

1 Introduction
Phthalocyanines are classical pigments that have been discovered accidentally for more than 70 years. Owing to the ease of preparation and modification, high stability, and extraordinary physical properties, this class of compounds have been widely studied and their applications have been extended to many other areas including advanced materials for various opto-electronic devices [1–6], catalysts for photo- and oxidative-degradation of pollutants [7, 8], and second-generation photosensitizers for photodynamic therapy [9–10]. Numerous studies have been performed to modify these macrocyclic compounds with the goal of tuning their properties and optimizing their efficacy for the above applications. Promising results have been achieved for research along this direction.
Non-aggregated phthalocyanines have received considerable attention. These compounds, normally with bulky substituents, possess good solubility, which can facilitate the purification and characterization processes. The non-aggregated nature can also prevent undesirable effects arising from stacking of molecules [11]. For the application of photodynamic therapy, for example, the phthalocyanines should remain in a monomeric state. Molecular aggregation, which is an intrinsic property of these large π–conjugated systems, provides an efficient non-radiative energy relaxation pathway leading to a weaker fluorescence emission, lower triplet population, and shorter lifetimes, thereby reducing the photosensitizing efficiency. We have been interested in this special class of phthalocyanines, in particular those which are soluble in water. Due to hydrophobic interactions, stacking of phthalocyanines in aqueous media is particularly strong. Hydrophilic and non-aggregated phthalocyanines are therefore extremely rare despite their great potential in photodynamic therapy and other photosensitizing applications.
To prepare this novel class of functional materials, we have incorporated sterically demanding dendritic substituents with hydrophilic surface groups onto the periphery of phthalocyanine rings. We have also investigated the aggregation behavior of the resulting dendritic phthalocyanines, mainly by absorption and fluorescence spectroscopy together with laser light scattering, and their interactions with various surfactants and poly(ethylene oxide), which act as disstacking agents. This brief account aims to summarize some of our recent findings in this area.
2 Synthesis of dendritic phthalocyanines
Dendritic phthalocyanines have been little studied [12–20]. During the course of our investigation, Kimura et al. reported the first examples, which contain four poly(amide-ether) dendritic substituents around a zinc(II) phthalocyanine core. The terminal ester groups could be hydrolyzed to give carboxylates making the phthalocyanines soluble in water [12]. The other hydrophilic series was reported by McKeown et al., who incorporated oligo(ethyleneoxy) surface groups on poly(aryl ether) dendrons attached to a metal-free or a silicon(IV) phthalocyanine core [18]. The properties of these compounds in water, however, were not reported. In our study [21], we employed the dendritic phenols 1 (n = 0, 1, 2) reported by Höger et al. [22] as starting materials. The first- and second-generation compounds 1 (n = 1, 2) were prepared from dimethyl 5-hydroxyisophthalate (1, n = 0), the zero-generation analogue, using the Mitsunobu reaction as a key step. Treatment of these phenols with 4-nitrophthalonitrile (2) in the presence of K2CO3 led to nucleophilic aromatic substitution giving the corresponding dendritic phthalonitriles 3 (n = 0, 1, 2) in good yields (Fig. 1). These dinitriles were then treated with Zn(OAc)2·2H2O and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in n–pentanol to give the respective zinc phthalocyanines as mixtures of constitutional isomers. Interestingly, the terminal methoxycarbonyl groups underwent transesterification in n–pentanol giving the n–pentoxycarbonyl analogues 4. Upon treatment with NaOH in a mixed solvent system of THF, MeOH, and water, these compounds were hydrolyzed affording the water-soluble phthalocyanines 5 in almost quantitative yields.
