.49 and 3.17 (8H, d, AB spin system, 2JAB = 13.2 Hz, ArCH2Ar), 3.97 (4H, t, 3J = 7.4 Hz, OCH2), 3.93 (4H, t, 3J = 7.4 Hz, OCH2), 3.83 (8H, t, 3J = 7.4 Hz, OCH2), 2.02–1.87 (16H, m, CH2CH3), 1.06 (12H, t, 3J = 7.4 Hz, CH3), 1.01 (6H, t, 3J = 7.4 Hz, CH3), 0.99 (6H, t, 3J = 7.4 Hz, CH3) ppm. 13C{1H} NMR (CDCl3, 125 MHz), δ 157.37 (arom. Cq–O), 157.07 (arom. Cq–O), 156.21 (2 × arom. Cq–O), 151.53 (NCN), 151.29 (arom. CH o-Py), 142.71 (arom. Cq–N), 139.73 (arom. Cq), 137.65 (arom. CH p-Py), 136.27 (arom. Cq), 135.82 (arom. Cq), 134.62 (arom. Cq), 134.34 (arom. Cq), 133.31 (arom. Cq), 129.51 (arom. CH), 128.39 (arom. CH), 128.24 (arom. CH), 128.07 (arom. CH), 127.27 (arom. CH), 127.18 (arom. CH), 124.42 (arom. CH), 123.77 (arom. CH), 123.48 (arom. CH), 122.19 (arom. CH), 122.00 (arom. CH), 76.92 (2 × OCH2), 76.81 (OCH2), 76.70 (OCH2), 31.24 (ArCH2Ar), 31.10 (ArCH2Ar), 23.45 (2 × CH2CH3), 23.33 (CH2CH3), 23.29 (CH2CH3), 10.61 (CH3), 10.32 (CH3) ppm. Found C, 71.84; H, 6.69; N, 2.43. Calcd for C100H109Cl2N3O8Pd·0.25 CH2Cl2 (Mr = 1658.31 + 21.23): C, 71.69; H, 6.52; N, 2.50%.
4.1.16 General procedure for palladium-catalysed Suzuki–Miyaura cross-coupling reactions
A mixture of the palladium complex (0.01 mmol), phenylboronic acid (0.146 g, 1.20 mmol) and K2CO3 (0.207 g, 1.50 mmol) was suspended in absolute ethanol (3 mL). After addition of p-tolyl chloride (0.126 g, 1 mmol), the mixture was vigorously stirred at 80 °C for 1 h. The hot mixture was filtered through Celite. 1,4-Dimethoxybenzene (0.069 g, 0.5 mmol), used as internal standard, was then added to the filtrate. The solvent was removed under reduced pressure, and the crude mixture was analysed by 1H NMR spectroscopy. The yields were determined by comparing the intensity of the methyl signal of the product [δ(Me) = 2.41 ppm] with that of the internal reference [δ(Me) = 3.78 ppm]. The isolated yield (concerning experiments with chromatographic product separation) turned out to be very close to that determined by using the internal reference (deviation less than 5%).
4.1.17 Crystal data for complex 9
Crystals suitable for X-ray analysis were obtained by slow diffusion of pentane into a chloroform solution of the complex. Crystal data: C100H109Cl2N3O8Pd·CHCl3, Mr = 1777.56 g mol−1, triclinic, space group , a = 13.7011(5) Å, b = 22.8400(8) Å, c = 35.5075(13) Å, α = 73.332(2)°, β = 83.700(2)°, γ = 79.678(2)°, V = 10,452.3(7) Å3, Z = 4, D = 1.130 g cm−3, μ = 3.004 mm−1, F(000) = 3728, T = 173(2) K. The sample was studied using a Kappa APEX II diffractometer (graphite monochromated Cu Kα radiation, λ = 1.54178 Å). The data collection (2θmax = 133.7°, omega omegascan frames by using 2°omega rotation and 10 s per frame, range hkl: h −16, 16; k −27, 23; l −42, 40) gave 204,167 reflections. The structure was solved using SHELXS-2013 [30], which revealed the non-hydrogen atoms of the molecule. After anisotropic refinement, all hydrogen atoms were found using a Fourier difference map. The structure was refined using SHELXL-2013 [31] by the full-matrix least-squares technique (use of F2 magnitude; x, y, z, βij for C, Cl, N and Pd atoms; x, y, z in riding mode for H atoms); 2138 variables and 27,946 observations with I > 2.0 σ(I); calcd w = 1/[σ2(Fo2) + (0.1202P)2 + 1.2609P] where P = (Fo2 + 2Fc2)/3, with the resulting R = 0.0601, RW = 0.1837 and SW = 1.081, Δρ < 1.149 eÅ−3. CCDC 1882970.
4.2 Computational details
All calculations were performed with the Gaussian 09 program [32], using the functional PBEPBE [33]. Dispersion corrections were included [34]. All atoms were described using the def2-SVP basis set [35] and associated pseudopotential for the palladium atom. To get an optimised structure, the geometry was simplified by replacing the propoxy groups of the calixarenes by methoxy groups. The structure was fully optimised and the wavefunction saved. The NCI analysis [36] was performed on this geometry. All calculations were performed in the gas phase.
Acknowledgements
H.A. gratefully acknowledges the "Association de spécialisation et d'orientation scientifique" (Lebanon) for financial support. We thank Mr. Vincent Bruno (Unistra) for assistance in obtaining NMR data.