

1 Introduction
De nombreuses méthodes de synthèse de thioglycosides sont décrites dans la littérature [1–3], et elles utilisent fréquemment, comme glycosyle donneur, des halogénures de glycosyles acétylés [4–7]. Pour ce type de réaction classique et générale, on oppose à l’halogénure de glycosyle un anion thiolate. Avec les anions thiolate aliphatiques, la réaction de ré-acylation est souvent nécessaire. Un autre intérêt de ces thioglycosides est leur utilisation dans les réactions de glycosylation [8], notamment en ce qui concerne les réactions de O-glycosylation, qui s’effectuent souvent en présence de sels métalliques – sels de mercure II [9–11], de palladium II [12–13], par exemple. Récemment, une approche électrochimique a été envisagée avec succès, pour laquelle la première étape correspond à une oxydation monoélectronique du thioglycoside [14–18]. Les thioglycosides contenant des groupes hydroxyles libres peuvent intervenir dans la synthèse d’oligosaccharides comme glycoside accepteur [19]. La synthèse de thioglycosides et leur utilisation en synthèse osidique présentent un intérêt certain, et il nous est apparu intéressant d’étudier, d’une part, la réactivité de toute une série d’anions thiolate préparés par voie électrochimique, vis-à-vis d’un bromure de glycosyle, précurseur de molécules d’intérêt biologique présentant, pour certaines d’entre elles une activité antithrombotique [20–22]. Comme nous le montrerons, la réactivité observée dépend directement du caractère nucléophile de l’anion thiolate mis en œuvre. D’autre part, nous indiquerons des résultats originaux concernant la réactivité d’anions thiolate vis à vis de thioglycosides.
2 Résultats et discussion
2.1 Électrosynthèse et propriétés redox d’anions thiolate
Les anions thiolate sont obtenus par réduction bi-électronique des disulfures correspondants [23] et, à titre d’exemple, la Fig. 1 donne le voltamogramme du diphényl disulfure PhSSPh 2 dans l’acétonitrile (MeCN) contenant 0,2 mol de Bu4NPF6 comme électrolyte support, sur une électrode de carbone, à température ambiante. En réduction, un pic irréversible A est observé à –1,33 V et, au balayage retour, un pic d’oxydation A′1. Dans le domaine anodique, PhSSPh s’oxyde à +1,58 V. L’électrolyse de 2, effectuée à –1,5 V (plateau de la vague A), consomme deux électrons (nexp = 2,3 F) et la solution, initialement incolore, devient orange. Par voltamétrie cyclique, la solution obtenue présente un pic d’oxydation irréversible A′1 (Fig. 1).

Voltamogramme cyclique du diphényl disulfure 2 dans une solution MeCN–Bu4NPF6 (0,2 M). Trait plein : 2 seul ; pointillé : après électrolyse à – 1,5 V/ECS. Potentiel initial : O V/ECS (trait plein) ; – 1 V/ECS (pointillé). Vitesse de balayage : 100 mV s–1.
Ces résultats sont en accord avec les réactions (1) et (2) :
Un comportement similaire est également observé pour les autres disulfures.
Le Tableau 1 donne les potentiels de réduction et d’oxydation des disulfures étudiés et le potentiel d’oxydation des anions thiolate correspondants.
Caractéristiques électrochimiques des disulfures et des anions thiolate dans une solution MeCN–Bu4NPF6 : (a) potentiels (en V/ECS) déterminés par voltamétrie cyclique à 0,2 V s–1 ; (b) les anions thiolate sont électrogénérés par réduction bi-électronique des disulfures sur toile de carbone au potentiel de la vague A ; (c) R′ = –C6H4–CO–C6H4–CN.
| Entrées | Disulfures | Ep(red) a | Ep(ox) a | Anion thiolate b | ||
| pic A | pic E′ | Ep(ox) a, pic A′1 | ||||
| 1 | R′SSR′ c | 1 | –0,95 | +1,76 | –0,16 | 1′ |
| 2 | PhSSPh | 2 | –1,33 | +1,58 | –0,36 | 2′ |
| 3 | (p-H3C–C6H4–S)2 | 3 | –1,66 | +1,62 | –0,32 | 3′ |
| 4 | (C6H5–CH2–S)2 | 4 | –2,26 | +1,50 | –0,56 | 4′ |
| 5 | (CH3S)2 | 5 | –2,54 | +1,38 | –0,46 | 5′ |
L’examen de ce tableau appelle plusieurs remarques.