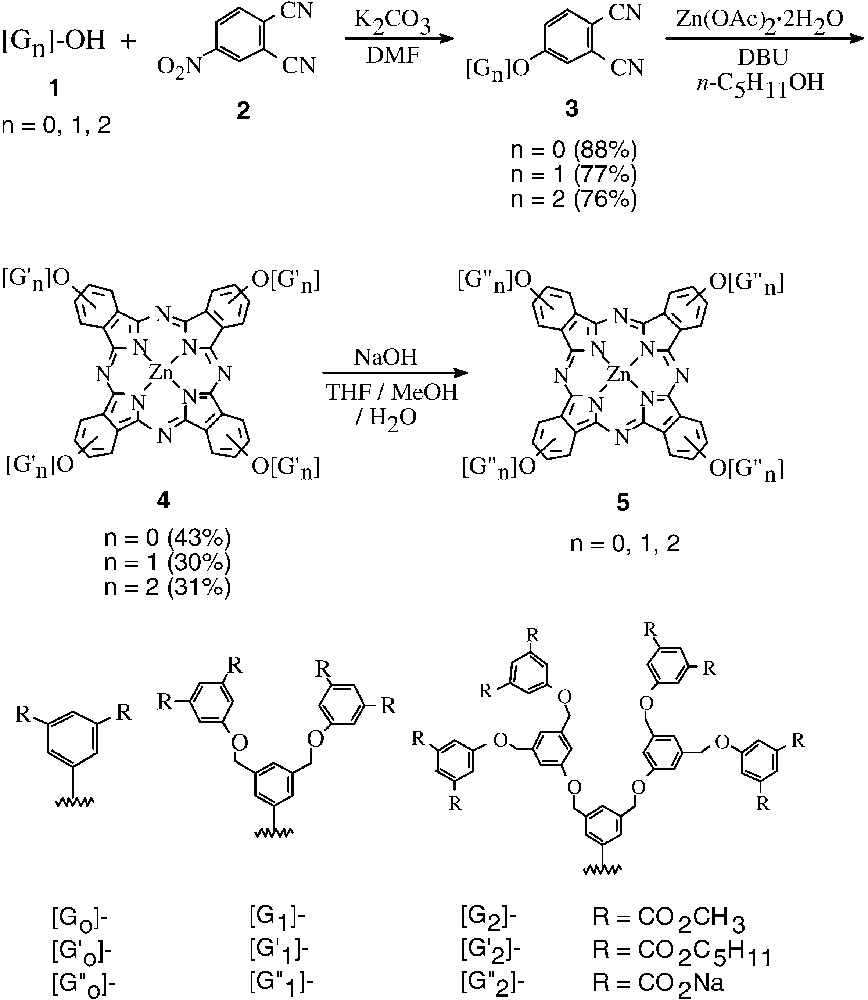
Preparation of dendritic phtalocyanines.
3 Spectroscopic and photophysical properties
The absorption spectra of phthalocyanines 4 in CHCl3 are typical for non-aggregated phthalocyanines, showing the B band at 350–352 nm, Q band at 677–680 nm, together with a vibronic band at 610–614 nm. Upon excitation at 610 nm, these compounds show a fluorescence emission at 685–688 nm with a quantum yield of 0.29–0.33 (with respect to unsubstituted zinc phthalocyanine in 1-chloronaphthalene, Φf = 0.30) and a lifetime of 4.7–4.9 ns. Different generations of dendritic substituents do not induce significant changes of the singlet state properties for this series of complexes. However, the rate of fluorescence quenching, using anthraquinone as the quencher, depends on the size of the dendrons. The Stern–Volmer quenching constant (KSV) is greater for a lower generation dendrimer, which imposes a less hindered environment for the quencher to reach the vicinity of the phthalocyanine core.
This series of dendritic phthalocyanines also exhibits an interesting photoinduced intramolecular energy transfer. Upon excitation at 310 nm, where the aryl dendrons absorb, these compounds emit strongly at 350 nm due to the dendrons, together with an emission at 690 nm due to a singlet-singlet energy transfer from the excited dendrons to the central phthalocyanine core (Fig. 2). Fig. 3 shows the excitation spectra of 4 (n = 0, 1, 2) and the model compound tetrakis(dodecyloxy)phthalocyaninatozinc(II) (6), which does not contain aromatic substituents. It can be seen that as the number of aryl groups increases, more photons are collected and transmitted to the phthalocyanine core. The energy transfer quantum yield increases slightly from 10.3% (for zero-generation analogue) to 13.3% (for second-generation analogue). Although the energy transfer efficiency is moderate for this series of compounds, they represent rare examples of light-harvesting phthalocyanine systems [23–25].
