

1 Introduction
L’étude de l’induction asymétrique-1,2 en série radicalaire acyclique 〚1–5〛 nous a permis de mettre en évidence l’isomérisation de l’oléfine (S)-4,5-O-isopropylidène-4,5-dihydroxy-2-penténoate d’éthyle 1-Z lors de l’addition du radical tris(triméthylsilyl)silane 〚6, 7〛. Sur cette dernière, nous nous sommes posé la question suivante : l’addition des radicaux silylés et stannylés sur les oléfines homochirales (1-Z et 1-E) 〚8〛 sont-elles sous contrôle cinétique ou thermodynamique ?
Beckwith et Raner 〚9〛 ont montré que le résultat de la stéréochimie cis/trans de la cycloaddition d’un radical alkyle sur une fonction aldéhyde dépendait de la concentration en n-Bu3SnH. À forte concentration, le rapport cis/trans est contrôlé par la stabilité thermodynamique du radical cycloalkyloxy. Dans cette note, nous allons montrer que l’hydrostannylation de l’oléfine 1-E est sous contrôle cinétique et non sous contrôle thermodynamique.
Par ailleurs, si l’addition de n-Bu3Sn• sur l’oléfine 1-Z conduit à l’oléfine 1-E, la formation de produits d’addition 2-syn et 2-anti à partir de l’oléfine 1-E dans les conditions utilisées 〚2〛 est sous contrôle cinétique (Fig. 1).
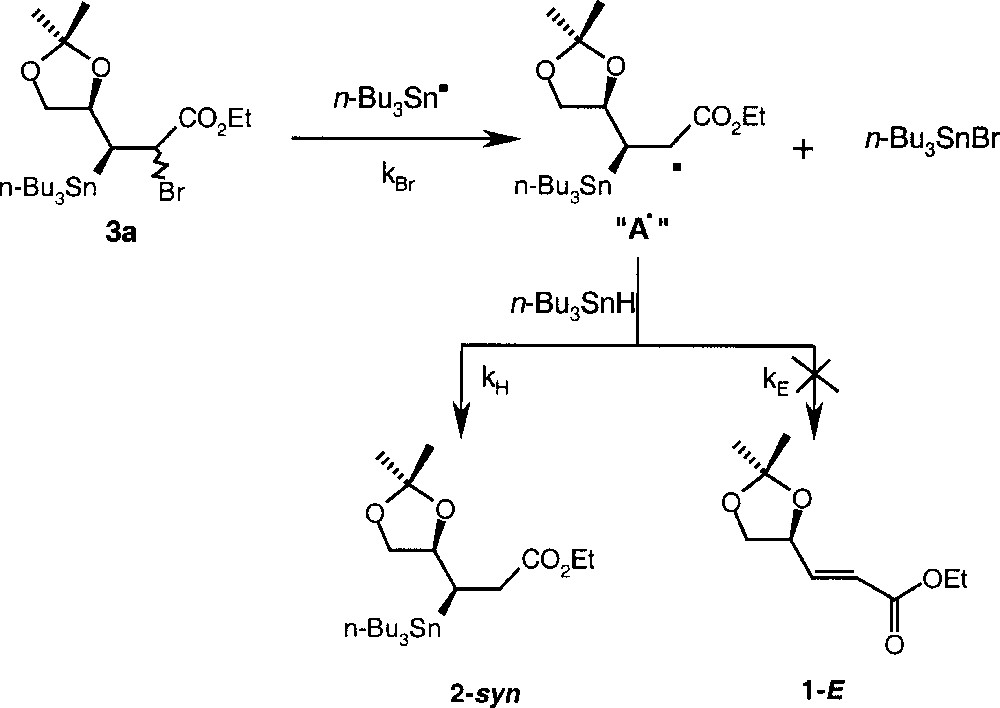
Addition de n-Bu3Sn• sur l’oléfine 1-Z conduisant à l’oléfine 1-E et au produit d’addition 2-syn.