- • Les disulfures aromatiques (entrées 1, 2 et 3) sont plus facilement réductibles que les disulfures aliphatiques. Le composé 1, qui possède un groupe carbonyle électroattracteur, est réduit à un potentiel moins négatif que celui du dérivé 2. La présence du groupe méthyle électrodonneur rend la réduction de 3 plus difficile que celle de 2. Le diméthyl disulfure 5 est le composé qui se réduit le plus difficilement (–2,54 V).
- • Les disulfures aromatiques s’oxydent plus difficilement que les disulfures aliphatiques, mais la différence de potentiel est beaucoup moins importante que celle relevée pour leur réduction (ΔEp ox = 0,38 V et ΔEp red = 1,59 V entre 1 et 5, par exemple).
- • Concernant les anions thiolate, on retrouve sensiblement la même séquence que précédemment : les anions dérivés des disulfures aromatiques s’oxydent à des potentiels plus anodiques que ceux obtenus au départ des disulfures aliphatiques. Cependant, la différence de potentiel est beaucoup plus faible, l’écart le plus important se situant entre les anions 1′ R′S– et 4′ C6H5–CH2–S– ; cette différence est égale à 0,400 V.
La connaissance des caractéristiques électrochimiques des différents couples disulfure/anion thiolate laisse entrevoir une réactivité chimique relativement différente, liée au caractère nucléophile et/ou au pouvoir réducteur plus ou moins marqué des anions thiolate, et permet d’envisager leur mise en œuvre dans des réactions de substitution ou de réduction en série osidique.
Nous avons examiné en détail, d’une part, la réactivité des différents anions thiolate 1′, 2′, 3′, 4′ et 5′ vis-à-vis du bromure de 5-thioxylopyranosyle Xyl–Br 6 (bromure de 2,3,4-tri-O-acetyl-5-thio-α-D-xylopyranosyle) [24] et, d’autre part, celle des anions thiolate 2′, 3′, 4′ et 5′ en présence du 1,5-dithioxylopyranoside Xyl–SR′ (R′ = –C6H4–CO–C6H4–CN) 10a ([4-(4′-cyanobenzophénone)] 2,3,4-tri-O-acétyl-1,5-dithio-α-d-xylopyranoside) et 10b ([4-(4′-cyanobenzophénone)] 2,3,4-tri-O-acétyl-1,5-dithio-β-d-xylopyranoside) [25] (Fig. 2).
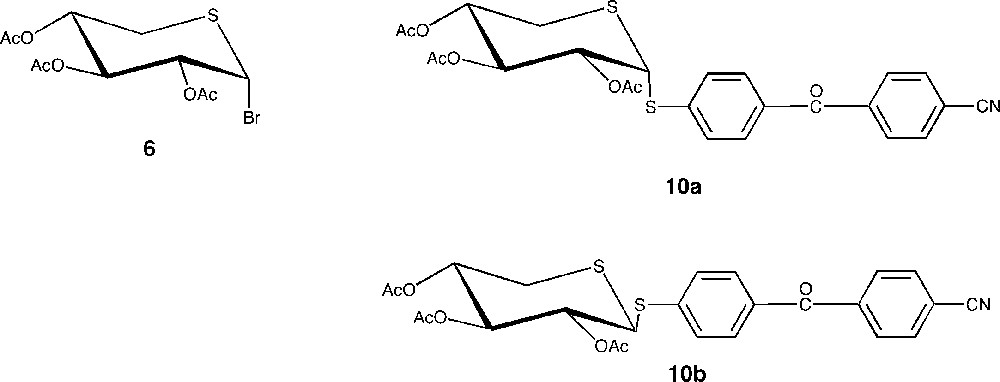
Composés 6, 10a et 10b.
2.2 Réactivité des anions thiolate 1′, 2′, 3′, 4′ et 5′ en présence de 6
L’addition d’un équivalent de 6 à une solution contenant l’anion 1′ ne modifie pas la vague d’oxydation A′1 de 1′, même après plusieurs heures, ce qui exclut toute réactivité directe. La réaction de substitution nucléophile, avec formation de Xyl–SR′ 10, est cependant possible, en opérant en présence d’un acide de Lewis qui active la liaison glycosidique [25].