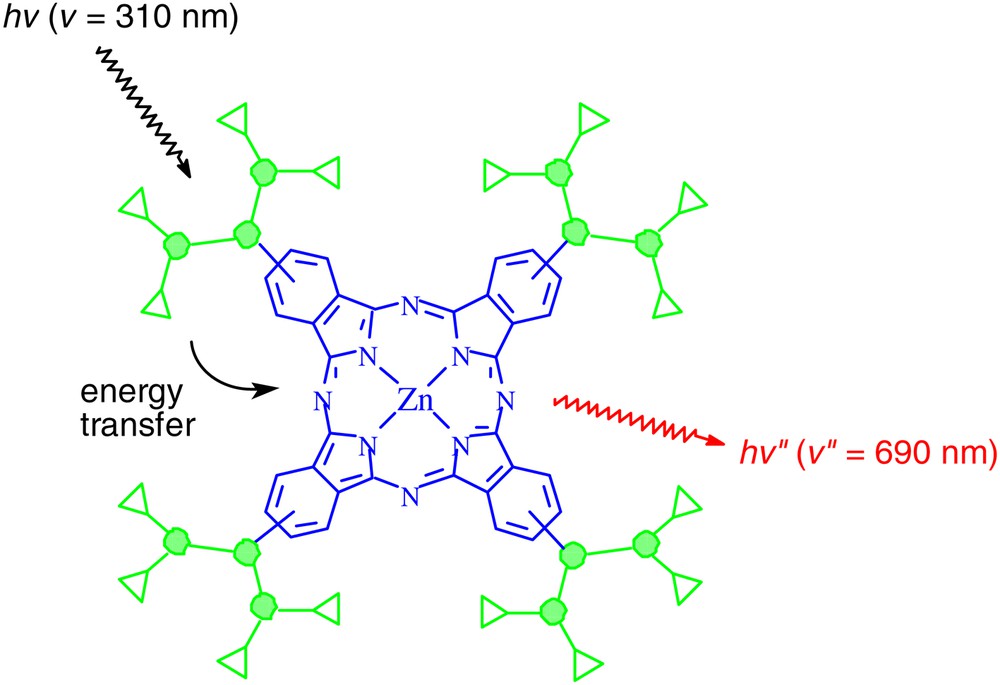
A schematic diagram showing the light-harvesting property of the dendritic phthalocyanines 4 (n = 0, 1, 2).
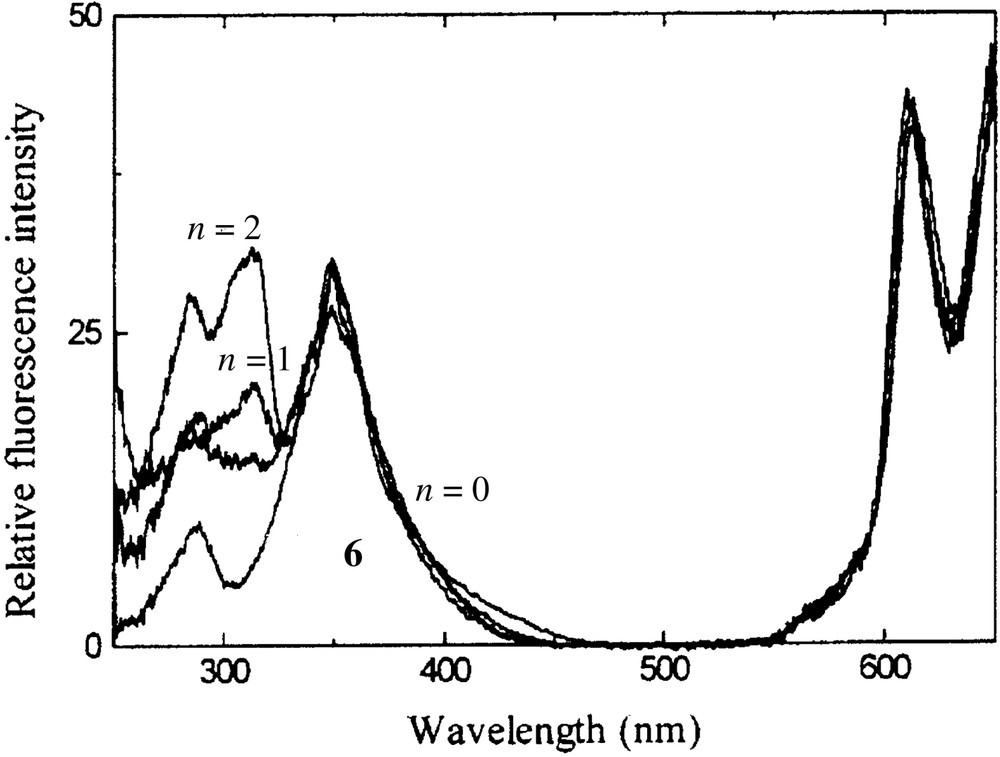
Excitation spectra of 4 (n = 0, 1, 2) and 6 in CHCl3, monitored at 690 nm. The spectra are normalized to a constant intensity at 610 nm.