En effet, l’étape essentielle est la réduction (kH) et doit être suffisamment rapide pour ne pas permettre à A• de donner le produit d’élimination (kE). Les études cinétiques 〚10, 11〛 menées avec l’hydrure de tributylétain sur des radicaux carbonés ont montré que le piégeage par l’hydrure était relativement indépendant de la nature du radical (kH ≈ 2 × 106 l mol–1 s–1). Le facteur kH 〚n-Bu3SnH〛 contrôle directement la durée de vie de A• et détermine donc la possibilité pour A• de subir une réduction ou une élimination.
Ces études ont montré, de plus, que la réaction d’élimination était rapide et pouvait être compétitive avec la rotation autour de la liaison σ. La constante de vitesse d’élimination kE est supérieure à 106 l mol–1 s–1. En jouant sur la concentration d’hydrure, il est possible de favoriser la réduction par rapport à l’élimination. Pratiquement, cet ajustement de la concentration en n-Bu3SnH se fait en opérant en milieu concentré.
On a :
2 Résultats et discussion
L’identification rigoureuse de la nature stéréochimique des produits d’addition radicalaire 〚2〛 a été réalisée en utilisant l’élimination de Peterson 〚12〛 en milieu acide.
Pour favoriser la réaction de réduction par rapport à la réaction d’élimination, on se place dans les conditions établies auparavant (kBr > kE ↔ 〚n-Bu3SnH〛 > 〚A–Br〛 afin d’étudier la réactivité des radicaux A• et B• obtenus par deux méthodes différentes : l’hydrostannylation de l’oléfine 1-E et la réduction du bromure correspondant (3a et 3b).
La réduction du mélange des bromures épimères 3a obtenus par bromation du produit 2-syn en présence de NBS/hν/CCl4 fournit exclusivement le produit 2-syn, sans aucune trace, ni de l’oléfine 1, ni du produit 2-anti, ce qui montre que le piégeage du radical A• est plus rapide que l’élimination (Fig. 2), dans les conditions de concentration (0,55 mol l–1) en n-Bu3SnH.
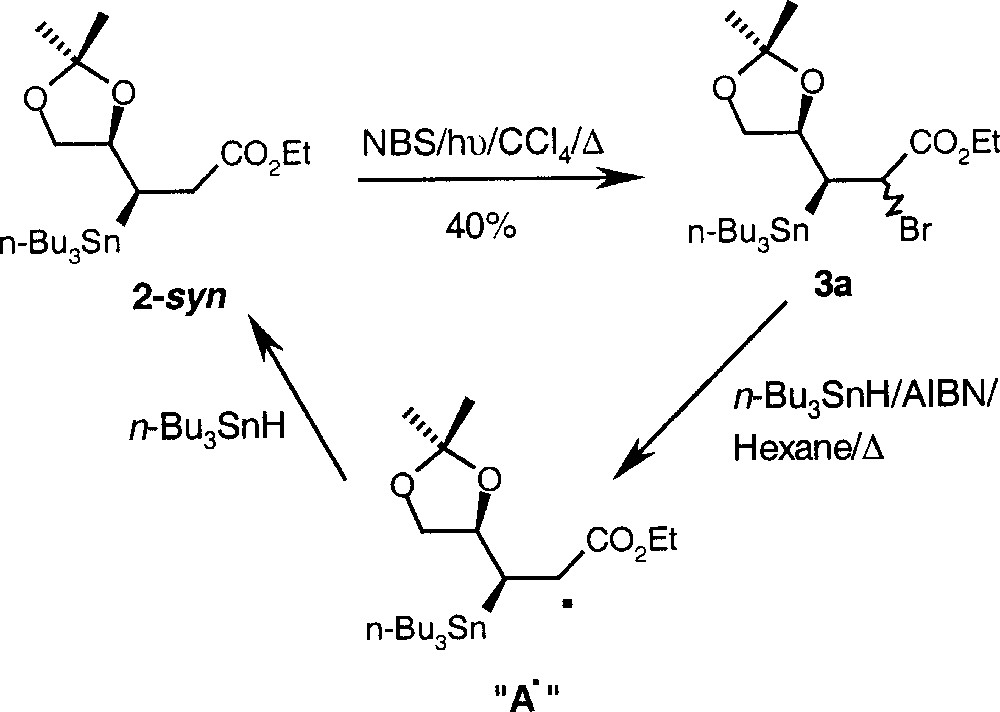
Réduction en présence de n-Bu3SnH du mélange des bromures épimères 3a, obtenus par bromation du produit 2-syn en présence de NBS/hν/CCl4, redonnant le produit 2-syn.