Contrairement au cas précédent, l’addition d’un équivalent de 6 à une solution de 2′ électrogénéré s’accompagne rapidement de la disparition de la vague d’oxydation A′1, de l’apparition d’une vague de réduction B et d’une vague A′2 à +0,78 V, caractéristique de l’oxydation de l’ion bromure Br– (Fig. 3). Dans les mêmes conditions opératoires, ACN/Bu4NPF6, Bu4NBr présente ce même pic d’oxydation A′2. Le pic B correspond à la réduction de Xyl–SPh 7, qui a été isolé et caractérisé sur la base des données spectroscopiques.

Voltamogramme cyclique de l'anion thiolate 2′ dans une solution MeCN–Bu4NPF6 (0,2 M). Trait plein : 2 seul ; pointillé : après ajout de 1 équiv de 6. Potentiel initial : – 1,2 V/ECS. Vitesse de balayage : 100 mV s–1.
La réaction entre l’anion 3′ et 6 conduit sensiblement au même résultat que précédemment et le composé Xyl–S–C6H4–CH3 8 correspondant a été caractérisé par spectroscopie RMN (équation (3)).
La réactivité des anions 2′ et 3′ vis-à-vis de 6 est tout à fait comparable, le caractère nucléophile et les propriétés rédox de ces deux anions étant proches.
Concernant les anions 4′ et 5′, dont le caractère nucléophile est davantage marqué par rapport à celui des autres anions, on assiste à une réactivité tout à fait inhabituelle et comparable, puisque dans les deux cas on obtient le même 5-thioxylopyranose 9 et le disulfure correspondant 4 et 5 (équation (4)).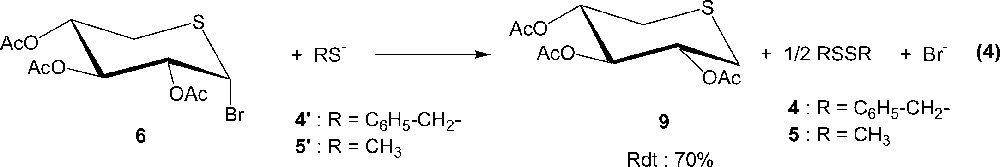
La formation de 9, qui correspond formellement au remplacement du brome par un hydrogène, s’explique par une réduction préalable de 6 par l’anion 4′ ou 5′. Nous avons montré que la réduction bi-électronique directe ou en présence de médiateurs rédox (dichlorure de niobocène, par exemple) [26] de 6 conduisait au sucre insaturé 3,4-di-O-acétyl-5-thioxylal, pour lequel la double liaison est localisée entre les atomes de carbone 1 et 2. Le transfert de deux électrons de deux équivalents de 4′ ou de 5′ sur un équivalent de 6 est donc interdit, puisque l’on obtiendrait le 5-thioxylal et, au contraire, l’hypothèse d’une réduction monoélectronique de 6 par 4′ ou 5′est tout à fait intéressante. Ce transfert électronique s’accompagne de la formation du radical anion correspondant (équation (5)), qui évolue rapidement par élimination de Br– au radical neutre (équation (6)). Ce radical, très réactif, est capable de capter un atome d’hydrogène (par exemple du solvant Solv–H), pour conduire à la formation de 9 (équation (7)). Ce type de réactivité de radicaux neutres aliphatiques ou aromatiques a déjà été mentionné dans la littérature [27–28].
2.3 Réactivité des anions thiolate 2′, 3′, 4′ et 5′ vis-à-vis de Xyl–SR′ 10
Ce type d’étude, original, nous a permis de mettre en évidence une réactivité intéressante et de préparer avec de très bons rendements des sulfures dissymétriques.
La réaction de l’anion 5′CH3S– sur le 1,5-dithio-β-d-xylopyranoside Xyl–SR′ 10b conduit au composé 11 (CH3–S–C6H4–CO–C6H4–CN), avec un rendement presque quantitatif (Rdt = 90 %), et au dérivé Xyl–SH 12b, de configuration β (Rdt = 70 %). Le même produit 11 (Rdt = 78 %) est obtenu en effectuant la réaction au départ du 1,5-dithio-α-d-xylopyranoside 10a et, dans ce cas, le 1,5-dithio-α-d-xylopyranoside 12a est aussi obtenu avec rétention de configuration.
La réaction entre l’anion 4′ et Xyl–SR′ 10a, 10b est très similaire à la précédente, puisque l’on obtient, d’une part, le sulfure mixte 13 C6H5–CH2–S–C6H4–CO–C6H4–CN, avec un rendement presque quantitatif (Rdt = 80 % et 95 %, respectivement) et, d’autre part, le 1,5-dithioxylose : 12a et 12b, respectivement, sont également obtenus avec rétention de configuration.