The zero-generation dendritic phthalocyanine 5 (n = 0), having eight carboxylate moieties, is highly aggregated in water, as shown by the broad and blue-shifted Q band at 638 nm and a very weak fluorescence emission [21]. As the size of the dendrons increases (from n = 0 to 2), the Q band becomes sharpened and shifts to the red. The fluorescence quantum yield also increases from 0.01 to 0.11, showing that the aggregation tendency decreases for higher generation analogues. It is worth noting that fluorescence of phthalocyanines is rarely observed in aqueous media due to their strong aggregation tendency (although some fluorescent dimers have been reported recently [26–28]). The strong fluorescence emission of 5 (n = 2) in water is really remarkable.
The rate of fluorescence quenching for this series of water-soluble phthalocyanines depends on the size of the dendritic substituents as described above and the nature of the quenchers. The rates are tremendously fast when the cationic quencher meso-tetrakis(1-methyl-4-pyridium)porphyrin [H2(TMePyP)]4+ is used. It is likely that a static quenching mechanism is taken place in which a non-luminescent complex is formed between the fluorophore and the quencher held by electrostatic forces. The fluorescence intensity of the second-generation analogue 5 (n = 2) remains virtually unchanged upon addition of sodium picrate, showing that the large number of negatively charged terminal groups in 5 (n = 2) almost completely block the approach of the anionic quencher.
4 Interactions with surfactants
The aggregation behavior of these water-soluble phthalocyanines varies upon interactions with surfactants [29]. The changes can be monitored readily by absorption and fluorescence spectroscopy as the monomeric and aggregated species exhibit very different spectral properties [11]. The first-generation compound 5 (n = 1), for example, is significantly aggregated in water as shown by the broad and blue-shifted Q band at 639 nm. This band shifts gradually to the red (680 nm) and becomes sharpened upon addition of n-hexadecyltrimethylammonium bromide (CTAB), eventually giving a typical spectrum for monomeric phthalocyanines. The fluorescence intensity of this compound also increases steadily with increasing the concentration of CTAB. These observations show that the cationic surfactant CTAB can prevent the molecular aggregation of 5 (n = 1) favoring the formation of monomeric species. Similar effects have been reported for tetra-sulfonated and carboxylated phthalocyanines [30–32], but a higher concentration of surfactant [usually above its critical micelle concentration (CMC)] is required. This indicates that the bulky dendritic substituents have supplementary effects to inhibit the aggregation of the phthalocyanine core.
The zero-generation analogue 5 (n = 0) behaves similarly, except that a higher concentration of CTAB is required to attain a similar effect. The spectral properties of the second-generation compound 5 (n = 2), however, are rather insensitive to CTAB. It is likely that the sterically hindered second-generation dendrons and the additional negative charges are effective to disrupt the π–π stacking of the macrocycles even in the absence of surfactants.
The non-ionic surfactant Triton X-100 is also capable of relieving the aggregation of 5 (n = 1), as shown by the stronger Q band and fluorescence emission during titration with this surfactant. The effects, however, are somewhat weaker than those induced by the cationic surfactant CTAB. The anionic surfactants sodium dodecyl sulfate (SDS) and sodium bis(2-ethylhexyl)sulfosuccinate (AOT) virtually do not alter the spectral properties of this compound. These results show that electrostatic interactions between the anionic terminal groups of the dendrimers and the cationic head of CTAB play an important role in promoting the monomer formation. This is supported by fluorescence quenching experiments using [H2(TMePyP)]4+ as the quencher. The quenching rate decreases remarkably with increasing concentration of CTAB, suggesting that there is a competition between the cationic quencher and CTAB for the anionic dendrimer surface so that fewer molecules of the quencher are adsorbed onto the dendrimer surface, resulting in a slower fluorescence quenching.
We have also studied the interactions of 5 (n = 1) and CTAB by dynamic laser light scattering [29]. For all the 5 (n = 1)/CTAB complexes formed in various concentrations of CTAB, the intensity-weighted distribution of the hydrodynamic radius shows two forms of aggregates. The larger aggregates are extremely small in number showing that they are much less thermodynamically stable. As shown in the number-weighted distribution (Fig. 4), the average hydrodynamic radius of the smaller aggregates decreases significantly (from 14.1 to 3.2 nm) with increasing the concentration of CTAB (from 0 to 3.2 × 10–3 mol dm–3). Fig. 5 shows a schematic diagram depicting the structural changes for the 5 (n = 1)/CTAB complexes upon addition of CTAB. Addition of a small amount of CTAB partially disrupts the phthalocyanine aggregates by electrostatic forces forming smaller aggregates. The π–π-stacking tendency of the rings is also relieved by the non-polar tail of the surfactant. When the concentration of CTAB reaches its CMC (ca. 2.6 × 10–4 mol dm–3), micelles of CTAB are formed of which the surfaces are adsorbed by the anionic macrocycles as shown in Fig. 5. This mode of interaction is supported by the fact that the average hydrodynamic radius of these aggregates (6.3 nm) is close to the sum of that of CTAB micelles [3.4 nm, measured in the absence of 5 (n = 1)] and the estimated dimension of 5 (n = 1) (ca. 3 nm by molecular modeling). Upon further addition of CTAB, more CTAB micelles are formed and the average number of phthalocyanine attached to each micelle decreases. The average hydrodynamic radius of these aggregates (3.2 nm) is very close to that of CTAB micelles, suggesting that the anionic macrocycles, mainly in monomeric form, are adsorbed onto the cationic micelle surfaces in a sideways manner (Fig. 5).

