

1 Introduction
Les complexes π des métaux de transition constituent aujourd'hui une classe de composés en plein essor, dont l'intérêt tout aussi bien en synthèse organique qu'organométallique a été maintes fois démontré [1]. Dans ce contexte, alors que la chimie des complexes (η6-arène)Cr(CO)3 a été largement explorée [2], les complexes isoélectroniques (η6-arène)Mn(CO)3+ cationiques ont connu, jusqu'à ces dernières années, un développement nettement moins rapide, principalement à cause des difficultés de préparation et de purification qu'ils présentent [3]. La complexation d'un arène par le greffon Mn(CO)3+ active de façon spectaculaire son caractère électrophile, jusqu'à permettre l'addition d'un nucléophile, menant aux complexes (η5-cyclohexadiényl)Mn(CO)3 neutres correspondants [1b,4]. Nous avons montré que les complexes η5 substitués par des groupes Cl, OMe, NR2 ou SPh donnaient lieu à des réactions d'hydrodéchloration, -déméthoxylation, –déamination ou -désulfurisation via des substitutions nucléophiles cine, tele-meta ou tele-para (Schéma 1, voie (a)) [5a]. De plus le champ de fonctionnalisation de ces complexes a été très étendu par notre équipe ces dernières années : l'arylation, la carbonylation, la substitution par des groupes oléfiniques, acétyléniques ou par des hétéroatomes peuvent êtres réalisées via des réactions de couplage palladocatalysé sous condition carbonylante (voie (b)), ou non (voie (c)) [1e,6], et la séquence de lithiation suivie du piégeage par un électrophile (voie (d)) [7] permet d'accéder aisément à une très grande variété de complexes diversement substitués. Parmi eux, les complexes (η5-cyclohexadiényl)Mn(CO)3 substitués par des groupes carbonyles forment une classe particulièrement intéressante, car ils présentent trois sites électrophiles : l'arène complexé, le groupe carbonyle et le greffon métallique sont susceptibles de réagir de façon compétitive avec des réactifs nucléophiles. Nous présentons ici l'étude de leur réactivité vis-à-vis d'hydrures métalliques.
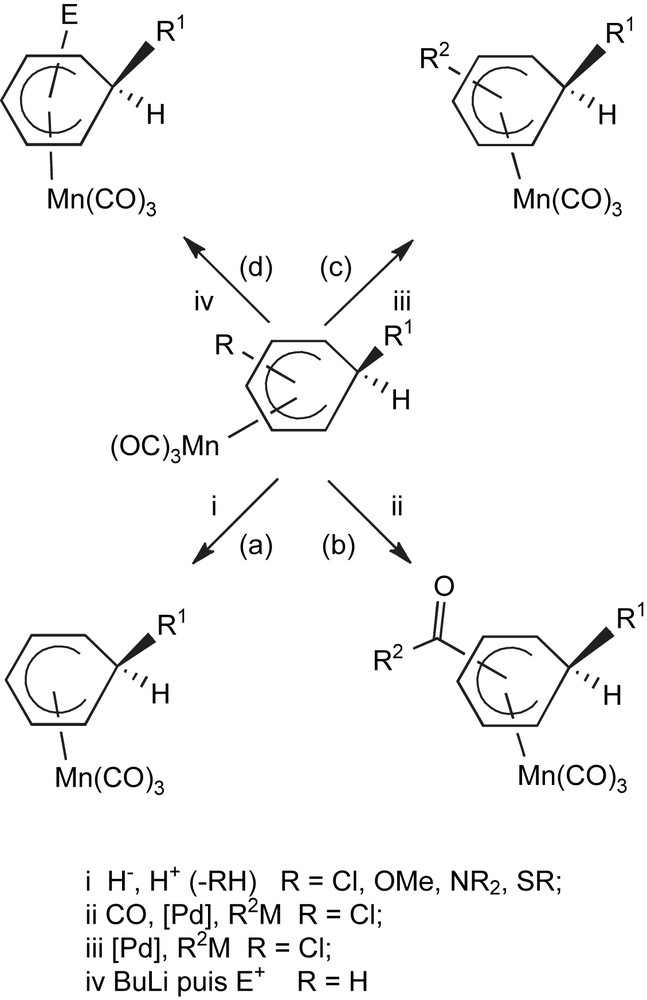
Réactivité générale des complexes (η5-cyclohexadiényl)Mn(CO)3.