La formation de 11 et de 13 s’explique par une attaque nucléophile de 5′ (ou de 4′) sur 10, qui correspond à une substitution nucléophile aromatique [29, 30]. Elle s’accompagne d’une rupture hétérolytique de la liaison soufre–carbone de l’aglycone S–R′ dans le 1,5-dithio-d-xylopyranoside 10, sans isomérisation, (équations (8)).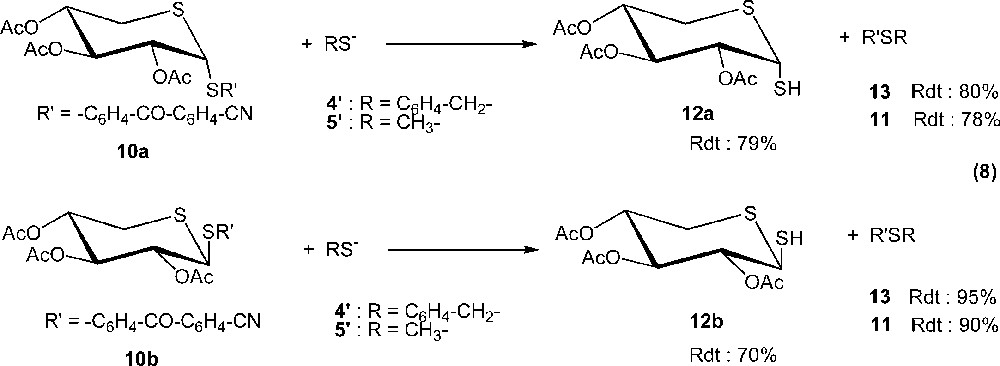
L’anion Xyl–S– formé initialement est, lors de l’extraction, protoné par l’eau ou par H+ (HPF6), et conduit au composé 12.
Aucune réactivité ne se manifeste entre les anions 1′2′, 3′et les thioglycosides Xyl–SR′ 10. Le caractère nucléophile de ces anions thiolate, beaucoup moins marqué que celui des anions 4′et 5′, explique cette différence importante de réactivité.
3 Conclusion
En conclusion, nous avons examiné la réactivité d’anions thiolate sur deux dérivés glycosidiques. Au départ du bromure de 5-thio-d-xylopyranosyle Xyl–Br, en fonction de la nucléophilie des anions mis en œuvre, nous obtenons, soit les 1,5-dithio-d-xylopyranosides Xyl–SR soit le sucre saturé 5-thioxylopyranose. Dans le cas du 1,5-dithio-d-xylopyranoside Xyl–SR′, une réactivité particulière est mise en évidence vis-à-vis d’anions thiolate à forte nucléophilie et s’accompagne de la formation de sulfures dissymétriques via un mécanisme de substitution nucléophile aromatique. Cette étude montre l’intérêt du recours à l’électrochimie, puisque les différences de réactivité observées peuvent être reliées directement aux propriétés redox des divers couples disulfure–anion thiolate et apporte une contribution intéressante dans le domaine de la glycochimie.
4 Partie expérimentale
4.1 Procédure générale
Les cellules sont conditionnées sous atmosphère d’azote, avec 20 ml d’acétonitrile et 1,6 g de Bu4NPF6 (0,2 M) comme électrolyte support. La technique expérimentale mise en œuvre et les appareils utilisés ont été décrits précédemment [31]. Les anions thiolate sont générés par électrolyse a potentiel contrôlé sur électrode de carbone à température ambiante. Après consommation de 2 F mol–1, on additionne le thioglycoside. Le milieu réactionnel est laissé sous agitation pendant 4 h. Il est ensuite évaporé, puis repris dans un mélange acétate d’éthyl-éther. L’électrolyte support est filtré et la phase organique est évaporée. Le produit est alors purifié par chromatographie sur gel de silice, avec, comme éluant, un mélange toluène–acétate d’éthyle.
4.2 Phényl 2,3,4-tri-O-acétyl-1,5-dithio-d-xylopyranoside 7
Le composé 7 est préparé à partir de 6 (109,5 mg ; 0,308 mmol) et 2 (33,5 mg ; 0,154 mmol), avec un rendement de 30 % (α/β : 46:54). Les données spectroscopiques sont en accord avec celles déjà publiées [32].