Number-weighted distribution of the hydrodynamic radius of the 5 (n = 1)/CTAB complexes at a fixed concentration of 5 (n = 1) at 1.0 × 10–4 mol dm–3 in 0.01 mol dm–3 NaBr aqueous solutions. [5 (n = 1)]:[CTAB] = 1:0 (□), 1:1 (○), 1:2.6 (▵), 1:10 (◊), 1:32 (▿); + denotes pure CTAB.
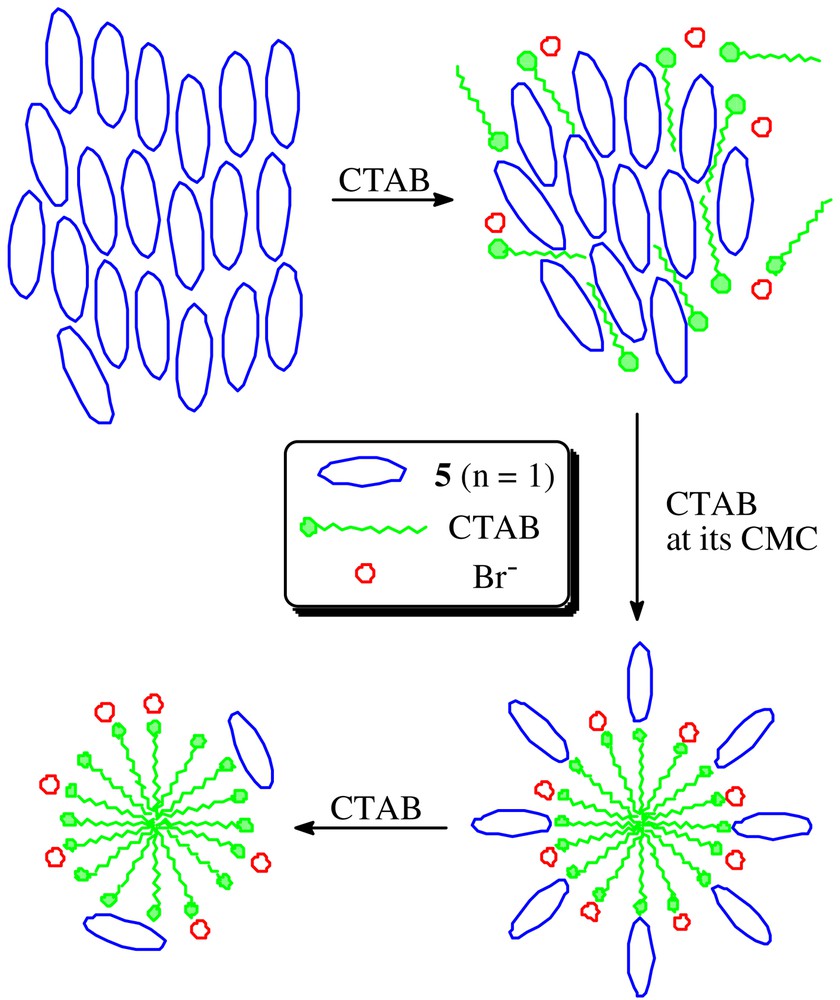
A schematic diagram showing the structural changes for the 5 (n = 1)/CTAB complexes upon addition of CTAB.