2 Contexte
Très récemment, il a été montré que les complexes 1 substitués par une fonction cétone à l'une des extrémités du système π et par un groupe phényle ou ortho-tolyle sur le carbone sp3 réagissaient avec le borohydrure de sodium de la manière suivante (Schéma 2) [8]. Après addition de 4 équivalents de NaBH4 le complexe 1a donne lieu à la formation de 3 composés.

Réaction des cétones (η5-cyclohexadiényl)Mn(CO)3 avec les hydrures.
Alors que les deux premiers ont été identifiés comme les produits de réduction 2a, 2a′ de la fonction cétone, résultant de l'addition d'hydrure sur le groupe COTh (82%, rapport des 2 diastéréoisomères : 58:42), le troisième produit 3a est un cyclohexadiène correspondant à l'addition d'hydrure sur le système cyclohexadiényle avec perte de l'entité Mn(CO)3 (rendement 13% en produit isolé). La même réactivité a été observée lorsque le complexe 1c, comportant un groupe o-tolyle sur le carbone sp3, est mis à réagir avec un excès de NaBH4, ainsi les complexes 2c, 2c′ diastéréoisomères (rapport 78:22) et le cyclohexadiène 3c (20%) ont été isolés avec des rendements respectifs de 66 et 20%. Bien que les raisons de la différence entre les 2 rapports de diastéréoisomères ne soient pas à ce jour entièrement élucidées, la nature du groupe porté par l'atome de carbone sp3 semble jouer un rôle certain dans la diastéréosélectivité de l'addition d'hydrure sur le groupe COTh.
Afin de déterminer le mécanisme de formation des cyclohexadiènes 3a,c, le complexe marqué 1b [8] a été préparé et engagé dans la même séquence réactionnelle. Traité par un excès de NaBH4, il conduit aux alcools diastéréoisomères 2b, 2b′ (69%, rapport 60:40) et au cyclohexadiène 3b dont l'analyse des données RMN 1H montre clairement la disparition des signaux correspondant aux protons H3 et H5 du composé 3a. En d'autres termes, la position des atomes de deutérium n'est pas modifiée lors de la formation du cyclohexadiène, excluant ainsi toute migration intramoléculaire de deutérure (ou d'hydrure) par l'intermédiaire du métal lors de la réaction.
Pour affiner ce résultat, le complexe 1a a été mis à réagir avec un excès de borodeutérure de sodium. Comme attendu, les deux complexes hydroxydeutérométhyl-thiényl(η5-cyclohexadiényl) Mn(CO)3 2d, 2d′ ont pu être isolés, ainsi que le cyclohexadiène 3d dont l'étude du spectre RMN 1H, entre autres, a permis de conclure sans ambiguïté quant à la position d'attaque du deutérure : le signal correspondant au proton H2 a complètement disparu. Cela prouve définitivement que l'addition du nucléophile a eu lieu en position 2 du système cyclohexadiényle.
3 Résultats et discussion
Nous avons alors étendu cette étude à l'addition d'hydrure sur le complexe η5 1e dont le carbone sp3 comporte 2 atomes d'hydrogène (Schéma 3). Ce complexe est préparé conformément à la procédure développée au laboratoire [6]. Après addition de LiAlH4 sur le complexe cationique 4, le complexe 5 se forme avec 54% de rendement correspondant à l'attaque régiosélective de l'hydrure en ortho du chlore (Schéma 3) [5a]. Ce complexe est alors engagé dans une réaction de Stille avec le 2-thiényltributylétain, sous atmosphère de CO, en présence du système catalytique Pd2dba3/AsPh3 dont nous avions prouvé la meilleure efficacité pour les couplages pallado-catalysés de complexes (η5-chloro cyclohexadiényl)Mn(CO)3. Le produit attendu 1e est alors obtenu et isolé avec 79% de rendement.