Données spectrales des épimères 3a : RMN 1H (CDCl3, 400 MHz) : 4,48–4,57 (1H, m) ; 4,16 (2H, J = 7,2 Hz, q) ; 3,64–3,75 (1H, m) ; 2,97–2,72 (2H, m) ; 1,37 (3H, s) ; 1,35 (3H, s) ; 0,90–1,71 (30H, m). RMN 13C (CDCl3, 100 MHz) : carbones primaires : 25,84 ; 25,75 ; 14,21 ; carbones secondaires : 67,82 ; 62,83 ; 29,65 ; 27,97 ; 10,32 ; carbones tertiaires : 78,17 ; 32,65 ; 32,41 ; 30,31 ; carbones quaternaires : 180,00 ; 175,61 ; 108,77. Données spectrales des épimères 3b : RMN 1H (CDCl3, 400 MHz) : 4,39–4,47 (1H, m) ; 3,77 (1H, J = 6,6 et 7,7 Hz, dd) ; 3,52 (2H, J = 7,1 Hz, q); 3,31 (1H, J = 7,4 Hz, t) ; 2,60 (0,5H, J = 2,8 Hz, d) ; 2,58 (0,5H, J = 11,1 Hz, d) ; 2,06 (1H, J = 7,2 et 10,5 Hz, dd) ; 1,37 (3H, s) ; 1,35 (3H, s) ; 0,90–1,70 (30H, m). RMN 13C (CDCl3, 100 MHz) : carbones primaires : 25,67 ; 14,20 ; 13,90 ; carbones secondaires : 68,60 ; 62,15 ; 29,64 ; 27,93 ; 10,14 ; carbones tertiaires : 77,64 ; 31,60 ; 31,38 ; 29,67 ; carbones quaternaires : 186,91 ; 179,92 ; 108,76.
La réduction du mélange des bromures épimères 3b obtenus par bromation du produit 2-anti montre qu’il y a une compétition entre la réaction de réduction et la réaction d’élimination, puisque l’on obtient 65% du produit de réduction 2-anti, 18% du produit d’élimination 1-E et 7% du produit 2-syn (non séparés, mais identifiés par RMN du proton). La majorité du produit 2-anti provient de la réduction directe du radical B• (étape 3, Fig. 3) ; en revanche, le reste provient de l’addition de n-Bu3Sn• sur l’oléfine 1-E issue de l’élimination (étape 5). Le diastéréomère 2-syn obtenu provient directement de l’addition de n-Bu3Sn• sur l’oléfine 1-E issue de l’élimination (étape 4).
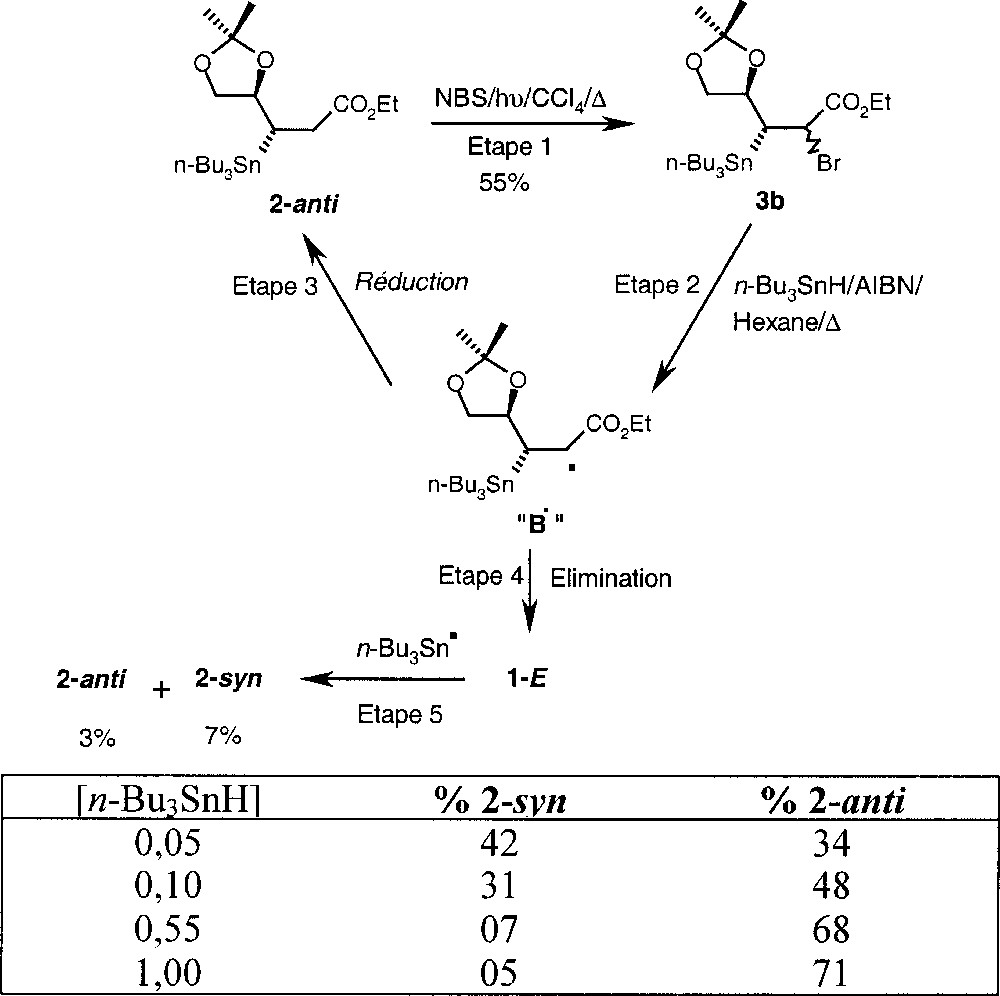
Réduction du mélange des bromures épimères 3b, obtenus par bromation du produit 2-anti.
Le piégeage par l’hydrure est relativement indépendant de la nature du radical ; donc, la différence de comportement des deux radicaux provient de leur stabilité induite par les centres stéréogènes présents dans la molécule.