5 Interactions with poly(ethylene oxide)
Although CTAB is an effective agent to promote monomerization of the water-soluble phthalocyanines 5 (n = 0, 1, 2), for biomedical applications, biocompatible disstacking agents are preferred to minimize undesirable side effects. We therefore have also investigated the interactions of the first-generation dendritic phthalocyanine 5 (n = 1) and poly(ethylene oxide) (PEO) [33,34], which is a well-known clinically used drug carrier [35]. Fig. 6 shows the hydrodynamic radius distributions f(Rh) of 5 (n = 1) and its PEO complexes. In the absence of PEO, two peaks can be observed showing the existence of two kinds of aggregates with different size. The smaller one (Rh ≈ 7 nm) can be attributed to a phthalocyanine dimer, which is a very common form of aggregate [28]. As shown in Fig. 6, the larger aggregates become smaller upon addition of PEO with different molecular weights. Generally, the longer the PEO chains, the higher the efficiency to disstack the aggregates. The narrowly distributed 5 (n = 1)/PEO10000 (PEO with a molecular weight of ≈ 10 000) complex is larger than the PEO2000 analogue simply because PEO10000 is about five times longer than PEO2000.
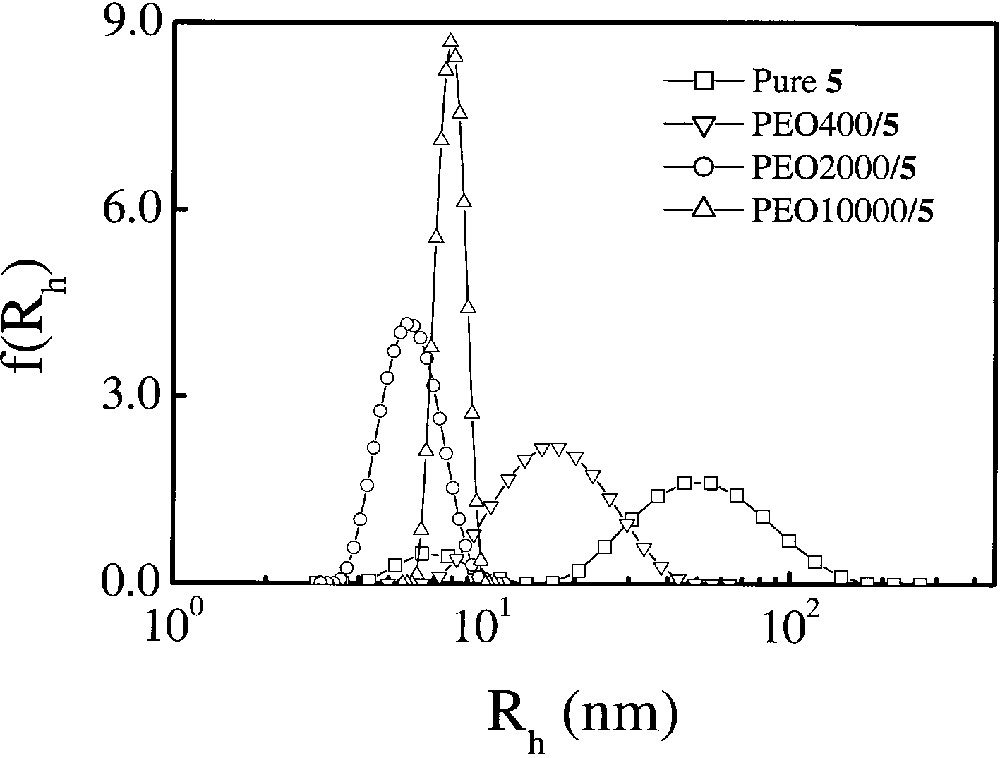
Hydrodynamic radius distributions of 5 (n = 1)/PEO complexes in water, where [5 (n = 1)] = 1.1 × 10–4 mol dm–3 and [PEO] = 5.0 mg ml–1.
The relief in aggregation tendency can also be monitored by absorption and fluorescence spectroscopy. Upon addition of long PEO (PEO2000 or PEO10000), the characteristic blue-shifted Q band at 639 nm due to the dimeric species diminishes, while the Q band at 685 nm ascribed to the monomeric species together with a vibronic band at 617 nm appear [33]. The fluorescence intensity also increases when these agents are added. The changes induced by PEO400 are much less significant. All these results show that PEOs, with sufficiently long chains, are able to disstack the aggregates of 5 (n = 1) in water, presumably by wrapping the macrocycles with their hydrophilic chains. Obviously, longer polymer chains would be more efficient than shorter ones for this purpose.