Synthèse et réactivité du complexe 1e.
Le complexe 1e soumis à l'action d'un excès de NaBD4 dans les mêmes conditions expérimentales que précédemment, fournit d'une part les deux alcools diastéréoisomères 2e, 2e′ qui sont purifiés par chromatographie sur colonne de silice (58%, rapport 75:25), d'autre part le cyclohexadiène 3e (19%) (Schéma 3). Les données RMN de ce dernier composé montrent sans ambiguité l'addition du deutérure en position 2 du système cyclohexadiényle : en effet le proton H3 couple seulement avec un des protons H5 et son signal correspond à un doublet à 5.18 ppm (4J3–5 = 1.9 Hz). Quant au carbone C2, il présente un signal à 128.3 ppm sous forme de triplet (J = 23 Hz).
Grâce à ces résultats de marquage isotopique, un mécanisme peut être proposé (Schéma 4 ; par souci de lisibilité, le système η5 est représenté comme un système η3/η2). L'addition exo du nucléophile par rapport au métal se fait en position 2 du système cyclohexadiényle, menant au complexe anionique η3/σ A. Après insertion du manganèse dans la liaison C–H2endo et décoordination de la liaison C1C2, l'intermédiaire anionique η3 B peut être formé et conduit par élimination réductrice au complexe anionique η4 C. Le cyclohexadiène est alors obtenu après perte du fragment métallique.

Mécanisme proposé pour la formation des cyclohexadiènes.
L'addition de nucléophiles carbonés stabilisés [9] ou d'hydrures forts [10] aux extrémités d'un ligand η5 de complexes (η5-cyclohexadiényl)Mn(CO)3 neutres non fonctionnalisés a déjà été étudiée et donne lieu à la formation de cyclohexadiènes démétallés ou des complexes η3 du manganèse à liaison agostique. A notre connaissance, les seuls exemples impliquant une addition en position interne du système η5 concernent des complexes (η5-pentadiényl)Mn(CO)3 ouverts [11] et des complexes cationiques (η5-cyclohexadiényl) Mn(NO)(L)2+ [12].
Dans notre étude, la nature du groupe électroattracteur COTh semble essentielle dans cette addition d'hydrure à la régiosélectivité inhabituelle. Afin de déterminer si le caractère conjugué du substituant cétone est important, le complexe 6, substitué par le groupe Cl essentiellement inductif attracteur, a été mis à réagir dans les mêmes conditions que précédemment avec un excès de NaBH4. Après hydrolyse, le complexe 7 a pu être isolé avec un rendement de 13%, aux côtés d'une grande quantité de produit de départ (Schéma 5).

Réaction du complexe 6 avec NaBH4.
Aucune trace d'un quelconque cyclohexadiène n'a pu être détectée lors de l'analyse du brut réactionnel. Cette transformation constitue un exemple de réaction d'hydrodéchloration, laquelle a été largement étudiée au laboratoire dans le cas de complexes (η5-cyclohexadiényle)Mn(CO)3 substitués par deux atomes d'hydrogène sur le carbone sp3 [5a]. La très faible conversion obtenue ici est à rapprocher du fait que des hydrures forts du type L-Selectride sont habituellement nécessaires pour effectuer ces réactions. C'est la première fois qu'une telle substitution nucléophile est rapportée dans la littérature, dans la série des complexes substitués en position exo sur l'atome de carbone sp3 par un groupe différent d'un atome d'hydrogène. Un mécanisme plausible peut être proposé (Schéma 6) : l'addition d'hydrure a lieu en position C1 du système cyclohexadiényle, menant au complexe η4 anionique D correspondant, lequel capte un proton lors du traitement du milieu réactionnel pour donner l'hydrure de manganèse neutre E à liaison Mn–H. Ce complexe peut se décrire comme étant un complexe neutre du Mn(I) à liaison agostique F en équilibre avec les complexes G et H obtenus par isomérisation qui peuvent, par élimination 1,2 ou 1,5 d'HCl mener au complexe 7. D'un point de vue global, le nucléophile s'additionne sur une position différente de celle où est fixé le groupe partant Cl, plus précisément en position C5 de celui-ci. Il s'agit donc d'un mécanisme de substitution nucléophile tele-meta [5].