Enfin, pour confirmer ce résultat, nous avons effectué la réaction de réduction des épimères 3b en présence de différentes concentrations en tributylétain. Les résultats obtenus montrent clairement que le piégeage par l’hydrure est intimement lié à la concentration de l’hydrure et à la nature des centres stéréogènes proches du centre radicalaire (Fig. 3).
Le piégeage du radical intermédiaire en α de l’ester par le deutérium se fait avec un excès diastéréomérique de 30% dans le cas du diastéréomère 2-syn et 78% pour le 2-anti (Fig. 4). Ce résultat montre que le piégeage par le deutérium est influencé par la présence des deux centres stéréogènes en position α et β ; en revanche, la diastéréosélectivité par rapport au premier centre est dans le même sens que ce qui a déjà été observé 〚1, 2〛.

Piégeage du radical intermédiaire en α de l’ester par le deutérium.
La régiosélectivité (attaque 1,4) et la stéréochimie des diastéréomères 5a et 5b ont été confirmées par l’action du di-isopropylamidure de lithium sur les diastéréomères 2-syn et 2-anti. L’anion résultant est piégé par l’éthanol deutéré (les produits obtenus sont les mêmes qu’avec n-Bu3SnD).
En conclusion, cette étude montre que la formation du radical A• lors de l’hydrostannylation de l’oléfine 1-E est sous contrôle cinétique, mais que celle du radical B• ne l’est que partiellement. Les produits d’hydrostannylation 2-syn et 2-anti de l’oléfine 1-E proviennent donc d’un contrôle cinétique et non d’un contrôle thermodynamique, dans les conditions de concentration 0,55 mol l–1 en n-Bu3SnH. En revanche, des faibles concentrations favorisent la β-élimination.
Enfin, nous avons montré que le piégeage du radical intermédiaire en α de l’ester par le deutérium est influencé par la présence des deux centres stéréogènes en positions α’ et β’.