Fig. 7 shows the variation of the relative Q band absorbance and the relative fluorescence intensity of 5 (n = 1) with the concentration of PEO. For PEO10000, both the absorbance and the fluorescence intensity reach their respective maximum when one molar equiv. of PEO is added. It seems that one PEO10000 chain is sufficiently long to wrap one phthalocyanine molecule to prevent its stacking. For shorter PEO2000, about 25 equiv of PEO is needed to attain similar effects. Hence, for a given PEO weight concentration, PEO10000 is still about five times more effective than PEO2000. The complexation of this phthalocyanine and the two kinds of PEOs is schematically shown in Fig. 8.
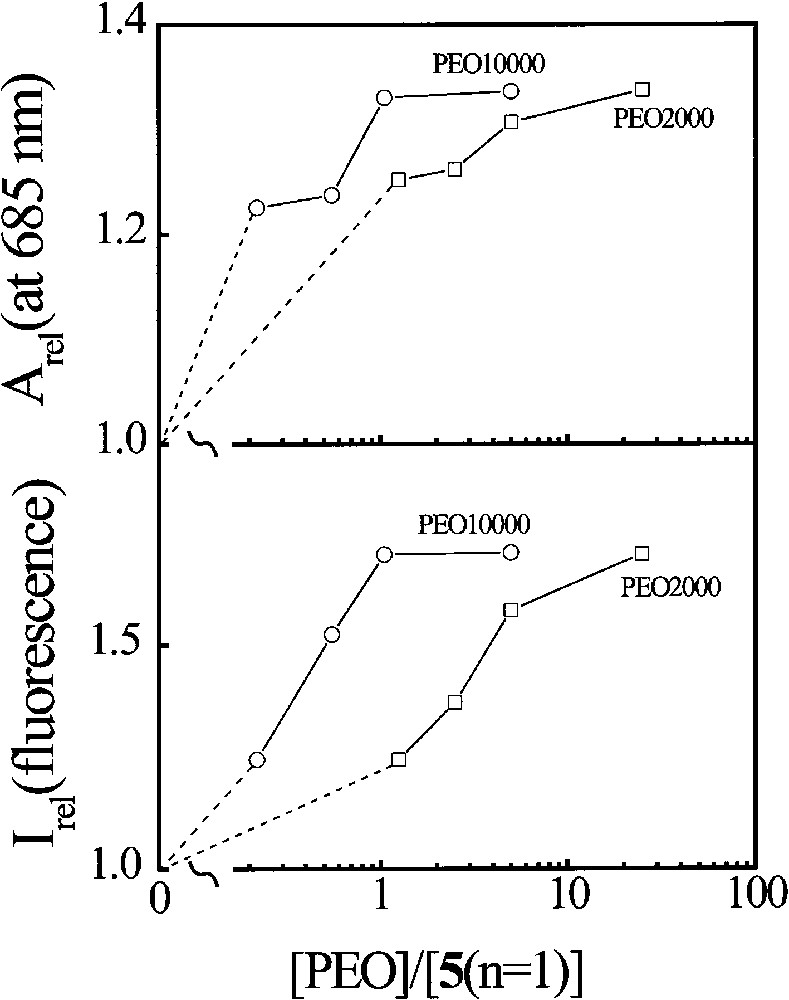
PEO concentration dependence of the relative absorbance at 685 nm and the relative fluorescence intensity of 5 (n = 1)/PEO complexes in water, where [5 (n = 1)] = 1.0 × 10–4 mol dm–3.
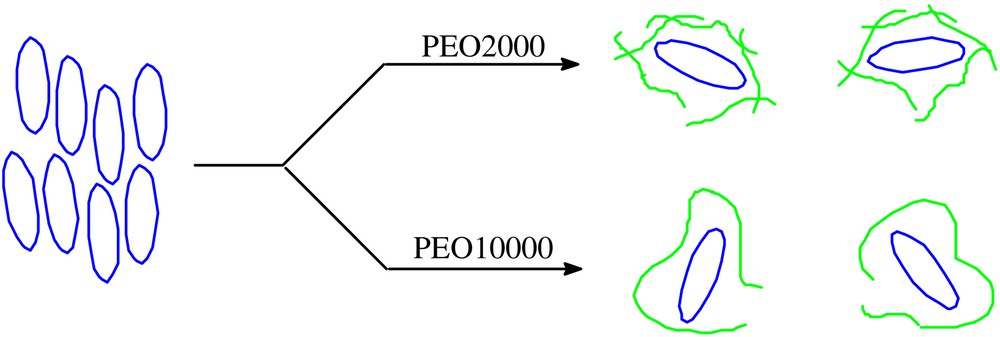
A schematic diagram showing the complexation of 5 (n = 1) and PEO.