Mécanisme proposé pour la réaction d'hydrodéchloration.
Le caractère inductif attracteur de Cl n'est donc pas suffisant pour induire l'addition de l'hydrure sur une position interne du système cyclohexadiényle. Dans le cas des complexes carbonylés, le groupe COTh, conjugué au ligand η5, peut donc modifier ses propriétés électroniques et permettre une telle attaque.
4 Conclusion
Lors de ce travail, la réactivité vis-à-vis d'hydrures de complexes (η5-cyclohexadiényl)Mn(CO)3 substitués par un groupe électroattracteur COTh a été étudiée. Les complexes (η5-hydroxythiénylcyclohexadiényl)Mn(CO)3 formés par réduction de la fonction cétone sont obtenus majoritairement, à coté de cyclohexadiènes inattendus, issus de l'addition minoritaire d'hydrure sur une position interne du système cyclohexadiényle en β de la cétone, suivie de perte de l'entité Mn(CO)3. Dans le cas d'un complexe substitué par un atome de chlore, aucun produit dû à l'addition de l'hydrure sur un carbone interne du système η5 n'est détecté mais le produit d'hydrodéchloration, issu de l'addition du nucléophile sur une position à l'extrêmité du ligand η5 a pu être isolé. Enfin, un mécanisme de formation des cyclohexadiènes a été proposé sur la base de différentes expériences de marquage isotopique au deutérium.
5 Partie expérimentale
5.1 Généralités
L'ensemble des réactions et des manipulations a été réalisé sous atmosphère inerte grâce à un système de rampe à vide. Le THF a été distillé sur sodium/benzophénone. Les spectres IR sont enregistrés sur un spectromètre Nicolet-Avatar 320 FT-IR. Les spectres RMN 1H et 13C sont respectivement enregistrés sur un appareil BRUCKER AC 200 MHz et BRUCKER ARX 400 MHz. Dans les deux cas, la référence interne est le tétraméthylsilane.
Les complexes 4 [5a], 5 [5a], 6 [5a], 7 [13], 1e [6] ont été préparés conformément aux procédures décrites dans la littérature.
5.2 Complexes 2e, 2e′ et 3e
Dans un bicol de 50 mL sont dissous le complexe 1e (0.193 g, 0.530 mmol, 1 équiv.) et NaBD4 (0.091 g, 2.12 mmol, 4 équiv.) dans 25 mL de THF. Le milieu réactionnel est porté au reflux du solvant pendant 2 h avant d'être hydrolysé avec 20 mL d'une solution saturée de NH4Cl et d'extraire la phase aqueuse avec 50 mL d'éther diéthylique. La phase organique est lavée avec 50 mL d'eau, 50 mL d'une solution saturée de K2CO3 et 50 mL d'une solution saturée de NaCl. Après filtration sur MgSO4, les solvants sont évaporés sous pression réduite, et le brut est chromatographié sur gel de silice (éther de pétrole) permettant d'isoler deux solides jaunes correspondant aux alcools diastéréoisomères 2e, 2e′ (0.136 g, 0.310 mmol, 58%) dans un rapport 25:75, et le cyclohexadiène 3e (0.022 g, 0.099 mmol, 19%).