4.3 Benzyl 2,3,4-tri-O-acétyl-1,5-dithio-d-xylopyranoside 8
Le composé 8 est préparé à partir de 6 (98 mg ; 0,276 mmol) et de 3 (34 mg ; 0,138 mmol), avec un rendement de 30 % (α/β : 46:54) [33].
4.4 2,3,4-tri-O-acétyl-xylotétrahydrothiopyrane 9
Le composé 9 est obtenu à partir de 4 (186 mg ; 0,276 mmol) et 6 (186 mg ; 0,524 mmol) avec un rendement de 70 %. Ce produit est également obtenu au départ de 5 [34].
4.5 2,3,4-tri-O-acétyl-d-1,5-dithioxylopyranoside 12
Le composé 12a est obtenu à partir de 4 (45 mg ; 0,183 mmol) et 10a (93,91 mg ; 0,183 mmol), avec un rendement de 79 %. Ce produit est également obtenu au départ de 5 [35].
Le composé 12b est obtenu à partir de 4 (45 mg ; 0,183 mmol) et 10b (93,91 mg ; 0,183 mmol), avec un rendement de 70 %. Ce même produit est également obtenu au départ de 5.
4.6 4-(4-Méthylsulfanyl-benzoyl)-benzonitrile 11
Le composé 11 est obtenu à partir de 5 (8μl ; 0,09 mmol) et 10b (92,37 mg ; 0,180 mmol) avec un rendement de 90 %. Ce composé est également obtenu par action de 10a sur 5, avec un rendement de 78 %. RMN 1H (CDCl3 ; 500 MHz) : δ (ppm) 7,82–7,84 (d, 2H, 8,52 Hz) ; 7,77–7,79 (d, 2H, 8,42 Hz) ; 7,70–7,72 (d, 2H, 8,56 Hz) ; 7,30–7,31 (d, 2H, 8,54 Hz) ; 2,53 (s, 3H, –SCH3). 13C (CDCl3 ; 125 MHz) : δ (ppm) 193 (CO) ; 125–145 (aromatiques) ; 14,66 (S–CH3). Masse (impact électronique) : m/z = 253 (M) ; 239 (M+1–CH3), 206 (M–CH3S), 151 (M–C6H4CN), 130 (M–CH3SC6H4), 123 (M–COC6H5CN), 102 (M–CH3SC6H4COC6H5). Analyse élémentaire : calc. C, 71,15 % ; H, 4,35 % ; S, 12,65 % ; N, 5,53 % ; exp. C, 71,03 % ; H, 4,28 % ; S, 12,45 % ; N, 5,45 %. IR : ν(CO) = 1650 cm–1 ; ν(CN) = 2227 cm–1.
4.7 4-(4-Benzylsulfanyl-benzoyl)-benzonitrile 13
Le composé 13 est obtenu à partir de 4 (45 mg ; 0,183 mmol) et 10b (187,82 mg ; 0,356 mmol), avec un rendement de 95 %. Ce composé 13 est également obtenu par action de 10a sur 4, avec un rendement de 80 %. RMN 1H (CDCl3 ; 300 MHz) : δ (ppm) 7,78–7,79 (d, 2H, 7,94 Hz) ; 7,74–7,77 (d, 2H, 8,40 Hz) ; 7,64–7,67 (d, 2H, 8,33 Hz) ; 7,23–7,39 (m, 7H) ; 4,22 (s, 2H, –CH2–) ; 13C (CDCl3 ; 75 MHz) : δ (ppm) 125–145 (aromatiques) ; 37,38 (S–CH2–). Masse (impact électronique) : m/z = 329 (M) ; 239 (M+1–C6H5CH2), 206 (M–CH3S), 130 (M–C6H5SC6H4), 102 (M– C6H5SC6H4COC6H5). Analyse élémentaire : calc. C, 76,60 % ; H, 4,56 % ; S, 9,73 % ; N, 4,26 % ; exp. C, 76,90 % ; H, 4,52 % ; S, 9,64 % ; N, 4,22 %. IR ν(CO) = 1669 cm–1 ; ν(CN) = 2230 cm–1.
Remerciements
Les auteurs remercient le conseil régional de Bourgogne et la société Fournier SA pour leur contribution financière (bourse D.B. et contrats d’études nos 00/5112/A1/00023 et 01/5162/S4/00023), ainsi que M. T. Compain pour sa très importante contribution expérimentale.