Apart from the excited singlet properties of these phthalocyanine/PEO complexes, we have also examined their triplet properties using transient spectroscopy [34]. Upon excitation with a 5-μs pulse laser at 355 nm, a rather weak transient absorption band at ca. 490 nm can be seen for 5 (n = 1) in water. Upon addition of PEO2000 and PEO10000, the intensity increases significantly. For a given PEO molar concentration, the increment is more pronounced for the longer PEO10000. The decay of this absorption is extremely fast in the presence of oxygen, showing that oxygen is an effective quencher for the excited state. This transient absorption is therefore due to the excited triplet state of 5 (n = 1), which exists mainly as monomeric state in the presence of these disstacking agents.
The decay rate also depends on the concentration of the phthalocyanine, showing that there is a self-quenching process: 3ZnPc* + ZnPc → 2 ZnPc. The small self-quenching rate constant (kq) (3.1 × 107 mol–1 dm3 s–1) indicates that the phthalocyanine in its ground state (ZnPc) is a weak quencher for its triplet excited state (3ZnPc*). For a given concentration of 5 (n = 1), both the triplet lifetime (τT) and the value of ΔI490, defined as the difference in intensity at 490 nm at t = t0 (when the concentration of triplet phthalocyanine reaches its maximum) and at t = ∞ (when the intensity becomes steady), increase with the concentration of PEO. It can be attributed to the disstacking of phthalocyanine aggregates upon interactions with PEO. It is likely that the PEO chains are able to wrap the phthalocyanine rings, giving monomeric species. The higher monomer content gives a stronger transient absorption, while the wrapping also hinders the self-quenching process resulting in a longer triplet lifetime. Similarly to the results from steady-state studies, both τT and ΔI490 reach their corresponding maxima when about one molar equivalent of PEO10000 is added. However, this study indicates that for shorter PEO2000 about 8 equiv of PEO are needed to reach the maximum values, suggesting that PEO10000 is only about 1.6 times more effective than PEO2000 to disstack the aggregates of 5 (n = 1).
To further demonstrate that the encapsulation of 5 (n = 1) by PEO can retard the quenching of the excited states, we have also studied the interactions of these systems with KI, which serves as a quencher. For a mixture of equal molar of 5 (n = 1) and PEO10000, addition of KI decreases the initial transient absorption at 490 nm (I490 at t0) and accelerates the relaxation. The decay can be analyzed by a double-exponential equation giving two lifetimes. The faster one (τT ≈ 170 μs) is due to an I––induced quenching, while the slower one (τT ≈ 20 μs) is attributed to the phthalocyanine self-quenching process.
Apart from quenching of the triplet state, we have also examined the quenching of the singlet state of 5 (n = 1) by fluorescence spectroscopy using KI as the quencher. In the absence of PEO10000, the fluorescence intensity decreases significantly upon addition of KI. However, for an equal molar mixture of 5 (n = 1) and PEO10000, the changes are much smaller. The fluorescence quenching data for both cases follow the Stern–Volmer equation, giving the Stern–Volmer quenching constants (KSV) of (6.3 ± 0.1) × 102 and 42 ± 4 mol–1 dm3, respectively. This clearly shows that PEO10000 can effectively wrap and shield the phthalocyanine rings, preventing them from quenching by KI.
6 Conclusion
Phthalocyanines are promising second-generation photosensitizers for photodynamic therapy because of their many desirable features. However, owing to their intrinsic aggregation tendency, in particular in aqueous media, their applications are greatly limited and in many cases various carrier systems are needed to enhance their solubility and prevent their aggregation. Study of hydrophilic and non-aggregated phthalocyanines is still in its infancy. We have developed a phthalocyanine-based dendritic system in which the hydrophilic terminal groups impart water-solubility to these compounds and the bulky substituents inhibit the aggregation to some extent. Monomerization can be achieved through their interactions with surfactants, in particular cationic ones, and PEO. The latter, being a certified biocompatible polymeric material by the Food and Drug Administration of US, is of particular interest. It is envisaged that this kind of material will find its use in photodynamic therapy and the study is in progress.
Acknowledgements
I am indebted to all my students and co-workers who have contributed to our work on dendritic phthalocyanines; their names appear in the references. Special thanks are due to Prof. Chi Wu and his group for their elegant work on the laser light scattering study and the interactions with PEO. Financial support from the Research Grants Council of the Hong Kong Special Administrative Region, China (RGC Ref. No. CUHK 4117/98P) and The Chinese University of Hong Kong (for partial support of a postdoctoral fellowship) is also gratefully acknowledged.