5.3 Diastéréoisomère minoritaire 2e
IR (CHCl3) : 1917, 2010 (Mn(CO)3). RMN 1H (CDCl3, 200 MHz) : 1.99 (1H, d, 2J = 12.5 Hz, H6exo) ; 2.58 (1H, dd, 2J = 12.5 Hz, 3J = 6.0 Hz, H6endo) ; 2.92 (1H, dd, 3J = 6.0 Hz, 4J = 2.1 Hz, H5) ; 3.48 (3H, s, OMe) ; 5.26 (1H, d, 3J = 5.9 Hz, H2) ; 5.77 (1H, dd, 3J = 5.9 Hz, 4J = 2.1 Hz, H3) ; 6.97–7.25 (3H, m, HAr). RMN 13C (CDCl3, 100 MHz) : 29.1 (C6) ; 35.7 (C5) ; 54.5 (COMe) ; 66.1 (C3) ; 70.0 (C1) ; 71.4 (t, C7) ; 89.1 (C2) ; 125.5–126.8 (CAr) ; 143.5 (C4) ; 144.4 (CAr).
5.4 Diastéréoisomère majoritaire 2e′
IR (CHCl3) : 1917, 2010 (Mn(CO)3). RMN 1H (CDCl3, 200 MHz) : 2.18 (1H, d, 2J = 12.6 Hz, H6exo) ; 2.93–3.01 (2H, m, H5 et H6endo) ; 3.46 (3H, s, OMe) ; 4.90 (1H, d, 3J = 5.8 Hz, H2) ; 5.72 (1H, dd, 3J = 5.8 Hz, 4J = 1.8 Hz, H3) ; 6.89–7.23 (3H, m, HAr). RMN 13C (CDCl3, 100 MHz) : 26.6 (C6) ; 37.0 (C5) ; 54.5 (COMe) ; 66.4 (C3) ; 70.9 (C1) ; 73.6 (t, C7) ; 93.0 (C2) ; 124.3–126.9 (CAr) ; 143.6 (C4) ; 144.4 (CAr). Anal. pour C15H12DMnO5S. Trouvée : C, 49.55 ; H, 4.09. Calculée : C, 49.86 ; H, 3.91.
5.5 Cyclohexadiène 3e
RMN 1H (CDCl3, 200 MHz) : 2.45 (2H, m, H5) ; 2.72 (2H, m, H6) ; 3.72 (3H, s, OMe) ; 5.18 (1H, d, 4J = 1.9 Hz, H3) ; 7.06–7.57 (3H, m, HAr). RMN 13C (CDCl3, 100 MHz) : 23.4 (C6) ; 29.6 (C5) ; 38.3 (CQ) ; 55.6 (COMe) ; 93.4 (C3) ; 127.4–131.7 (CAr) ; 128.3 (t, C2) ; 139.4 (CQ) ; 143.9 (CQ) ; 167.2 (CO). SM (MALDI-TOF) : 222.0542 (MH+).
5.6 Complexe 7
Dans un bicol de 50 mL, le complexe 6 (0.466 g, 1.30 mmol, 1 equiv.) est dissous avec NaBH4 (0.197 g, 5.20 mmol, 4 equiv.) dans 12 mL de THF. Le milieu réactionnel est porté au reflux du solvant pendant 4 h avant d'être hydrolysé avec 20 mL d'une solution saturée de NH4Cl et d'extraire la phase aqueuse avec 50 mL d'éther diéthylique. La phase organique est lavée avec 50 mL d'eau, 50 mL d'une solution saturée de K2CO3 et 50 mL d'une solution saturée de NaCl. Après filtration sur MgSO4, les solvants sont évaporés sous pression réduite, et le brut réactionnel est chromatographié sur gel de silice (éther de pétrole/diéthyléther) permettant d'isoler un solide jaune (0.055 g, 0.17 mmol, 13%) dont les données spectroscopiques sont compatibles avec celles du produit 7 précédemment décrit dans la littérature [13].
Remerciements
Ce travail a été financé par le CNRS. Nous remercions D. Jonathan pour des résultats préliminaires et l'École normale supérieure (Paris) pour le financement d'A.E.